


Abstract
Centrally active muscarinic agonists display pronounced analgesic effects. Identification of the specific muscarinic acetylcholine receptor (mAChR) subtype(s) mediating this activity is of considerable therapeutic interest. To examine the roles of the M2 and M4 receptor subtypes, the two Gi/Go-coupled mAChRs, in mediating agonist-dependent antinociception, we generated a mutant mouse line deficient in both M2 and M4 mAChRs [M2/M4 double-knockout (KO) mice]. In wild-type mice, systemic, intrathecal, or intracerebroventricular administration of centrally active muscarinic agonists resulted in robust analgesic effects, indicating that muscarinic analgesia can be mediated by both spinal and supraspinal mechanisms. Strikingly, muscarinic agonist-induced antinociception was totally abolished in M2/M4 double-KO mice, independent of the route of application. The nonselective muscarinic agonist oxotremorine showed reduced analgesic potency in M2 receptor single-KO mice, but retained full analgesic activity in M4 receptor single-KO mice. In contrast, two novel muscarinic agonists chemically derived from epibatidine, CMI-936 and CMI-1145, displayed reduced analgesic activity in both M2 and M4 receptor single-KO mice, independent of the route of application. Radioligand binding studies indicated that the two CMI compounds, in contrast to oxotremorine, showed >6-fold higher affinity for M4 than for M2 receptors, providing a molecular basis for the observed differences in agonist activity profiles. These data provide unambiguous evidence that muscarinic analgesia is exclusively mediated by a combination of M2 and M4 mAChRs at both spinal and supraspinal sites. These findings should be of considerable relevance for the development of receptor subtype-selective muscarinic agonists as novel analgesic drugs.
A large body of evidence indicates that muscarinic acetylcholine receptor (mAChR) agonists as well as cholinesterase inhibitors can produce powerful analgesic effects in many different species including man (reviewed by Green and Kitchen, 1986; Pert, 1987; Hartvig et al., 1989;Eisenach, 1999). The antinociceptive effects of these agents are predicted to be mediated by activation of mAChRs present at both spinal and supraspinal sites (Green and Kitchen, 1986; Pert, 1987; Hartvig et al., 1989; Eisenach, 1999). Muscarinic agonists have been shown to be as efficacious as full opioid agonists in stringent antinociceptive tests, including the mouse tail-flick assay (Harris et al., 1969; Howes et al., 1969; Green and Kitchen, 1986; Pert, 1987). In addition, several studies (Widman et al., 1985; Swedberg et al., 1997; Petry et al., 1998) suggest that the use of muscarinic analgesics is less likely to lead to addiction, physical dependence, and tolerance, severe side effects that limit the use of classic opioid analgesics. Identification of the mAChR subtype(s) mediating these potent analgesic effects is therefore of considerable therapeutic interest.
Molecular cloning studies have revealed the existence of five molecularly distinct mAChR subtypes (M1–M5; Caulfield, 1993;Wess, 1996). Multiple mAChRs have been detected in spinal cord, thalamus, and several other regions of the CNS known to be involved in the transmission of pain impulses (Levey et al., 1991; Levey, 1993;Vilaro et al., 1993; Wei et al., 1994; Wolfe and Yasuda, 1995;Höglund and Baghdoyan, 1997). Pharmacological studies have yielded contradictory results regarding the molecular identity of the mAChR subtypes mediating the analgesic effects of muscarinic agonists. For example, it has been proposed that muscarinic antinociception is mediated by M1 (Bartolini et al., 1992;Ghelardini et al., 2000), M1 and/or M2 (Gillberg et al., 1989; Iwamoto and Marion, 1993), M1 and/or M3 (Naguib and Yaksh, 1997), M2 (Ma et al., 2001), M3 (Honda et al., 2000), or M4 (Shannon et al., 1997; Ellis et al., 1999) mAChRs. It is likely that the limited receptor subtype selectivity of the currently available muscarinic agonists and antagonists that were used in these in vivo studies is the primary reason for these discrepant results (Caulfield, 1993; Wess, 1996). To overcome these difficulties, we and others generated mutant mouse strains in which specific mAChR genes were inactivated via gene targeting techniques (Hamilton et al., 1997; Gomeza et al., 1999a,b; Matsui et al., 2000;Yamada et al., 2001).
We recently demonstrated that the centrally active, nonsubtype-selective muscarinic agonist oxotremorine (administered s.c.) showed clearly reduced analgesic potency in M2 receptor-deficient mice (Gomeza et al., 1999a), suggesting that the M2 receptor subtype plays a key role in mediating muscarinic antinociception. However, we also noted that oxotremorine-dependent antinociception was not abolished in the M2 receptor KO mice (Gomeza et al., 1999a), indicating that non-M2 mAChRs also participate in this activity. Because the M4receptor subtype couples to similar G proteins (Gi/Go family) as the M2 receptor (Caulfield, 1993; Wess, 1996), we speculated that the analgesic responses remaining in the M2 receptor KO mice might be mediated by M4 mAChRs. To test this hypothesis, we generated a mutant mouse strain lacking both M2 and M4 mAChRs (M2/M4 double-KO mice).
In the present study, we demonstrate that M2/M4 double-KO mice do not display any significant analgesic effects after systemic (s.c.), intrathecal (i.t.), or intracerebroventricular (i.c.v.) administration of muscarinic agonists. In contrast, considerable analgesic activity remained in M2 and M4receptor single-KO mice, independent of the route of application. We also noted that the magnitude of analgesic responses remaining in the M2 and M4 single-KO mice was critically dependent on the M2/M4 receptor selectivity profile of the individual agonists used. These results provide unambiguous evidence that activation of both M2and M4 receptors fully accounts for muscarinic-agonist dependent antinociception at both spinal and supraspinal sites.
Materials and Methods
Generation of M2/M4 mAChR Double-KO Mice.
The generation of homozygous M2receptor KO [genetic background, 129/J1 (50%) × CF1 (50%)] and M4 receptor KO [129SvEv (50%) × CF1 (50%)] mice has been described previously (Gomeza et al., 1999a,b). To generate mice deficient in both M2 and M4 mAChRs, homozygous M2receptor KO mice were mated with homozygous M4receptor KO mice. The resulting F1 compound heterozygotes were then intercrossed to generate F2 mice of nine possible M2/M4 genotypes. According to Mendelian inheritance, 1 of 16 of the F2 pups were predicted to be homozygous for both the M2 and the M4 receptor gene disruptions (M2/M4 double-KO mice). Similarly, 1 of 16 of the F2 pups were predicted to carry two copies of the wild-type M2 and M4receptor genes. The M2/M4double-KO mice were interbred to generate mice used for the experiments described in this study. In parallel, the wild-type F2 mice were interbred to obtain wild-type control mice. Thus, both M2/M4 double-KO mice and the corresponding wild-type control mice had an equivalent genetic background [129/J1 (25%) × 129SvEv (25%) × CF1 (50%)]. Mouse genotyping was carried out by Southern blotting and polymerase chain reaction analysis of mouse tail DNA as described previously (Gomeza et al., 1999a,b).
