


Abstract
The flavin-containing monooxygenases (FMOs) are important for the oxidation of a variety of environmental toxicants, natural products, and therapeutics. Consisting of six family members (FMO1–5), these enzymes exhibit distinct but broad and overlapping substrate specificity and are expressed in a highly tissue- and species-selective manner. Corresponding to previously identified regulatory domains, a YY1 binding site was identified at the major rabbit FMO1promoter, position −8 to −2, two overlapping HNF1α sites, position −132 to −105, and two HNF4α sites, position −467 to −454 and −195 to −182. Cotransfection studies with HNF1α and HNF4α expression vectors demonstrated a major role for each of these factors in enhancing FMO1 promoter activity. In contrast, YY1 was shown by site-directed mutagenesis to be dispensable for basal promoter activity but suppressed the ability of the upstream domains to enhance transcription. Finally, comparisons between rabbit and humanFMO1 demonstrated conservation of each of these regulatory elements. With the exception of the most distal HNF4α site, each of the orthologous human sequences also was able to compete with rabbit FMO1 cis-elements for specific protein binding. These data are consistent with these same elements being important for regulating human FMO1 developmental- and tissue-specific expression.
The flavin-containing monooxygenases (FMOs) (EC 1.14.13.8) are a family of microsomal enzymes important for the oxidative metabolism of a wide range of compounds that possess soft nitrogen, sulfur, selenium, and phosphorous nucleophilic centers. Substrates include dietary components such as trimethylamine and methionine, pesticides such as fonfos and phorate, therapeutic agents such as imipramine, cimetidine, and ketoconazole, and plant alkaloids such as nicotine (for review, see Rettie and Fisher, 1999). Five mammalian FMO isoforms have been identified (FMO1–5), each exhibiting a distinct but unusually broad and overlapping substrate specificity that is partly attributable to the unique catalytic mechanism of the FMO (Poulsen and Ziegler, 1995). These enzymes are expressed at high levels in several tissues and in all animal species examined (Poulsen, 1991). In the human, up to a 10-fold range in interindividual FMO activity has been reported (Overby et al., 1997). However, unlike the cytochrome P450-dependent monooxygenase family, such interindividual variation is unlikely to be caused by differential environmental exposure; with rare exception,FMO expression is not affected by exogenous agents. Expression of the different FMO isoforms is highly tissue- and species-selective, can be affected by endogenous steroids (Rettie and Fisher, 1999), and also is influenced by genetic variability (Cashman et al., 2000; Whetstine et al., 2000). An extensive number of reports have appeared on FMO protein chemistry and overall expression pattern (for review, see Rettie and Fisher, 1999). In contrast and despite the impact differential expression must have on target- and species-selective therapeutic efficacy and toxicant susceptibility, little is known regarding specific molecular mechanisms regulating FMO expression.
Much of the previous work on FMO has been conducted using the rabbit as an experimental model. In this species and in most other mammals, the major adult hepatic isoform is FMO1 (Rettie and Fisher, 1999). In the human, however, FMO1 expression seems restricted to the fetal liver. Yet in both rabbit and human, FMO1 represents a major xenobiotic metabolizing enzyme in the adult kidney and intestine (Yeung et al., 2000). Despite these differences, we have posited that rabbit and human FMO1 share at least some tissue-selective regulatory mechanisms. In the current study, the homeodomain-containing factor HNF1α, the orphan nuclear receptor HNF4α, and the zinc finger protein YY1 are demonstrated to have important roles in controlling rabbit FMO1 expression. Consistent with our hypothesis, we also provide evidence that these same factors are equally important in regulating human FMO1expression.
Experimental Procedures
Materials.
Chemical reagents, cell culture medium, and fetal bovine serum were purchased from Sigma (St. Louis, MO). Restriction endonucleases and DNA modifying enzymes were obtained from Invitrogen (Carlsbad, CA) or New England Biolabs (Beverly, MA). The pGL3 luciferase reporter system and luciferase assay kit were purchased from Promega (Madison, WI). The LumiGal assay system for β-galactosidase activity was obtained from CLONTECH Laboratories, Inc. (Palo Alto, CA). Oligonucleotides were custom synthesized by Integrated DNA Technologies, Inc. (Coralville, IA). [α-32P]Deoxyribonucleotide triphosphates (3000 Ci/mmol) were purchased from PerkinElmer Life Sciences (Boston, MA). The YY1 (C-20), HNF1α (C-19), HNF1β (C-20), HNF4α (S-20), and COUP/TF (T-19) antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The HNF4 expression plasmid pCMVHNF4α (Stoffel and Duncan, 1997) was provided by Dr. Stephen A. Duncan (Medical College of Wisconsin, Milwaukee, WI). The HNF1 expression plasmids pBJ5HNF1α (Kuo et al., 1990), pBJ5HNF1β (Mendel et al., 1991a), and pBJ5DCoH (Mendel et al., 1991b) were generous gifts from Dr. Gerald R. Crabtree (Stanford University School of Medicine, Stanford, CA).
Plasmids.
pRNH493, pRNH498, and the nested deletions derived from pRNH498 contain various rabbit FMO1P0 promoter (the major rabbit FMO1promoter) fragments cloned upstream of the luciferase reporter gene in pGL3Basic and have been described previously (Luo and Hines, 1997). pRNH687 was made by deleting a SstI fragment (FMO1 position −60 to −43)1 from pRNH493. pRNH636 is identical with pRNH687 with the exception that FMO1positions −8 to −6 were mutagenized from CCA to GGT by replacing theNheI/SstI fragment with a synthesized double-stranded oligonucleotide containing the variant sequence. An identical procedure was used to generate pRNH654 from pRNH498 (FMO1 position −2120 to +53), pRNH654.a from pRNH498.a (FMO1 position −176 to +53), and pRNH654.c from pRNH498.c (FMO1 position −348 to +53). To generate probes for electrophoretic mobility shift and chemical modification assays, fragments spanning from FMO1 position −38 to +11 (NheI/SstI), position −161 to −42 (SfcI/SstI), and position −559 to −416 (DraI/BstXI) were isolated from pRNH498, treated with T4 DNA polymerase to prepare flush ends, and cloned into theEcoRV site of pBluescriptII KS−(Stratagene, La Jolla, CA) to generate pRNH584, pRNH593, and pRNH634, respectively. Plasmid pRNH527, containing a rabbit FMO1fragment from position −336 to −162, was described previously (Luo and Hines, 1997).
Cell Culture and Transient Transfections.
The HepG2 human hepatoblastoma cell line (Aden et al., 1979) was a gift from Dr. Barbara Knowles (Jackson Laboratories, Bar Harbor, ME) and was cultured in Eagle's minimal essential medium supplemented with 10% fetal bovine serum. The H441 human bronchioalveolar carcinoma cell line was obtained from the American Type Culture Collection (Manassas, VA) and was cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum. Routine maintenance of both cell lines, transient transfection, and subsequent luciferase and β-galactosidase assays were performed essentially as described previously (Luo and Hines, 1996). Cotransfection with expression plasmids was conducted to test the effect of HNF1 and HNF4 transcription factors on FMO1promoter activity. As controls, parallel transfections were conducted using the empty, parent vectors for these same expression plasmids. For HNF1α and HNF1β cotransfection experiments, an expression plasmid for the HNF1 dimerization cofactor (Mendel et al., 1991b), was included at a molar ratio of 2:1 (HNF1/dimerization cofactor of HNF1). The significance of differences observed in the transient expression assays was tested using analysis of variance followed by a Tukey-Kramer multiple comparisons post hoc test. A p value less than 0.05 was accepted as significant.
Sequence Scanning and Alignment.
Selected sequences were scanned for potential transcription factor recognition sequences using the MatInspector program (version 2.2) and the TRANSFAC 4.0 database (http://transfac.gbf.de) (Quandt et al., 1995). Alignment and determination of sequence identity between the human and rabbitFMO1 sequences was performed with the Align Plus V 4.1 program (Scientific and Educational Software, Durham, NC).
Electrophoretic Mobility Shift Assays.
Nuclear extract was prepared and EMSA was performed as described by Boucher et al. (1993)with slight modifications. Instead of using poly(dI-dC) to block nonspecific binding, 1 to 5 μg of sheared salmon sperm DNA was used for some assays. Approximately 0.1 ng of radiolabeled probe (70,000–80,000 cpm) and 6.5 μg of nuclear extract protein were used in each reaction. Unlabeled, double-stranded oligonucleotides (Table1) were added as competing DNA at a 200-fold molar excess. For supershift assays, 2 μg of antibody was included in the DNA/protein binding reaction. DNA probes were generated by isolating BamHI/HincII orHindIII/PstI fragments from pRNH584, pRNH593, and pRNH634, or a PstI/EarI fragment from pRNH527 (FMO1 position −267 to −158). The fragments were end-labeled by filling-in using Klenow DNA polymerase with [α-32P]dATP or [α-32P]dCTP or both. All EMSA were performed under conditions of probe excess.
Oligonucleotide probes
Chemical Modification Assays.
DNA probes were generated as described above. For modification by methylation using dimethyl sulfate (DMS), 10 ng of DNA was modified as described by Boucher et al. (1993). For modification by ethylation using diethylpyrocarbonate (DEPC), 10 ng of DNA was modified as described by Sturm et al. (1987). DEPC-modified DNA fragments were allowed to anneal in 5 μl of Tris-EDTA buffer, pH 8.0, at 4°C overnight. The modified fragment was then end-labeled as above and interference assays carried out essentially as described by Boucher et al. (1993).
Results
Basal Promoter Analysis.
In a previous study, we were able to localize the rabbit FMO1 P0 minimal promoter to a 37-bp sequence immediately upstream of the most 5′ transcription start site (Luo and Hines, 1997). Conventional TATA box elements or Sp1 sites are not found within this basal promoter, however, sequence analysis revealed two consensus initiator elements (Inr) (YYANWYY) (Javahery et al., 1994), one at the major transcription start site, position +14 to +20, and a second immediately upstream at position −8 to −2. The latter also matches the consensus sequence for the YY1 transcription factor (i.e., 5′-CCATNTT-3′). YY1 is capable of behaving both as a positively and negatively acting transcription factor (Thomas and Seto, 1999). Furthermore, it is able to modulate a diverse number of promoters through binding to both distal regulatory and Inr elements (Shi et al., 1991; Usheva and Shenk, 1996). In the latter role, YY1 is critical for the basal promoter activity of several genes (Basu et al., 1997; Seelan and Grossman, 1997; Janssens et al., 1999; Wong-Riley et al., 2000). However, consistent with its dual role as both a positively and negatively acting factor, YY1 binding to the adeno-associated virus (AAV) P5 Inr represses transcription (Shi et al., 1991). To determine what role YY1 may be playing in regulating rabbit FMO1 expression, we first resolved whether specific YY1 binding was possible on the FMO1P0 promoter using EMSA. When a DNA fragment containing rabbit FMO1 sequences from position −38 to +11 was used as a probe, a single specific DNA/protein complex was observed with nuclear extract from either HepG2 or H441 cells (Fig.1A, lanes 2, 3, and 8). Nuclear protein binding to the probe was competed by an oligonucleotide representing the AAV YY1-dependent initiator element (P5 + 1) (Shi et al., 1991) (Fig. 1A, lane 4), but not by an oligonucleotide representing a mutated YY1 site (Fig. 1A, lane 6). Furthermore, a supershifted complex was observed when YY1 antibody was included in the DNA protein binding reaction (Fig. 1A, lane 5). A DMS modification interference assay was performed to confirm the precise location of the YY1 binding site. As shown in Fig. 1B, methylation of a GG pair on the antisense strand corresponding to the consensus YY1 binding site interfered with the formation of this specific DNA/protein complex.

DNA-protein interactions on the basal rabbit FMO1 P0 promoter. A, an EMSA was used to characterize specific HepG2 and H441 nuclear protein binding to a radiolabeled fragment representing position −38 to +11 of the rabbit FMO1gene. The addition of a 200-fold molar excess of competitive DNA or antibody to the binding reaction is indicated above each lane. A solid arrow indicates the single, specific DNA/protein complex observed, whereas an open arrow indicates the supershifted complex. B, a DMS chemical modification interference assay was used to locate nucleotide residues within the rabbit FMO1 −38 to +11 fragment that are critical for protein binding. Lane G represents the results from a chemical sequencing reaction of the same fragment (guanine residues only). Lane F shows the fractionation pattern observed with the unbound probe recovered from the EMSA gel, whereas lane B depicts the fractionation pattern observed with the protein-bound DNA probe. Residues interfering with protein binding are indicated by black dots and correspond to the boxed residues on the sequence shown.
To determine whether YY1 is necessary for FMO1 basal promoter activity, point mutations were introduced into a plasmid in which the minimal FMO1 P0 promoter (position −41 to +11) directs the luciferase reporter gene (i.e., mutation of CCA (position −8 to −6) to GGT at the YY1 binding site). These changes abolished the ability of this sequence to compete for YY1 binding (Fig. 1 A, lane 6). Interestingly, these mutations had no significant effect on FMO1 P0 basal promoter activity (Fig. 2A). In contrast, when additional 5′ flanking sequences were included in the reporter construct, mutation of the YY1 site resulted in a significant increase in activity (p < 0.01) (Fig. 2B). These data suggest that YY1 is dispensable for basal promoter activity but serves to dampen the ability of upstream elements to enhance FMO1transcription.