M2 and M4 receptor single-KO mice were maintained by interbreeding homozygous mutant mice of the F2 generation. In parallel, wild-type littermates of the F2 generation were interbred to generate wild-type control mice of the matching genetic background [M2, wild-type and KO, 129/J1 (50%) × CF1 (50%); M4, wild-type and KO, 129SvEv (50%) × CF1 (50%)].
Unless stated otherwise, all experiments were performed during the light cycle using male mice that were at least 8 weeks old. In all experiments, KO mice and age-matched wild-type mice of the proper genetic background were run in parallel. All animal studies were conducted according to the National Institutes of Health guidelines for standard animal care and usage.
Analgesia Assays.
Antinociceptive responses were quantitated by using the tail-flick and hot plate methods, after s.c., i.t., and i.c.v. administration of muscarinic agonists. For analgesia measurements after s.c. administration of drugs, a cumulative dose-response protocol was employed (Wenger, 1980; Adams et al., 1990;Duttaroy et al., 1997). Intrathecal or i.c.v. injections were carried out using a single dose of agonist that induced maximum analgesia in all mice tested (see below) (dose per mouse: oxotremorine, 10 μg; CMI-936 and CMI-1145, 20 μg). These doses were chosen based on pilot studies using several different agonist doses (doses per mouse: oxotremorine, 1, 10, and 20 μg; CMI-936 and CMI-1145, 10, 20, and 50 μg).
The tail-flick test was carried out using a modified version (radiant heat method) of the procedure described by D'Amour and Smith (1941). Briefly, the mouse was placed on the surface of the tail-flick analgesia meter (Pamotor, Burlingame, CA), and radiant heat was applied from a halogen lamp focused on the dorsal surface of its tail (2–3 cm from the base of the tail). Movement of the tail activated a photocell that turned off the stimulus light and stopped a reaction timer that recorded the time. The intensity of the radiant heat was adjusted so that baseline tail-flick occurred within 2 to 4 s. Withdrawal latency was measured immediately before (baseline) and 30 min after administration of drugs (Ellis et al., 1999; Gomeza et al., 1999a,b); a 10-s cut-off time was imposed to prevent tissue damage. The hot plate test (Woolfe and Macdonald, 1944) was performed on the same set of mice using an electronically controlled hot plate analgesia meter (Columbus Instrument, Columbus, OH). Responses were measured after placing mice on a 55°C hot plate before (baseline, 5–10 s) and 30-min after drug injection; the cut-off time was 30 s. Response latency to the first hind-paw response was recorded. The hind-paw response was either a paw lick or a foot shake, whichever occurred first. Mice that failed to respond within the respective cut-off times were defined as “analgesic”.
In the case of the cumulative dose-response procedure, mice were injected s.c. with a low starting dose of drugs (oxotremorine, 0.01 mg/kg; CMI-936 and CMI-1145, 10 mg/kg) and tested 30 min after injection. Mice that were not analgesic (i.e., that responded within the specified cut-off time) were injected with another dose of drug (increment dose) and retested 30 min later, essentially as described byDuttaroy et al. (1997). This procedure was continued until all mice were analgesic (except for the M2/M4 double-KO mice, which showed no antinociception, and for the M4 KO mice after CMI-936 administration due to the toxicity of cumulative CMI-936 doses greater than 680 mg/kg). Cumulative dose (s.c.) ranges were: oxotremorine, 0.01 to 2.62 mg/kg; CMI-936, 10 to 680 mg/kg.; CMI-1145, 20 to 1,000 mg/kg. ED50 values (± S.D.) for cumulative dose-response curves were calculated via nonlinear regression analysis (Prism v. 3.0; GraphPad Software, San Diego, CA).
For control purposes, we also carried out a series of analgesia studies using a more conventional single dose-response protocol. In this protocol, groups of mice (n = 5–8/dose/genotype) were injected (s.c.) once with a single dose of oxotremorine (dose range, 0.01–1.0 mg/kg) or vehicle and tested for antinociception 30 min later in the tail-flick and hot plate tests. ED50values (± 95% confidence intervals) obtained from a single experiment were calculated via nonlinear regression analysis (Prism).
After i.t. and i.c.v. injection of drugs, antinociceptive responses were expressed as percentage of maximum possible effect (%MPE): tail-flick assay, %MPE = 100 × [(postdrug latency − baseline latency)/(10 − baseline latency)]; hot plate assay, %MPE = 100 × [(postdrug latency − baseline latency)/(30 − baseline latency).
Drugs and Drug Administration.
Oxotremorine sesquifumarate was purchased from Sigma (St Louis, MO). CMI-936 and CMI-1145 were synthesized as described previously (Ellis et al., 1999). Oxotremorine was dissolved in 0.9% NaCl, whereas CMI-936 and CMI-1145 were first dissolved in DMSO and then diluted in 0.9% NaCl to reduce the final concentration of DMSO to less than 0.1%. Subcutaneous injections (injection volume, body weight in grams × 0.01 ml) were performed using a 1-cc syringe attached to a 26.5-gauge needle. Intrathecal injections (5 μl/mouse) into the L5/L6 region of the mouse spinal cord were carried out as described previously (Hylden and Wilcox, 1980), using a 30.5-gauge needle attached to 25-gauge tubing that was connected to a Hamilton syringe (0.5 ml). Intracerebroventricular injections (2.5 μl/mouse) were given freehand (Haley and McCormick, 1957) 0.3 mm posterior, 1.0 mm lateral, and 2.5 mm ventral to the bregma below the outer surface of the skull into the right lateral ventricle using a similarly sized needle and syringe as used for i.t. injections. Mice were anesthetized with pentobarbital (25 mg/kg s.c.) and injected i.t. or i.c.v. with muscarinic agonists 25 to 30 min after pentobarbital injection. Mice recovered from the pentobarbital anesthesia within 10 to 15 min after the i.t. or i.c.v. injections of muscarinic agonists. In control experiments, wild-type mice (n = 5/group) were injected i.t. or i.c.v. with vehicle. These control studies showed that the i.t. or i.c.v. injections themselves had no significant effect on response latencies in the two analgesia assays (data not shown).