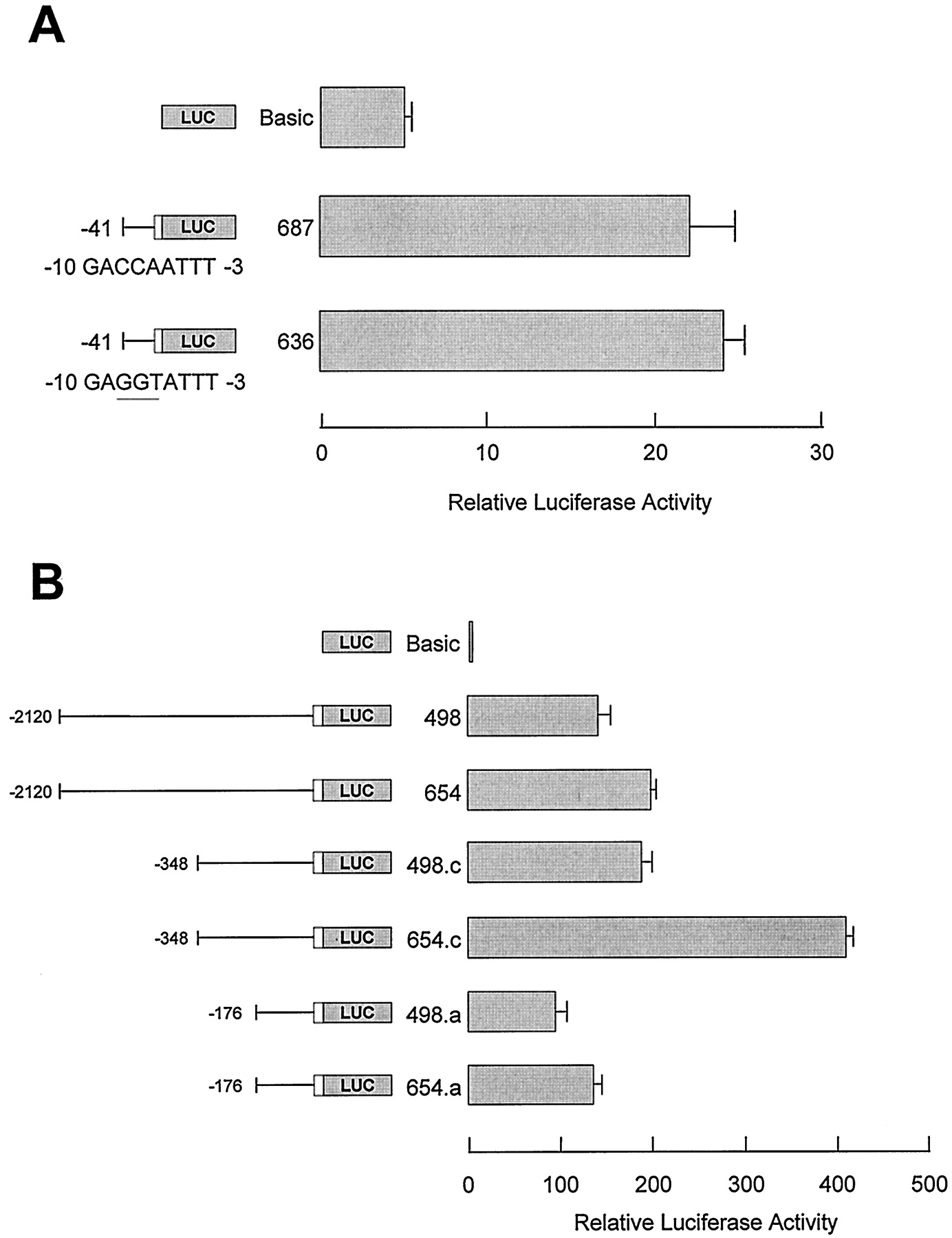
Mutation analysis of rabbit FMO1 P0 basal promoter. A, plasmid constructs containing the luciferase reporter gene directed by either the normal, minimal FMO1P0 promoter (position −41 to +53) or mutated minimalFMO1 P0 promoter (position −8 to −6, CCA–GGT) were analyzed by transient expression in HepG2 cells. B, the effect of the basal promoter mutations on upstream enhancer activity was determined by extending the FMO1 sequences directing luciferase expression to position −2120, −348, or −176 and comparing the activity of the normal (pRNH498 series) or mutated basal promoter (pRNH654 series) constructs after transient expression in HepG2 cells. Salient features of each reporter plasmid construct are shown on the left side of the bar graphs. The activity reported has been normalized for transfection efficiency and protein content and represents the mean ± S.D. from triplicate experiments.
Analysis of the FMO1 5′ Enhancer Domain.
Nested deletion analysis of the 750-bp sequence immediately upstream of the P0 promoter previously demonstrated the presence of multiple positively acting elements important for FMO1regulation (Luo and Hines, 1997). Because FMO1 expression has been characterized as liver-selective, it was hypothesized that one or more liver-selective transcription factors would act through this domain. Indeed, sequence scanning revealed multiple, putative binding sites for members of the HNF and C/EBP transcription factor families (Luo and Hines, 1997). EMSA and chemical modification assays were used to further explore a potential role for these or other factors in regulating FMO1 expression.
Scanning using the MatInspector software program in conjunction with the TRANSFAC 4.0 database suggested the presence of two overlapping, HNF1 binding sites at position −132 to −114, and −119 to −105. The location of these elements is consistent with a substantial increase in activity observed in FMO1 P0 promoter activity when sequences between −176 and −60 were analyzed in a transient expression reporter gene assay (Luo and Hines, 1997). To further explore the importance of these elements, EMSAs were performed with a DNA probe containing FMO1 sequences from position −161 to −42 (Fig. 3A). Using nuclear extract from HepG2 cells, two minor and one major specific DNA/protein complexes were observed (Fig. 3A, lanes 2 and 3). Only the two minor specific DNA/protein complexes were observed with nuclear extract isolated from H441 cells (Fig. 3A, lane 8). Competition for the major specific complex was demonstrated with a double-stranded oligonucleotide representing an HNF1 consensus sequence (Table 1; Fig.3A, lane 4), but not with those representing HNF3, HNF4, HNF6, or C/EBP consensus sequences (Table 1) (data not shown). Furthermore, incubation with an anti-HNF1α antibody resulted in a supershift of this DNA/protein complex. No supershift was observed when this same experiment was repeated with an HNF1β antibody (Fig. 3A, lanes 5 and 6). The precise location of this HNF1α binding site was further refined employing chemical modification interference assays. Interference was observed when adenine residues located at positions −128, −127, −122, −120, −113, and −109 were modified with DEPC on the antisense strand (Fig. 3B, left). Also on the antisense strand, modification with DMS resulted in interference at a single guanine residue within this same element (i.e., at position −132) (Fig. 3B, right).

DNA/protein interaction within the rabbit FMO1 upstream regulatory domain, position −161 to −42. A, an EMSA was used to characterize specific HepG2 and H441 nuclear protein binding to a radiolabeled fragment representing position −161 to −42 of the rabbitFMO1 gene. The addition of a 200-fold molar excess of competitive DNA or antibody to the binding reaction is indicated above each lane. A solid arrow indicates the single, specific DNA/protein complex observed, whereas an open arrow indicates the supershifted complex. B, DEPC (left) and DMS (right) chemical modification interference assays were used to locate nucleotide residues within the rabbit FMO1 −161 to −42 fragment that are critical for protein binding. Lane F shows the fractionation pattern observed with the unbound probe recovered from the EMSA gel, whereas lane B depicts the fractionation pattern observed with the protein-bound DNA probe. Residues interfering with protein binding are indicated by black dots and correspond to the boxed residues on the sequence shown. With DEPC modification (left), we consistently observed double bands for each modified residue. Although this clearly represents ann-1 probe population within the binding reaction, several attempts to eliminate the problem were unsuccessful and the cause remains unknown.
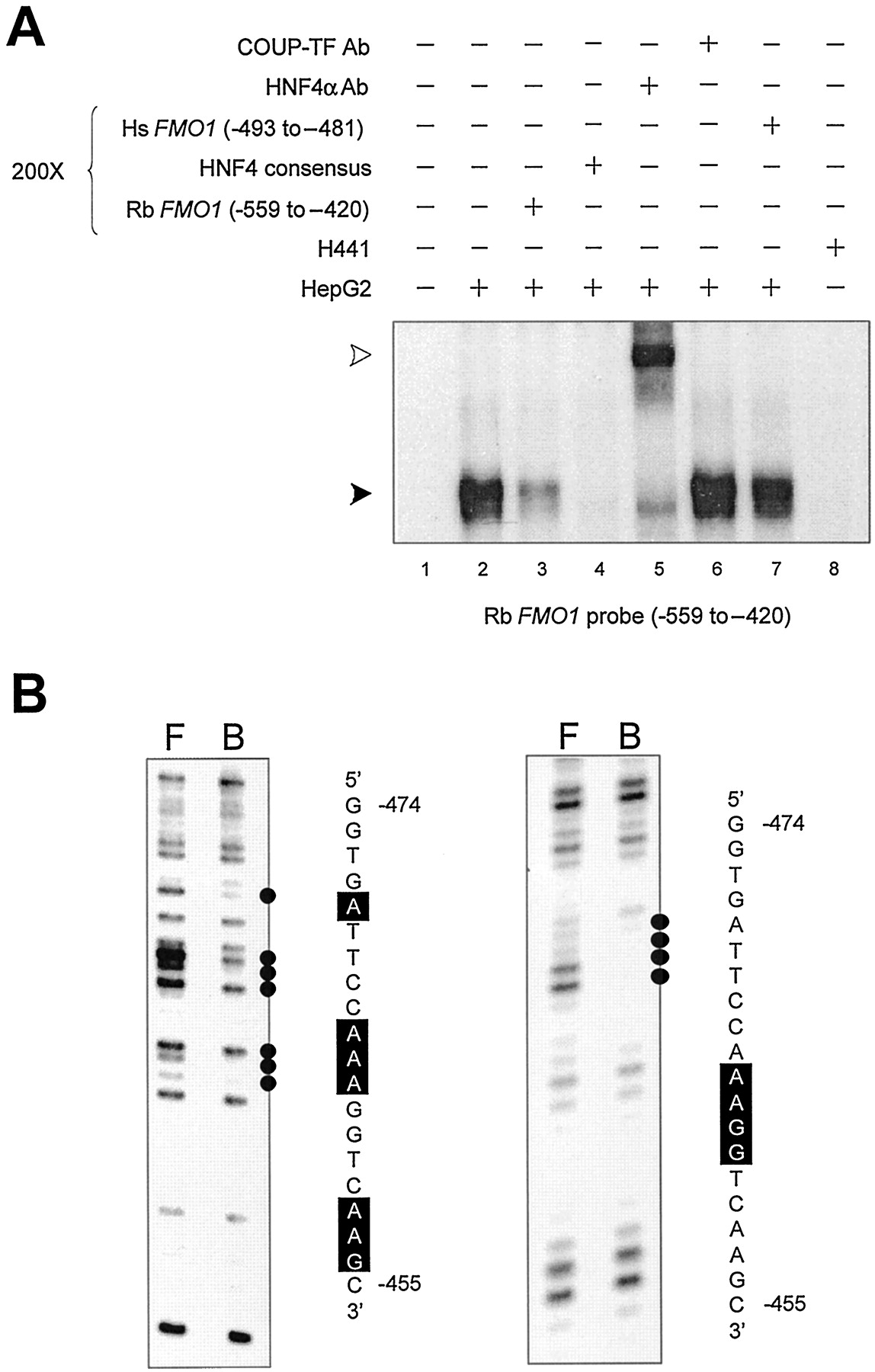
In addition to the HNF1 binding site described above, two HNF4 binding sites were identified within the 750-bp enhancer domain. An EMSA probe representing FMO1 sequences from position −267 to −158 formed a single specific complex that was competed with a 200-fold molar excess of an unlabeled double-stranded oligonucleotide representing an HNF4 consensus sequence (Table 1; Fig.4A, lanes 2–4), but not with oligonucleotides representing HNF1, HNF3, or C/EBP consensus sequences (Table 1) (data not shown). The complex also was supershifted by inclusion of an antibody against HNF4α in the DNA/protein binding reaction but not when a similar incubation was performed with a COUP/TF antibody (Fig. 4A, lanes 5 and 6). No specific DNA/protein complexes were observed using nuclear extract isolated from H441 cells (Fig. 4A, lane 8). DEPC chemical modification of adenine residues at positions −182, −184, and −187 to −189 on the sense strand interfered with specific protein binding (Fig. 4B), consistent with the previous identification of a putative consensus HNF4 binding site at position −195 to −182 using computer assisted sequence scanning. Using a similar approach, a second HNF4 binding site was found using an EMSA probe representing FMO1 sequences from position −559 to −420. Once more, a single specific complex was observed with nuclear extract from HepG2 cells (Fig. 5A, lanes 2 and 3) but not with nuclear extract isolated from H441 cells (Fig.5A, lane 8). The specific DNA/protein complex was competed with an HNF4 consensus oligonucleotide (Table 1; Fig. 5A, lane 4), but not with those representing HNF1, HNF3, or C/EBP (Table 1) (data not shown). The complex also was supershifted by inclusion of the HNF4α antibody in the binding reaction, but not by the COUP/TF antibody (Fig. 5A, lanes 5 and 6). Using a DMS chemical modification interference assay, adenine residues at positions −464 and −463 and guanine residues at positions −462 and −461 on the sense strand were shown to be important for DNA/protein binding (Fig. 5B, right). Interference also was observed at adenine residues, position −470, −465 to −463, −458, and −457, and the guanine residue at −456 using a DEPC chemical modification interference assay (Fig. 5B, left). Thus, combined with previously described reporter gene studies, these in vitro DNA/protein binding assays are consistent with two overlapping HNF1α sites at position −132 to −114, and −119 to −105 and two HNF4α sites at positions −195 to −182 and −467 to −454 being important for regulating rabbitFMO1 expression.

DNA/protein interaction within the rabbit FMO1 upstream regulatory domain, position −267 to −158. A, an EMSA was used to characterize specific HepG2 and H441 nuclear protein binding to a radiolabeled fragment representing position −267 to −158 of the rabbit FMO1 gene. The addition of a 200-fold molar excess of competitive DNA or antibody to the binding reaction is indicated above each lane. A solid arrow indicates the single, specific DNA/protein complex observed, while an open arrow indicates the supershifted complex. B, a DEPC chemical modification interference assay was used locate nucleotide residues within the rabbitFMO1 −267 to −158 fragment that are critical for protein binding. Lane F shows the fractionation pattern observed with the unbound probe recovered from the EMSA gel, whereas lane B depicts the fractionation pattern observed with the protein-bound DNA probe. Residues interfering with protein binding are indicated by black dots and correspond to the boxed residues on the sequence shown.