For control purposes, we also carried out a series of i.c.v. and i.t. injection experiments using inhalational anesthesia (isoflurane/oxygen; 4:96) (Fortec model; Cyprane, Keighley, UK). Specifically, we assessed the analgesic effects of oxotremorine, administered i.t. or i.c.v. (10 μg per mouse), in M2 single-KO and M2/M4 double-KO mice and the two corresponding wild-type control groups (n = 5/group), using isoflurane/oxygen as the anesthetizing agent. For the gas anesthesia procedure, mice were placed in a plastic chamber provided with a nozzle and attached to a vacuum line that delivered isoflurane/oxygen (4:96) at a constant rate. Once anesthetized, mice were removed from the chamber and placed on a plane surface to perform the i.c.v. or i.t. injections. Mice recovered from the gas anesthesia within 3 to 5 min after the i.t. or i.c.v. injections of oxotremorine.
Radioligand Binding Studies.
To determine the overall density of mAChRs in mouse spinal cord, whole spinal cord tissue was removed and frozen immediately on dry ice. Tissue samples were homogenized by hand with 20 strokes of a Dounce tissue grinder in 0.32 M sucrose, 5 mM Tris-HCl, pH 7.5, and 1 mM phenylmethylsulfonyl fluoride. Membranes were prepared, and ligand binding experiments were carried out using a saturating concentration (2 nM) of the nonselective muscarinic antagonist,N-[3H]methylscopolamine ([3H]NMS; 82 Ci/mmol; PerkinElmer Life Science, Boston, MA), essentially as described previously (Dörje et al., 1991; Gomeza et al., 1999a,b). In addition, competition-binding assays were carried out to determine the binding affinities of different muscarinic agonists (oxotremorine, CMI-936, and CMI-1145) at cloned M2 and M4 mAChRs. Membranes were prepared from CHO-K1 cell lines individually expressing the cloned human M2 and M4mAChRs and incubated with eight different concentrations of muscarinic agonist in the presence of 50 pM [3H]NMS, following the protocol described by Dörje et al. (1991). In saturation binding assays, six different concentrations of the radioligand, [3H]NMS, ranging from 10 to 3000 pM, were tested. Binding buffer consisted of 25 mM sodium phosphate, pH 7.4, and 5 mM MgCl2. Incubations were carried out for 3 h at room temperature (22°C). Nonspecific binding was determined in the presence of 10 μM atropine. Competition binding data were analyzed using a nonlinear regression curve fitting procedure (Prism: sigmoidal dose-response, variable slope, bottom plateau = 0). IC50 values were converted toK i values by using the Cheng-Prusoff equation (Cheng and Prusoff, 1973).
Immunoprecipitation Assays.
For immunoprecipitation studies, mAChR subtype-specific rabbit polyclonal antisera were raised against nonconserved regions of the third cytoplasmic loops of the mouse mAChR proteins (Gomeza et al., 1999a,b; Yamada et al., 2001), following a procedure similar to that described by Levey et al. (1991). Membrane homogenates were prepared (see above) from different areas of the mouse brain and then incubated for 1 h with 2 nM of the nonselective muscarinic antagonist, [3H]quinuclidinyl benzilate ([3H]QNB; 81.5 Ci/mmol; PerkinElmer Life Science), washed thoroughly, and solubilized with 1% digitonin, followed by immunoprecipitation of solubilized [3H]QNB-labeled receptors with receptor subtype-selective antisera, essentially as described previously (Gomeza et al., 1999a,b; Yamada et al., 2001).
Immunohistochemistry.
Mice were fixed by perfusion with 4% paraformaldehyde. Frozen sections of cervical spinal cord were processed for immuno-peroxidase staining using an M2 receptor-specific rat monoclonal antibody (1:500; Levey et al., 1995) or an M4receptor-specific rabbit polyclonal antibody (0.75 mg/ml; Levey et al., 1991). After incubation with biotinylated secondary antibody (rabbit anti-rat or goat anti-rabbit) and avidin-biotinylated enzyme complex reagent (Vector Laboratories, Burlingame, CA), sections were developed with diaminobenzidine as described in detail previously (Levey et al., 1991, 1995).
Statistics.
Statistical significance between two or more groups was determined by Student's t tests or one-way analysis of variance using post hoc t tests (Bonferroni's method).
Results
Generation of M2/M4 mAChR Double-KO Mice.
To study the roles of M2 and M4 mAChRs in muscarinic agonist-induced analgesia, we generated mice containing targeted disruptions of both the M2 and M4 mAChR genes. Initially, we crossed homozygous M2receptor KO mice (Gomeza et al., 1999a) with homozygous M4 receptor KO mice (Gomeza et al., 1999b) to generate compound heterozygotes (M2 ±, M4 ±). The compound heterozygotes were then intermated to obtain homozygous M2 and M4 receptor mutant mice (M2/M4 double-KO mice). M2/M4 double-KO mice were obtained at a frequency of about 6% (the predicted Mendelian frequency is 1 of 16 or 6.25%), indicating that the absence of functional M2 and M4 receptors did not lead to an increase in embryonic or postnatal mortality. The inactivation of the M2 and M4 receptor genes and the absence of M2 and M4 receptor protein in the M2/M4 double-KO mice was confirmed by Southern and polymerase chain reaction analyses and immunoprecipitation studies (data not shown), respectively, in a fashion identical to that described for the M2and M4 single-KO mice (Gomeza et al., 1999a,b). The M2/M4 double-KO mice showed no obvious morphological or behavioral abnormalities and did not differ from their wild-type littermates in overall health, fertility, and longevity.
We carried out a set of immunoprecipitation studies to verify that inactivation of the M2 and M4 mAChR genes did not lead to compensatory changes in the expression levels of the M1 and M3 receptor subtypes, the two major Gq-coupled mAChRs [M5receptor protein is barely detectable in rat (Yasuda et al., 1993) or mouse brain (M. Yamada and J. Wess, unpublished results)]. Specifically, membrane preparations derived from different areas of the mouse brain (wild-type and M2/M4 double-KO) were incubated with a saturating concentration (2 nM) of the nonselective muscarinic antagonist [3H]QNB to radioactively label all mAChRs. [3H]QNB-labeled receptors were solubilized with 1% digitonin and then immunoprecipitated using M1 and M3 receptor subtype-selective antisera (Yamada et al., 2001). As shown in Fig.1, both antisera immunoprecipitated similar amounts of M1 and M3 receptors in tissues derived from wild-type and M2/M4 double-KO mice, indicating that the lack of functional M2 and M4 receptors did not lead to compensatory changes in the expression levels of the M1 and M3 receptor proteins.

Expression levels of M1 and M3 mAChRs in wild-type and M2/M4double-KO mice as determined in immunoprecipitation studies. Membranes were prepared from the indicated brain regions derived from wild-type and M2/M4 double-KO mice, followed by labeling of mAChRs with 2 nM [3H]QNB and solubilization of labeled receptors with 1% digitonin. [3H]QNB-labeled M1 or M3 mAChRs were immunoprecipitated with receptor subtype-specific rabbit antisera as described underMaterials and Methods. Cx, cerebral cortex; Hc, hippocampus; Str, striatum; OB, olfactory bulb; Cer, cerebellum; BS; brain stem. Data are presented as means ± S.D. (n = 3).