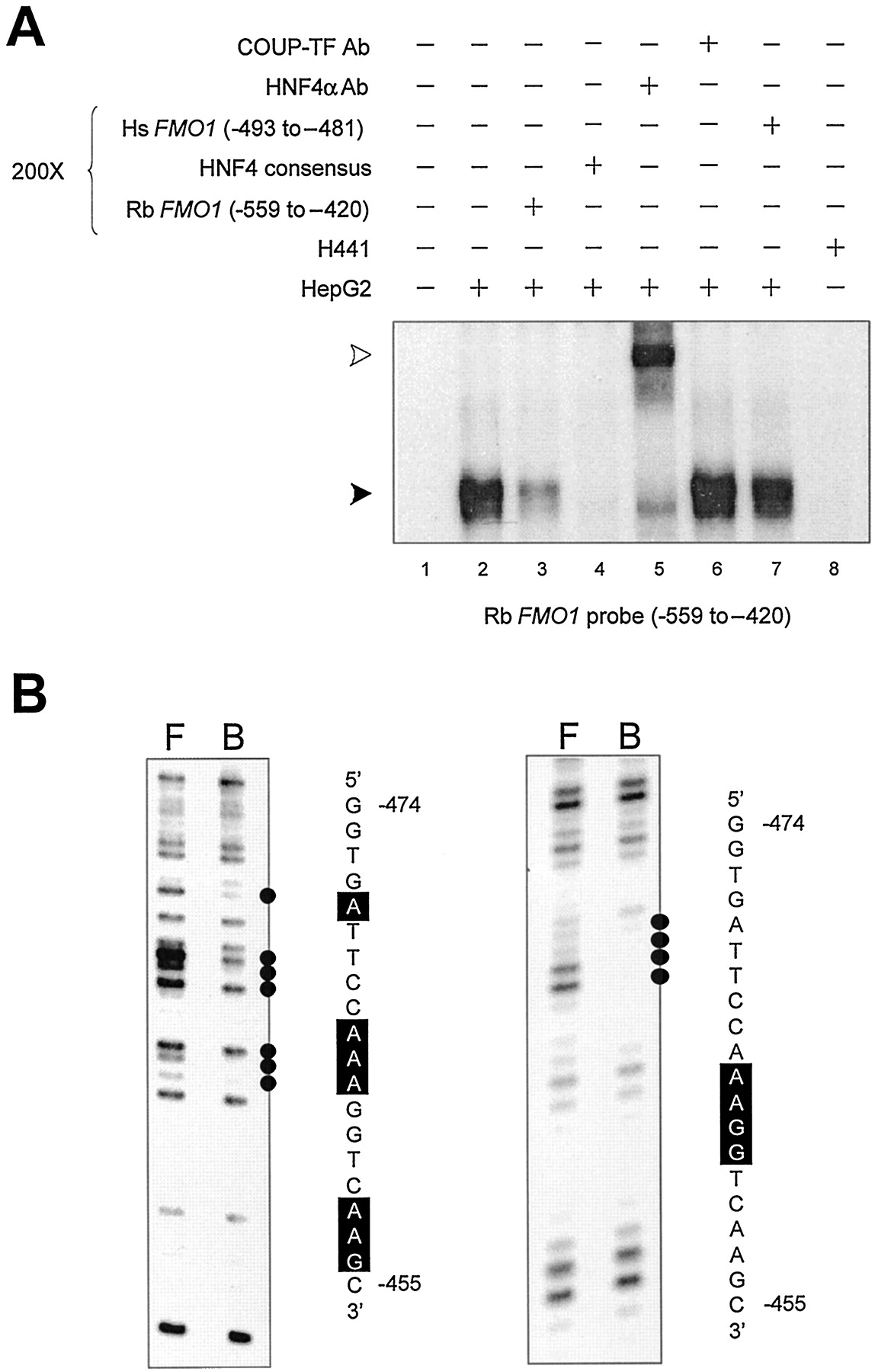
DNA/protein interaction within the rabbit FMO1 upstream regulatory domain, position −559 to −420. A, an EMSA was used to characterize specific HepG2 and H441 nuclear protein binding to a radiolabeled fragment representing position −559 to −420 of the rabbit FMO1 gene. The addition of a 200-fold molar excess of competitive DNA or antibody to the binding reaction is indicated above each lane. A solid arrow indicates the single, specific DNA/protein complex observed, whereas an open arrow indicates the supershifted complex. B, DEPC (left) and DMS (right) chemical modification interference assays were used locate nucleotide residues within the rabbit FMO1 −559 to −420 fragment that are critical for protein binding. Lane F shows the fractionation pattern observed with the unbound probe recovered from the EMSA gel, whereas lane B depicts the fractionation pattern observed with the protein-bound DNA probe. Residues interfering with protein binding are indicated by black dots and correspond to the boxed residues on the sequence shown.
Functional Analysis of Putative HNF Elements.
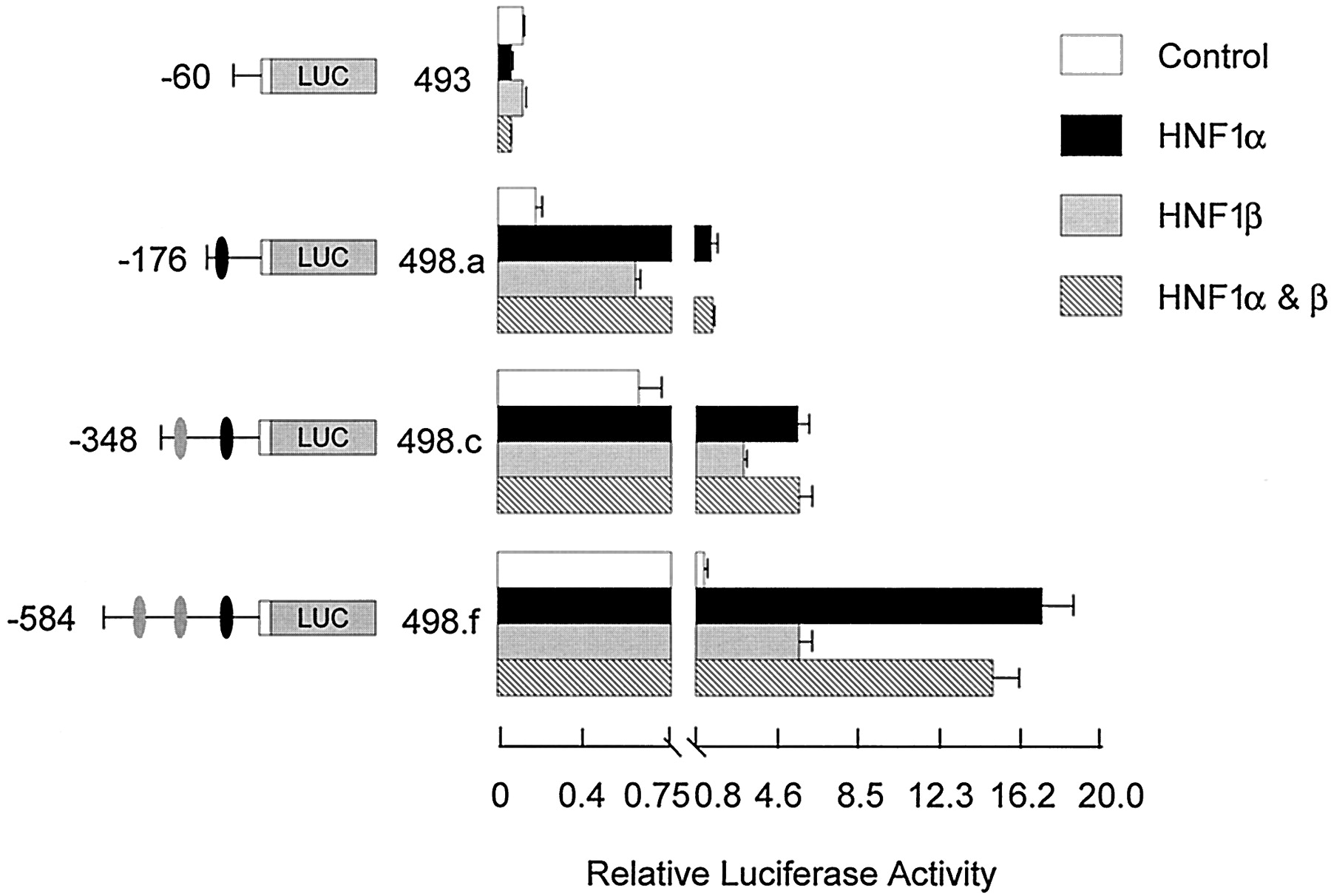
To verify the functional significance of the identified HNF1α and HNF4α elements in regulating rabbit FMO1 expression, transient expression studies were performed after cotransfecting HepG2 cells with various reporter gene constructs and HNF expression plasmids. Results obtained for both HNF1α and HNF1β are shown in Fig.6. Neither HNF1α, HNF1β, nor both factors together had any effect on the activity of the FMO1P0 basal promoter (pRNH493, FMO1sequences from position −60 to +53). In contrast, extending theFMO1 sequences to position −176 (pRNH498.a, FMO1sequences from −176 to +53), which includes the HNF1α sites identified by in vitro DNA/protein binding assays, resulted in a significant 9-fold stimulation of luciferase expression with HNF1α (p < 0.001) but only a 4-fold stimulation with HNF1β (p < 0.05). Because HNF1α and HNF1β can form heterodimers, cotransfection with both expression plasmids was performed, but no further increase in activity was observed over that seen with HNF1α alone. Extending the FMO1 sequences to position −348 [i.e., including both the putative HNF1α and proximal HNF4α sites (pRNH498.c)] resulted in a 3.5-fold increase in control activity, most probably due to the endogenous HNF4α present in HepG2 cells. However, no further stimulation was observed by cotransfection with either HNF1 expression vector (compare pRNH498.c and pRNH498.a plus and minus cotransfection with HNF1α). Inclusion of the putative distal HNF4α site (pRNH498.f; FMO1 sequences from position −584 to +53) did result in a further increase in activity with HNF1α [i.e., 14-fold over the control (p < 0.001)].

Functional analysis of the FMO1 regulatory domains in response to cotransfection with HNF1 expression vectors. The functional significance of identified HNF1 binding sites was analyzed by cotransfection of specific FMO1 luciferase reporter constructs and various HNF1 expression vectors into HepG2 cells with subsequent transient expression analysis. Salient features of each reporter plasmid construct are shown on the left side of the bar graph. The black oval represents the putative HNF1α elements, position −132 to −105, whereas the two gray ovals represent the putative HNF4α elements, position −467 to −454 and −195 to −182. Cotransfection with the parent expression vector (control), HNF1α, HNF1β, or both HNF1α and HNF1β expression vector is indicated by the legend. The activity reported has been normalized for transfection efficiency and protein content and represents the mean ± S.D. from triplicate experiments. Note the change in scale after the break on thex-axis.
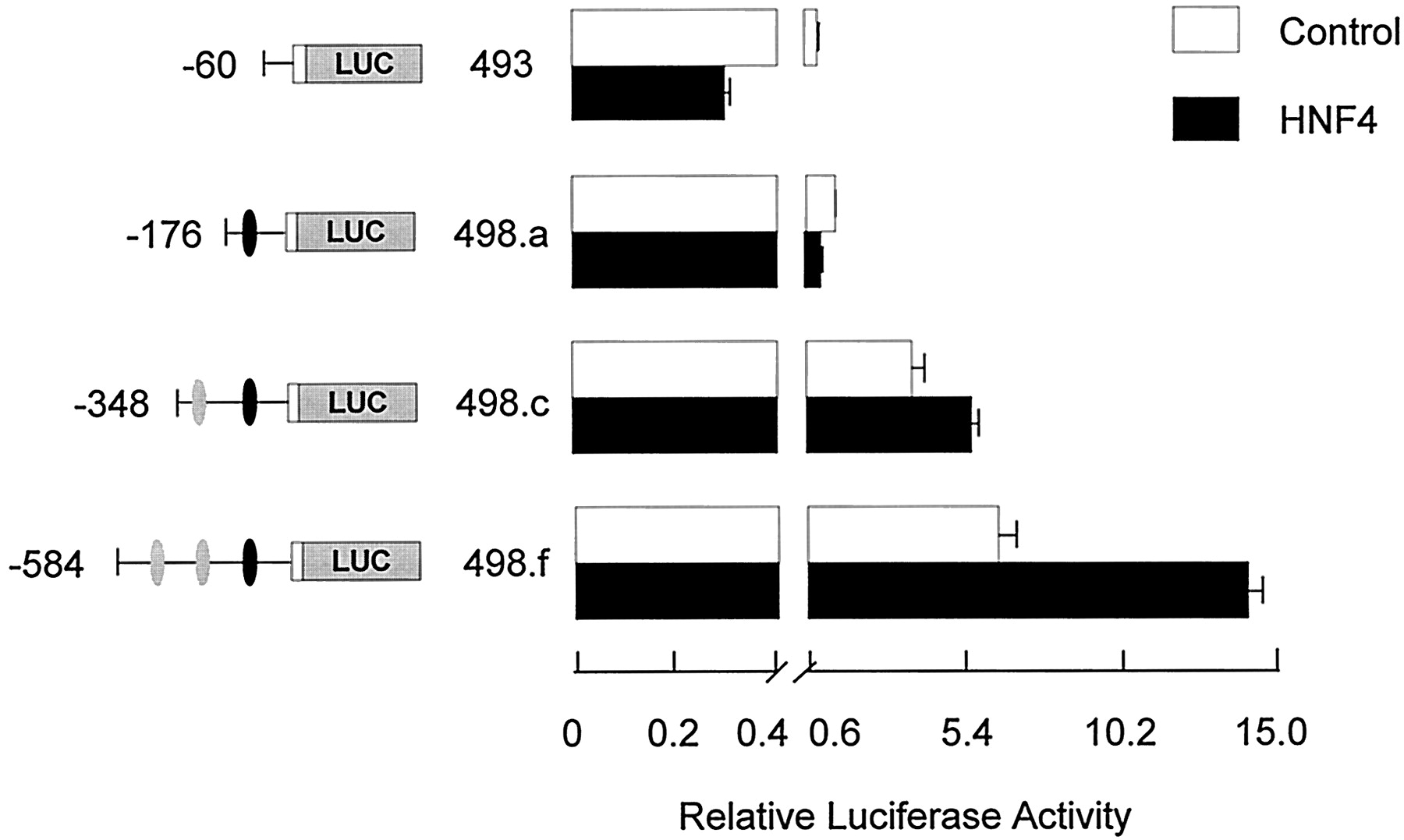
Similar experiments were conducted with an HNF4α expression vector to test the functional significance of the putative elements at position −195 to −182 and −467 to −454 (Fig.7). Cotransfection with the HNF4α expression plasmid had no effect on luciferase activity driven by theFMO1 basal P0 promoter (pRNH493;FMO1 sequences from −60 to +53) or that observed by including the HNF1α sites at position −132 to −105 (pRNH498.a;FMO1 sequences from position −176 to +53). A 1.5-fold enhancing effect was observed with HNF4α by including the putative proximal HNF4α site at −195 to −182 (pRNH498.c; FMO1sequences from position −348 to +53) (p < 0.01), which was increased by an additional 2.4-fold by inclusion of the second, putative HNF4α site at −467 to −454 (pRNH498.d;FMO1 sequences from position −491 to +53) (p < 0.001).