Analgesic Effects after Systemic Administration of Muscarinic Agonists.
In the first set of experiments, wild-type and mAChR mutant mice received s.c. injections of different centrally active muscarinic agonists. Drug-induced antinociceptive responses were quantitated using the tail-flick and hot plate assays (D'Amour and Smith, 1941; Woolfe and Macdonald, 1944), employing a cumulative dose-response protocol (Wenger, 1980; Adams et al., 1990; Duttaroy et al., 1997). The antinociceptive properties of muscarinic agonists were assessed in M2 single-KO, M4 single-KO, and M2/M4 double-KO mice. In each case, wild-type animals of the corresponding genetic background served as controls (see Materials and Methods).
In wild-type mice, the nonselective muscarinic agonist oxotremorine produced profound analgesic effects in both the tail-flick and hot plate tests (ED50, ∼ 0.03–0.18 mg/kg s.c.; Fig. 2; Table1). In M2 KO mice, as reported previously (Gomeza et al., 1999a), the analgesic potency of oxotremorine was significantly reduced in the tail-flick and hot plate assays (13- and 3-fold, respectively) (Fig. 2, A and D; Table1). However, oxotremorine retained the ability to elicit maximum analgesic responses in the M2 KO mice in both tests (Fig. 2, A and D). In contrast, oxotremorine showed similar analgesic potencies in M4 KO mice and the corresponding wild-type control mice (Fig. 2, B and E; Table 1). Strikingly, oxotremorine was completely devoid of antinociceptive activity in M2/M4 double-KO mice in both analgesia tests used (Fig. 2, C and F; Table 1).

Antinociceptive responses of wild-type and mAChR KO mice after systemic (s.c.) administration of oxotremorine. A-C, tail-flick assay. D-F, hot plate assay. M2 KO (A, D), M4 KO (B, E), and M2/M4 double-KO mice (C, F) and the corresponding wild-type (WT) control mice (n = 5–6/group) were injected s.c. with increasing doses of oxotremorine, using a cumulative dose-response protocol. Data are plotted as cumulative oxotremorine dose versus percent mice displaying full analgesia, as described under Materials and Methods. The dose-response curves shown are representative of two independent experiments which gave similar results.
Analgesic potencies of muscarinic agonists administered systemically (s.c.) to wild-type and mAChR mutant mice
To examine whether the results obtained with oxotremorine could be mimicked by other centrally active muscarinic agonists, we carried out analogous studies using two novel muscarinic agonists, CMI-936 and CMI-1145, which are chemically derived from epibatidine (Ellis et al., 1999). A recent study (Ellis et al., 1999) showed that the analgesic effects of these compounds could be partially prevented by injecting mice with the MT-3 snake toxin, which can block M4 receptors with relatively high selectivity (Jolkkonen et al., 1994; Liang et al., 1996), suggesting that the CMI compounds might induce analgesia preferentially via activation of M4 mAChRs. However, the in vitro receptor selectivity profile of these compounds was not reported and the possibility cannot be excluded that metabolic degradation of the snake toxin in vivo might have resulted in peptide fragments capable of blocking non-M4 mAChRs (e.g., M2). To address this question, we analyzed the analgesic properties of CMI-936 and CMI-1145 in M2 KO, M4 KO, and M2/M4 double-KO mice and the corresponding wild-type control animals. In addition, we also characterized the receptor subtype selectivity profiles of the two CMI compounds at cloned M2 and M4 mAChRs (see below).
In wild-type mice, s.c. administration of CMI-936 and CMI-1145 caused dose-dependent antinociceptive effects in both analgesia assays used (range of ED50 values, 48–180 mg/kg s.c.) (Figs.3 and 4; Table 1). In M2 KO mice, as observed with oxotremorine, both drugs showed significantly reduced (∼2–6-fold) antinociceptive potencies (Figs. 3, A and D, and 4, A and D; Table 1). However, in contrast to oxotremorine, which displayed essentially unchanged analgesic activity in M4 KO mice (Fig.2, B and E), CMI-936 and CMI-1145 also exhibited significantly reduced analgesic potencies (∼2–5-fold) in M4 KO mice (Figs. 3, B and E, and 4, B and E; Table 1). Strikingly, the two epibatidine analogs, like oxotremorine, were completely devoid of analgesic activity in M2/M4double-KO mice in both the tail-flick and hot plate assays (Figs. 3, C and F, and 4, C and F; Table 1).

Analgesic responses of wild-type and mAChR KO mice to systemic (s.c.) administration of CMI-936. A-C, tail-flick assay. D-F, hot plate assay. M2 KO (A, D), M4 KO (B, E), and M2/M4 double-KO mice (C, F) and the corresponding wild-type (WT) control mice (n = 5–6/group) were injected s.c. with increasing doses of CMI-936, using a cumulative dose-response protocol. Data are plotted as cumulative CMI-936 dose versus percent mice displaying full analgesia, as described under Materials and Methods. The dose-response curves shown are representative of two independent experiments that gave similar results.