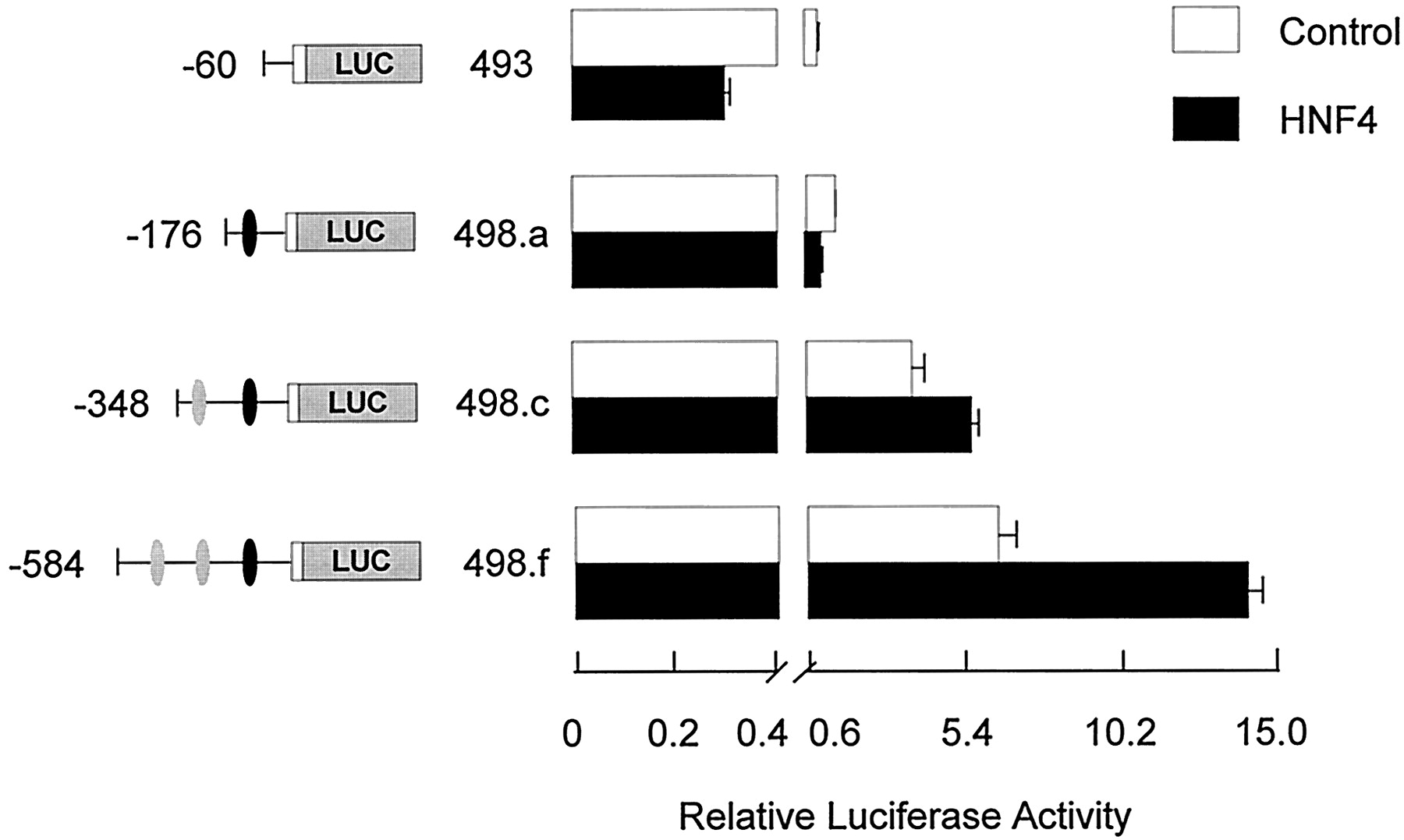
Functional analysis of the FMO1 regulatory domains in response to cotransfection with a HNF4α expression vector. The functional significance of identified HNF4α binding sites were analyzed by cotransfection of specific FMO1 luciferase reporter constructs a HNF4α expression vector into HepG2 cells with subsequent transient expression analysis. Salient features of each reporter plasmid construct are shown on the left side of the bar graph. The black oval represents the putative HNF1α elements, position −132 to −105, whereas the two gray ovals represent the putative HNF4α elements, position −467 to −454 and −195 to −182. Cotransfection with the parent expression vector (control) or HNF4α expression vector is indicated by the legend. The activity reported has been normalized for transfection efficiency and protein content and represents the mean ± S.D. from triplicate experiments. Note the change in scale after the break on the x-axis.
The above -fold increases were determined relative to the expression level observed with the same reporter gene construct minus cotransfection with the different HNF expression vectors. However, this does not take into account the contribution of endogenous HNF1α or HNF4α in the host HepG2 cells. If one assumes the -fold increase is caused solely by the inclusion of the HNF-responsive elements in the reporter gene constructs and that inclusion of exogenous HNF saturates these sites, then the degree of enhancement would be more accurately determined by comparing the expression observed with the pRNH498 constructs plus HNF1α or HNF4α to that observed with shorter constructs that are missing the respective elements. For HNF1α, comparing the activity observed with pRNH498.a plus HNF1α to pRNH493 minus HNF1α, a 14-fold stimulation is observed (Fig. 6). Making similar assumptions, a more accurate assessment of the enhancement observed with the proximal HNF4α site alone is 4-fold (comparing activity observed with pRNH498c plus HNF4α with that observed with pRNH498a alone) and 11-fold when both HNF4α sites are included (comparing activity observed with pRNH498f plus HNF4α with that observed with pRNH498a alone) (Fig. 7). One also can assess whether or not any synergism is observed between the HNF1α elements and the more distal HNF4α sites. In Fig. 6, the enhanced expression caused by inclusion of one or two upstream HNF4α sites can be assessed by the increased luciferase activity observed after transfection of pRNH498.c and pRNH498.f under control conditions relative to that observed with pRNH493 (i.e., 4- and 7-fold, respectively). If the combination of the HNF1α and HNF4α sites were merely additive, one would predict a 52-fold stimulation with pRNH498.c and 98-fold stimulation with pRNH498.f. The observed stimulation (pRNH498 plasmids plus HNF1α relative to pRNH493 control) was 51- and 155-fold, consistent with synergism between the more distal but not proximal HNF4α element and the overlapping HNF1α elements.