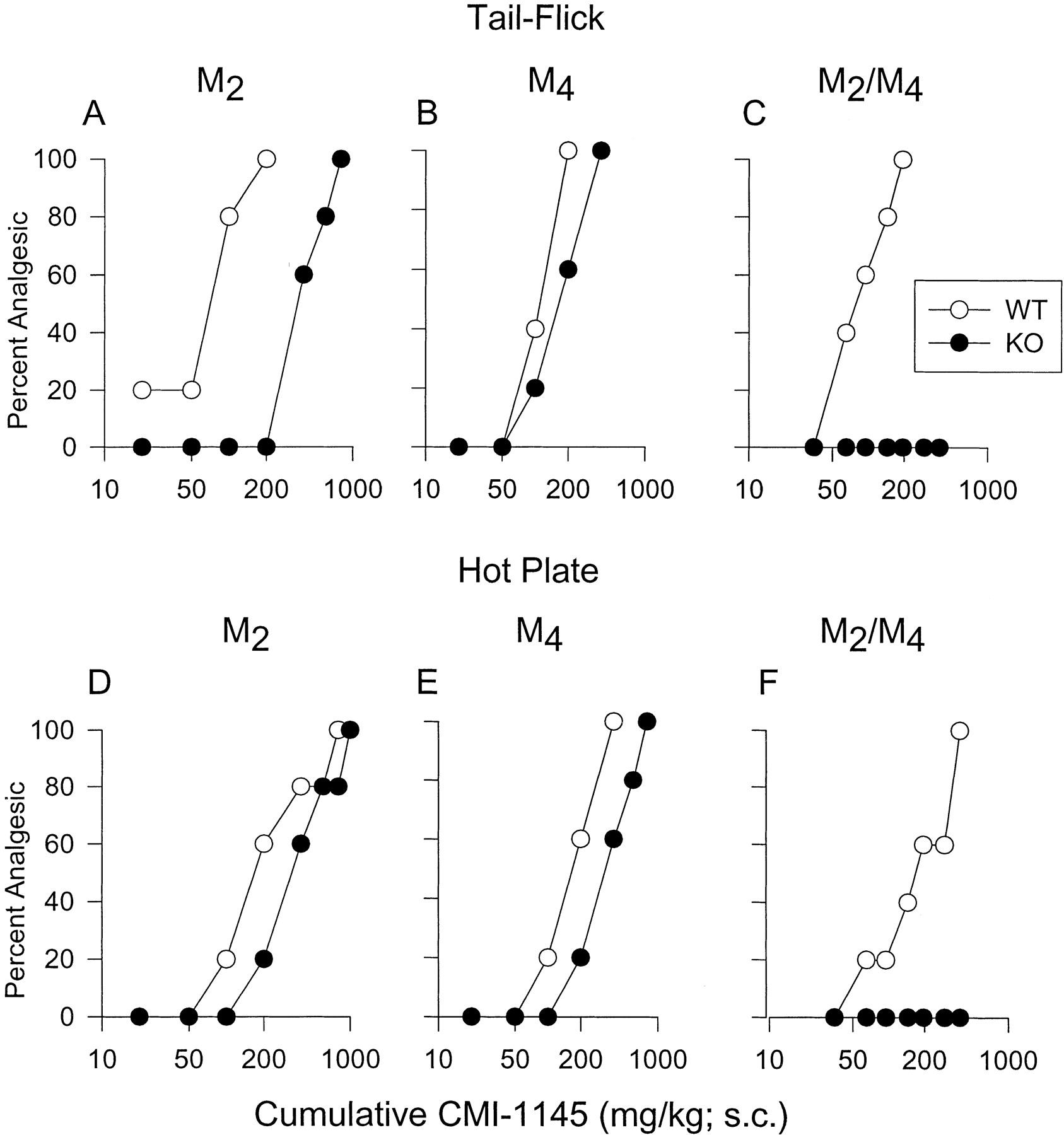
Antinociceptive responses of wild-type and mAChR KO mice after systemic (s.c.) administration of CMI-1145. A-C, tail-flick assay. D-F, hot plate assay. M2 KO (A, D), M4KO (B, E), and M2/M4 double-KO mice (C, F) and the corresponding wild-type (WT) control mice (n = 5–6/group) were injected s.c. with increasing doses of CMI-1145, using a cumulative dose-response protocol. Data are plotted as cumulative CMI-1145 dose versus percent mice displaying full analgesia, as described under Materials and Methods. The dose-response curves shown are representative of two independent experiments which gave similar results.
To verify experimentally that the key results obtained after systemic administration of muscarinic agonists were not affected by the use of the cumulative dose-response protocol, we carried out an additional set of studies using the more conventional single dose-response protocol. Specifically, we employed a standard single dose protocol to assess oxotremorine-mediated analgesic responses in M2single-KO and M2/M4double-KO mice and the two corresponding wild-type groups. For these studies, groups of mice (n = 5–7/dose/genotype) were injected s.c. with different doses of oxotremorine (0.01, 0.03, 0.1, 0.3, and 1.0 mg/kg) or vehicle, and analgesia responses were recorded 30 min later. This analysis showed that oxotremorine was significantly less potent in both the tail-flick (∼13-fold reduction in potency) and hot plate tests (∼4-fold reduction in potency) in the M2 single-KO mice, compared with the corresponding wild-type group. The following ED50values were obtained: tail-flick assay, M2 WT, 0.025 mg/kg [95% confidence interval (CI), 0.011 to 0.086], M2 KO, 0.32 mg/kg (95% CI, 0.21 to 0.63); hot plate assay, M2 WT, 0.12 mg/kg (95% CI, 0.07 to 0.22), M2 KO, 0.49 mg/kg (95% CI, 0.26 to 0.55). However, at the highest dose used (1 mg/kg s.c.), oxotremorine was still able to induce maximum analgesia (100% MPE) in the M2 KO mice. On the other hand, oxotremorine-mediated analgesia responses were totally abolished in M2/M4 double-KO mice (data not shown). These findings obtained by using a standard single dose-response protocol closely mimicked those obtained by application of the cumulative dose-response protocol (Fig. 2; Table 1), indicating that the outcome of the analgesia tests was not affected by the nature of the dosing protocol used.
Moreover, to exclude the possibility that the hybrid background of the mAChR wild-type and KO mice may have affected the outcome of the analgesia experiments, we compared oxotremorine-mediated analgesic responses in the 129SvEv (Taconic, Germantown, NY) and CF-1 (Charles River Laboratories, Wilmington, MA) parental mouse strains, using a standard single dose-response protocol (n = 6–8/dose). These studies showed that oxotremorine ED50values did not differ significantly between the two parental mouse strains in either the tail-flick test [CF-1, 0.037 mg/kg (95% CI, 0.021 to 0.086); 129SvEv, 0.029 mg/kg (95% CI, 0.019 to 0.075)] or hot plate assay [CF-1, 0.059 (95% CI, 0.037 to 0.094); 129SvEv, 0.057 mg/kg (95% CI, 0.041 to 0.091)]. Because the two parental strains did not differ in their sensitivity to muscarinic agonist-induced antinociception, it is very unlikely that the key conclusions drawn from the analysis of the mAChR mutant mice used in the present study were significantly affected by the mixed genetic background of the mice used.
Analgesic Effects after Intrathecal and Intracerebroventricular Administration of Muscarinic Agonists.
We next wanted to assess the relative contribution of spinal and supraspinal mechanisms to muscarinic agonist-induced analgesic effects. Toward this goal, oxotremorine, CMI-936, and CMI-1145 were administered either i.t. or i.c.v. to wild-type and mAChR KO mice, and analgesic responses were measured using the tail-flick and hot plate tests. In wild-type mice, all three agonists (oxotremorine, 10 μg/mouse; CMI-936 and CMI-1145, 20 μg/mouse) produced maximum analgesia in both the i.t. and i.c.v. injection experiments (Figs.5–7). After i.t. or i.c.v. administration, the analgesic activity of oxotremorine was significantly reduced (by about 50–90%) in M2 KO mice in both assay systems used (Fig. 5A), as observed after s.c. administration (see above). On the other hand, oxotremorine-induced antinociceptive responses were not significantly affected in M4 KO mice, independent of the route of application (i.t. or i.c.v.; Fig. 5B). In M2/M4 receptor double-KO mice, oxotremorine, administered i.t. or i.c.v., was completely devoid of analgesic activity (Fig. 5C). The very small residual responses seen in Fig. 5C were not significantly different from the corresponding preinjection values (data not shown).

Antinociceptive responses of wild-type and mAChR KO mice after i.t. or i.c.v. administration of oxotremorine. A, M2 KO and wild-type control mice. B, M4 KO and wild-type control mice. C, M2/M4 double-KO and wild-type control mice. Mice (n = 5–6/group) were injected i.t. or i.c.v. with a single dose (10 μg/mouse) of oxotremorine, and analgesic effects were determined using the tail-flick (TF) and hot plate (HP) assays. Antinociceptive effects were expressed as %MPE as described under Materials and Methods. Two independent experiments gave similar results. Data are plotted as means ± S.D. (*, p < 0.05).
As observed with oxotremorine, i.t. or i.c.v. administration of CMI-936 and CMI-1145 resulted in a significant reduction in the magnitude of analgesic responses (by ∼40–60%) in M2 KO mice in both the tail-flick and hot plate tests (Figs. 6A and 7A). However, in contrast to oxotremorine, the two epibatidine derivatives, when administered i.t. or i.c.v., also showed pronounced reductions in analgesic activity (by ∼50–80%) in M4 KO mice in both assays used (Figs. 6B and 7B). As found with oxotremorine, the analgesic responses to i.t. or i.c.v. CMI-936 and CMI-1145 were totally abolished in M2/M4double-KO mice (Figs. 6C and 7C).

Analgesic responses of wild-type and mAChR KO mice after i.t. or i.c.v. administration of CMI-936. A, M2 KO and wild-type control mice. B, M4 KO and wild-type control mice. C, M2/M4 double-KO and wild-type control mice. Mice (n = 5–6/group) were injected i.t. or i.c.v. with a single dose (20 μg/mouse) of CMI-936, and analgesic effects were determined using the tail-flick (TF) and hot plate (HP) assays. Antinociceptive effects were expressed as %MPE, as described under Materials and Methods. Two independent experiments gave similar results. Data are plotted as means ± S.D. (*,p < 0.05).

Analgesic responses of wild-type and mAChR KO mice to i.t. or i.c.v. administration of CMI-1145. A, M2 KO and wild-type control mice. B, M4 KO and wild-type control mice. C, M2/M4 double-KO and wild-type control mice. Mice (n = 5–6/group) were injected i.t. or i.c.v. with a single dose (20 μg/mouse) of CMI-1145, and analgesic effects were determined using the tail-flick (TF) and hot plate (HP) assays. Antinociceptive effects were expressed as %MPE as described under Materials and Methods. Two independent experiments gave similar results. Data are plotted as means ± S.D. (*,p < 0.05).
To exclude the possibility that the use of pentobarbital as the anesthetic agent had major effects on the outcome of the i.c.v. and i.t. injection experiments, we repeated several key experiments using inhalational anesthesia (isoflurane/oxygen; 4:96). Specifically, we assessed the analgesic effects of oxotremorine, administered i.t. or i.c.v. (10 μg per mouse), in M2 single-KO and M2/M4 double-KO mice and the two corresponding wild-type control groups (n = 5/group), using isoflurane/oxygen as the anesthetizing agent. These experiments showed that the analgesic activity of oxotremorine was significantly reduced (∼65–75% reduction in MPE) in M2 KO mice in both tail-flick and hot plate tests. The following responses were obtained (WT versus M2 single KO): a) tail-flick, i.c.v., 96.5 ± 5.2/36.6 ± 12.2% MPE; i.t., 91.6 ± 10.2/24.3 ± 11.1% MPE; b) hot plate, i.c.v., 100 ± 0/35.0 ± 9.3% MPE; i.t., 87.2 ± 9.4/26.0 ± 15.2% MPE. In M2/M4 double-KO mice, oxotremorine showed only residual analgesic responses (reduction of MPE by ≥90%) which were not significantly different from the corresponding preinjection values (data not shown). The following responses were obtained (WT versus M2/M4 double-KO): a) tail-flick, i.c.v., 100 ± 0/7.3 ± 6.9% MPE; i.t., 96.8 ± 12.2/8.6 ± 5.7% MPE. b) hot plate, i.c.v., 100 ± 0/10.0 ± 4.6% MPE; i.t., 95.7 ± 11.4/6.9 ± 8.0% MPE. These results were similar to those obtained in studies in which pentobarbital was used as the anesthetic agent (Fig. 5), indicating that the key findings obtained in the i.c.v. and i.t. injection experiments were independent of the nature of the anesthetic agent used (pentobarbital versus isoflurane/oxygen).
Agonist Affinities for Cloned M2 and M4mAChRs.
Unlike oxotremorine, CMI-936 and CMI-1145 showed decreased analgesic effects not only in M2 KO but also in M4 KO mice, perhaps because of preferential binding of the CMI agents to M4 receptors. To test this hypothesis, we determined the binding affinities of oxotremorine, CMI-936, and CMI-1145 for human M2and M4 mAChRs individually expressed in stable CHO-K1 cell lines (Dörje et al., 1991). This analysis showed that oxotremorine displayed similar affinities for M2and M4 receptors (Fig.8A; Table2). In contrast, CMI-936 and CMI-1145 displayed markedly higher affinities (15.5- and 6.5-fold, respectively) for M4 than for M2receptors (Fig. 8, B and C; Table 2).

Displacement of specific [3H]NMS binding to cloned M2 and M4 mAChRs by the three muscarinic agonists used in this study. A, oxotremorine. B, CMI-936. C, CMI-1145. Competition binding assays were carried out with membranes prepared from CHO-K1 cell lines stably expressing the cloned human M2 and M4 mAChRs, using 50 pM of [3H]NMS (for details, see Materials and Methods). The specific binding determined in the absence of agonists was defined as 100%. Data are presented as means ± S.D. from two or three separate experiments carried out in duplicate.
Binding affinities of oxotremorine, CMI-936, and CMI-1145 for the M2 and M4 mAChRs
[3H]NMS Binding Studies with Spinal Cord Tissue.
The results of the tail-flick and i.t. injection experiments indicated that spinal cord M2 and M4mAChRs play important roles in muscarinic agonist-induced analgesic responses. To quantitate spinal M2 and M4 receptor densities, membrane homogenates prepared from mouse whole spinal cord were incubated with a saturating concentration (2 nM) of the nonselective muscarinic antagonist [3H]NMS. As shown in Fig.9, spinal cord preparations from M2 KO and M2/M4 double-KO mice showed a striking reduction (by about 90%) in the number of specific [3H]NMS binding sites, compared with preparations from the corresponding wild-type strains. On the other hand, the number of [3H]NMS binding sites detectable in spinal cord tissue from M4 KO mice did not differ significantly from the corresponding wild-type value (Fig. 9). These observations clearly indicate that the M2 subtype represents the predominant mAChR species in mouse spinal cord. In contrast, the levels of spinal cord M4 receptors seem to be too low (<10% of the total mAChR population in mouse spinal cord) to be reliably detectable in [3H]NMS binding assays.

Number of [3H]NMS binding sites in spinal cord tissue from wild-type and mAChR KO mice. Membranes prepared from whole spinal cord of M2 KO, M4 KO, and M2/M4 double-KO mice and their corresponding wild-type (WT) control mice were incubated with a saturating concentration (2 nM) of the nonselective muscarinic antagonist, [3H]NMS, as described under Materials and Methods. Data are presented as means ± S.D. from three separate experiments carried out in triplicate (*,P < 0.05).
Immunohistochemical Analysis of mAChR Expression in Mouse Spinal Cord.
To complement the [3H]NMS binding assays, we also carried out immunohistochemical studies to map the anatomical localization of M2 and, if possible, M4 receptors in the mouse spinal cord. As shown in Fig. 10, use of an M2 receptor-specific rat monoclonal antibody (Levey et al., 1995) indicated that M2 receptors were widely expressed in the spinal cord of wild-type and M4 single-KO mice. M2 receptor-specific fibers and puncta were located in neuropil throughout the gray matter, but staining was most intense in lamina II of the dorsal horn (substantia gelatinosa). In lamina IX of the ventral horn, large M2receptor-specific puncta covered the outer surface of motor neurons. The plasma membrane of these neurons was also immunoreactive. As expected, spinal cord preparations derived from M2 KO and M2/M4 double-KO mice gave only faint background staining, confirming the specificity of the M2 receptor antibody (Fig. 10). In contrast, use of an M4 receptor-specific rabbit polyclonal antibody (Levey et al., 1991), which readily detected M4 receptors in mouse striatum and other forebrain regions (Hohmann et al., 1995), did not reproducibly lead to M4 receptor-specific staining of mouse spinal cord tissue (data not shown). In agreement with the results of the [3H]NMS binding assays (see previous paragraph), this observation indicates that M4receptors are expressed at very low levels in the mouse spinal cord.

Immunohistochemical localization of the M2 mAChR in mouse spinal cord. Slices of mouse cervical spinal cord were incubated with an M2 receptor-specific rat monoclonal antibody (Levey et al., 1995) and processed for immuno-peroxidase staining as described under Materials and Methods (Levey et al., 1995). In preparations from wild-type (WT) and M4 KO mice, M2 receptor-specific staining was detected throughout the dorsal and ventral gray matter. M2 receptor immunoreactivity was particularly rich in the substantia gelatinosa of the dorsal horn (SG) and the outer surface of motor neurons (MN) of lamina IX of the ventral horn. As expected, only background staining was observed in samples derived from M2KO and M2/M4 double-KO mice.
Discussion
To assess the roles of the M2 and M4 mAChRs in muscarinic agonist-induced analgesia, we studied the antinociceptive effects of several centrally active muscarinic agonists in M2 KO, M4 KO, and M2/M4 double-KO mice. Analgesia measurements were carried out using the tail-flick and hot plate assays. Strikingly, the nonselective muscarinic agonist oxotremorine when administered s.c., was completely devoid of analgesic activity in M2/M4 double-KO mice in both assays. As reported previously (Gomeza et al., 1999a), the analgesic potency of oxotremorine was significantly reduced in M2 single-KO mice (Fig. 2, A and D). However, maximum analgesia could still be elicited in M2KO mice by increased doses of oxotremorine (Fig. 2, A and D), indicating that non-M2 mAChRs can also mediate profound antinociception. The complete absence of muscarinic agonist-induced antinociception in M2/M4 double-KO mice indicates that both M2 and M4 mAChRs are involved in mediating muscarinic analgesia and that non-M2/M4 mAChRs do not contribute to this activity. As observed by Gomeza et al. (1999b), oxotremorine showed similar analgesic potencies in M4 single KO and wild-type mice (Fig. 2, B and E; Table 1). This observation is consistent with the concept that oxotremorine-induced analgesia preferentially involves M2 mAChRs and that the presence of the intact M2 receptor signaling pathway in the M4 KO mice is sufficient to trigger potent analgesic effects.
To examine whether the results obtained with oxotremorine are also applicable to other classes of muscarinic agonists, we carried out analogous studies with two novel muscarinic agonists, CMI-936 and CMI-1145 (Ellis et al., 1999), which are oxadiazole derivatives of the nicotinic receptor agonist, epibatidine. When injected s.c. into mice, both compounds displayed robust analgesic effects that could be partially blocked by i.t. administration of the snake toxin, MT-3 (Ellis et al., 1999). Because the MT-3 toxin displays at least 40-fold higher affinity for M4 than for the other mAChR subtypes (Jolkkonen et al., 1994; Liang et al., 1996), this observation suggested (but did not prove) that CMI-936 and CMI-1145 might be endowed with a receptor selectivity profile different from that of oxotremorine. In the present study, we therefore carried out radioligand binding studies to determine the affinities of CMI-936 and CMI-1145 for the cloned M2 and M4 mAChRs (Dörje et al., 1991). In contrast to oxotremorine, which displayed little receptor subtype preference, CMI-936 and CMI-1145 bound to M4 receptors with markedly higher affinities (15.5 and 6.5-fold, respectively) than to M2 receptors (Fig. 8, Table 2). Although the degree of M4 selectivity displayed by these agents is relatively modest, the two CMI compounds nevertheless are important novel pharmacological tools. It should be noted in this context that another muscarinic agonist, McN-A-343, has also been reported to exhibit a certain degree of M4receptor selectivity (Lazareno et al., 1993; Richards and van Giersbergen, 1995).
In contrast to oxotremorine, which displayed a significant reduction in analgesic potency only in the M2 single-KO mice, CMI-936 and CMI-1145, when given s.c., showed reduced analgesic potencies (by about 2- to 6-fold) in both M2 and M4 single-KO mice (Figs. 3 and 4, Table 1), probably reflecting their ability to preferentially bind to M4 receptors. However, like oxotremorine, the two CMI compounds were completely devoid of analgesic activity in the M2/M4 double-KO mice (Figs.3, C and F, and 4, C and F). These data confirm that both M2 and M4 receptors participate in muscarinic agonist-mediated analgesic responses and indicate that the receptor selectivity profile of the specific muscarinic agonist under investigation determines to which extent the two receptors contribute to the observed analgesia response.
To study the relative contribution of spinal and supraspinal pathways to muscarinic agonist-dependent antinociception, we carried out analogous analgesia measurements after i.t. or i.c.v. administration of oxotremorine, CMI-936, and CMI-1145. The results of these studies closely mirrored those obtained after systemic (s.c.) administration of these three agonists. Oxotremorine, when given i.t. or i.c.v. (10 μg/mouse), showed a significantly reduced analgesic response (by ∼50–90%) in M2 KO mice but retained full analgesic activity in M4 KO mice (Fig. 5, A and B). In contrast, the two CMI compounds, administered i.t. or i.c.v. (20 μg/mouse), showed a significant decrease in analgesic activity (by ∼40–80%) in both M2 and M4 single-KO mice (Figs. 6, A and B, and 7, A and B). As observed after systemic administration, all three agonists lacked significant analgesic activity in M2/M4 double-KO mice when administered i.t. or i.c.v. (Figs. 5C, 6C, and 7C). These findings indicate that both M2 and M4 receptors participate in mediating spinal as well as supraspinal analgesic effects and that other mAChRs are not involved in this activity. However, our data do not completely rule out the possibility that stimulation of M1, M3, or M5 mAChRs can also reduce pain sensitivity (at least theoretically) by activating a cholinergic pathway which ultimately leads to stimulation of M2 and/or M4 receptors.
[3H]NMS radioligand binding studies with spinal cord preparations from wild-type, M2 KO, M4 KO, or M2/M4 double-KO mice showed that M2 receptors represent the majority (∼ 90%) of spinal cord mAChRs (Fig. 9). Immunohistochemical studies revealed that M2 receptors are abundantly expressed in lamina II of the dorsal horn (Fig. 10), a region in which the primary nociceptive afferent fibers (Aδ and C fibers) terminate (Yaksh, 1988; Gillberg et al., 1990; Levine, 1998; Snider and McMahon, 1998, 1999). This observation is consistent with immunohistochemical and radioligand binding studies carried out with rat (Höglund and Baghdoyan, 1997; Yung and Lo, 1997) and human (Potter et al., 1996) tissues demonstrating high levels of M2 receptor expression in the superficial layers of the dorsal horn. In both [3H]NMS radioligand binding and immunohistochemical studies, we were unable to reproducibly detect M4 receptors in mouse spinal cord tissue, suggesting that spinal M4 receptors are expressed at very low abundance. In agreement with this observation, pharmacological studies indicate that M4receptors represent only a minor fraction of the total mAChR population in rat (Höglund and Baghdoyan, 1997) and human (Potter et al., 1996) spinal cord. The low abundance of M4receptors in mouse spinal cord may explain why the nonselective muscarinic agonist, oxotremorine, displayed virtually unchanged analgesic activity in M4 single-KO mice (see discussion above). Analogously, the high density of spinal M2 receptors is likely to be responsible for the very pronounced reduction in analgesic potency of systemically administered oxotremorine observed with M2single-KO mice in the tail-flick experiments (Fig. 2, A and Table 1).
Lesion studies suggest that mAChRs in the spinal dorsal horn are localized to the nerve terminals of primary afferent pain fibers (Gillberg and Wiksten, 1986). In agreement with this observation, light and electron microscopic studies have detected M2and M4 receptor immunostaining in small type neurons of rat dorsal ganglia believed to be involved in the transmission of pain stimuli to the spinal cord (Bernardini et al., 1999; Haberberger et al., 1999). In addition, electrophysiological experiments (Bleazard and Morris, 1993), combined with electron microscopic studies (Ribeiro da Silva and Cuello, 1990), strongly suggest that presynaptic mAChRs present on pain fibers terminating in lamina II of the dorsal horn function to inhibit the release of excitatory neurotransmitters required for the efficient propagation of pain signals. However, electrophysiological studies also indicate that inhibitory postsynaptic mAChRs located on spinal dorsal horn projection neurons (Urban et al., 1989) or excitatory mAChRs present on spinal GABA-ergic interneurons (Baba et al., 1998) may also contribute to the inhibition of spinal nociceptive pathways. Taken together, these studies suggest that both presynaptic and postsynaptic mechanisms contribute to the antinociceptive effects mediated by spinal M2 and/or M4 mAChRs. It has been proposed that spinal μ and δ opioid and α2 adrenergic receptors mediate their analgesic effects through similar mechanisms (Siddall and Cousins, 1995; Yaksh, 1999).
The distribution of supraspinal (brain) M2 and M4 mAChRs has been mapped in great detail in previous studies (see, for example, Levey et al., 1991; Levey, 1993;Hohmann et al., 1995; Gomeza et al., 1999a,b). These studies have shown that M2 and M4 receptors are widely expressed throughout the brain, including all major regions predicted to be involved in pain transmission, modulation, and perception (pons/medulla, midbrain, thalamus, cerebral cortex, etc.).
Although spinal M4 receptors are predicted to be expressed at much lower levels than spinal M2receptors, our results suggest that this rather small population of M4 receptors nevertheless can mediate pronounced analgesic effects. M4 receptors, unlike M2 receptors, which are widely expressed both in the body periphery and in the CNS, are primarily expressed in the brain (Levey et al., 1991; Levey, 1993; Vilaro et al., 1993; Yasuda et al., 1993; Wolfe and Yasuda, 1995; Gomeza et al., 1999a,b). Thus, from a clinical point of view, the development of selective M4 receptor agonists as novel analgesic drugs seems particularly attractive. Such agents are predicted to lack significant muscarinic side effects, because the clinically most bothersome unwanted effects of muscarinic agonists, such as changes in heart rate, dry mouth, and impaired smooth muscle activity, are predicted to be mediated primarily by M2 and M3 mAChRs (Caulfield, 1993; Wess, 1996; Eglen et al., 1999). In addition, several lines of evidence (Widman et al., 1985; Swedberg et al., 1997; Petry et al., 1998) suggest that the use of muscarinic agonists for the management of chronic pain may be less likely to lead to tolerance, addiction, and physical dependence, severe side effects associated with the use of classic opioid analgesics. As discussed above, the two epibatidine derivatives, CMI-936 and CMI-1145, showed a moderate degree of M4 receptor selectivity (Table 2). Systematic structural modification of these or other lead compounds should eventually lead to more selective M4 receptor agonists suitable for clinical use.
Acknowledgments
We thank H. Rees for expert technical assistance.
Footnotes
- Received February 21, 2002.
- Accepted July 23, 2002.
-
↵1 Current address: Human Genome Sciences, Inc., Rockville, MD 20850.
-
This research was supported by a Cooperative Research and Development Agreement between the National Institute of Diabetes and Digestive and Kidney Diseases and the Eli Lilly Research Laboratories.
Abbreviations
- mAChR
- muscarinic acetylcholine receptor
- KO
- knockout
- %MPE
- percentage of maximum possible effect
- CMI-936
- 2-exo{5-(3-methyl-1,2,4-oxadiazolyl)}-[2.2.1.]-7-azabicycloheptane
- CMI-1145
- 2-exo{5-(3-amino-1,2,4-oxadiazolyl)}-[2.2.1.]-7-azabicycloheptane
- CHO
- Chinese hamster ovary
- NMS
- N-methylscopolamine
- QNB
- quinuclidinyl benzilate
- CI
- confidence interval
- U.S. Government





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}