Comparison with Human FMO1.
In both the rabbit and human, FMO1 expression is largely restricted to the liver, intestine, and kidney (Shehin-Johnson et al., 1996; Yeung et al., 2000). A major difference is that FMO1 is a major liver enzyme in the adult rabbit, but seems to be restricted to the fetal liver in the human (Dolphin et al., 1996; Yeung et al., 2000). Despite this difference in developmental expression, we questioned whether a sequence comparison between the two species might lend further support to the importance of the regulatory elements identified in this study. Sequences within a human chromosome 1 YAC fragment, accession number HS127D3,2exhibiting 77% identity to rabbit FMO1 exon 0 (Luo and Hines, 1996) and identical with the most 5′ human FMO1 cDNA sequence were identified using the Basic Local Alignment Search Tool to search against the human genome. Comparison of the 750-bp sequences immediately upstream of this human FMO1 exon to the 750 bp of rabbit FMO1 5′ flanking sequence revealed an overall identity of 65%. However, a much higher degree of identity was observed for each of the regulatory elements identified in this study (Fig. 8) (YY1, 81%; overlapping HNF1α, 92%; proximal HNF4α, 93%; and distal HNF4α, 71%). As a first step in confirming the regulatory roles of these human sequences, the ability of double-stranded oligonucleotides representing each to compete with the rabbit FMO1 probes was determined using EMSA. As shown in part A, lane 7, of Figs. 1, 3, 4, and 5, each of the human sequences except that corresponding to the distal HNF4α site was able to compete with the corresponding rabbit probe for specific nuclear protein binding. These results strongly suggest that YY1 plays a comparable role in controlling human FMO1 expression and that similar to the rabbit, both HNF1α and HNF4α are important for the tissue-selective expression of this gene in the human.

Sequence alignment of the rabbit and human FMO1 promoter region. Shaded nucleotide residues indicate sequence identity between the rabbit and human FMO1 genes. The HNF1α, HNF4α, and YY-1 binding sites identified in this study are boxed. Arrows indicate the previously identified multiple transcription start sites, whereas the double arrow indicates the major transcription start site (Luo and Hines, 1996).
Discussion
Studies on the mammalian FMO gene family have focused on the catalytic mechanism and substrate specificity of the encoded enzymes, gene structure and variability, and overall expression patterns. From the latter, it is clear that regulation at the transcriptional level significantly contributes to interindividual species- and tissue-specific metabolic capacity and, as such, differential susceptibility to therapeutics and environmental toxicants. This report is the first describing specific molecular mechanisms controlling FMO expression. We have identified a YY1 Inr element within the major rabbit FMO1 promoter but provide evidence that this transcription factor is dispensable for basal FMO1 expression. Rather, YY1 dampens the ability of upstream elements to enhance promoter activity. We also identified binding sites for tissue-selective transcription factors within previously identified FMO1 positive regulatory domains (Luo and Hines, 1997). In vitro DNA binding assays were consistent with an HNF1α element spanning a relatively large domain (i.e., position −132 to −105). Sequence analysis using MatInspector and the TRANSFAC database, however, suggests that this site actually represents two overlapping HNF1α binding elements. Further upstream, HNF4α sites were identified at positions −467 to −454 and −195 to −182. Cotransfection assays with appropriate expression plasmids confirmed that each of these HNF sites functions to significantly enhanceFMO1 expression. Evidence for synergism between the HNF1α sites at −132 to −105 and either the distal HNF4α element or another element(s) located in the −584 to −348 domain is evidenced by the more than additive stimulated expression observed when these sequences were included in the HNF1α cotransfection experiments. DNA/protein binding assays eliminated the possibility of a distal HNF1 element within this same domain (data not shown). The human and rabbitFMO1 genes share extensive sequence identity within a 750-bp region immediately upstream of the major promoter and extending into the first noncoding exon. Perhaps more importantly, specific human sequences corresponding to the rabbit FMO1 regulatory elements described herein share even higher identity and, with the exception of the distal HNF4α site, also are able to compete with the rabbit sequences for specific nuclear protein binding. Thus, it is likely that these same elements are important for regulating humanFMO1 expression.
Transcription factors such as HNF1, -3, and -4, C/EBP, and D-element binding protein have been identified as important for the liver-selective expression of many genes (Cereghini, 1996), including several xenobiotic metabolizing enzymes (e.g., Ueno and Gonzalez, 1990;Cairns et al., 1996; Hansen et al., 1997; Metz et al., 2000). However, the expression of these factors is not strictly limited to liver. For example, HNF1 and HNF4 expression also has been observed in kidney, intestine, and stomach (Kuo et al., 1990; Sladek et al., 1990), whereas HNF3 expression has been documented in the brain, kidney, lung, and intestine (Clevidence et al., 1993). Studies from our laboratory (Shehin-Johnson et al., 1995) and others (Dolphin et al., 1996; Yeung et al., 2000) have shown that FMO1 expression, although highest in adult rabbit and human fetal liver, also is observed in the adult intestine and kidney of both species but not in the lung. Thus, there is good correlation between the tissue-selective expression patterns of HNF1, HNF4, and FMO1. Given the location of these elements in what was previously identified as a major, positive regulatory domain for the FMO1 gene (Luo and Hines, 1997) and the ability of these factors to significantly enhance the expression of FMO1 reporter gene constructs in HepG2 cells, our observations support the idea that HNF1 and HNF4 are important and perhaps solely responsible for the FMO1 tissue-selective expression pattern. This conclusion is further supported by our inability to detect any specific nuclear protein binding to theFMO1 HNF1α and HNF4α elements using nuclear extract prepared from the H441 human bronchioalveolar carcinoma cell line, although two minor specific DNA/protein complexes were observed with the proximal promoter fragment (position −162 to −42, Fig. 3A). Furthermore, transfection of the full-length FMO1 reporter gene constructs into H441 cells, which have been used by several groups to study lung-specific gene expression (e.g., Yan et al., 1995), failed to demonstrate anything other than basal expression levels (data not shown).
Our results fail to offer any insight into the molecular mechanism controlling an important species difference in FMO1expression. In the human, this enzyme is essentially nondetectable in adult liver. Rather, FMO3 is the major human adult hepatic FMO enzyme, whereas FMO1 but not FMO3 is expressed in the human fetal liver (Dolphin et al., 1996; Yeung et al., 2000). Given the recognized role of HNF1 and HNF4 in development (for review, see Cereghini, 1996), both factors may well be important for FMO1 expression in the human fetal liver. However, given the conservation between the human and rabbit FMO1 genes reported herein, it is unlikely these regulatory factors are critical for the switch to FMO3observed in humans.
Significant interindividual variability in the expression of some human FMOs has been described. Although polymorphisms in FMO3(Cashman et al., 2000) and FMO2 (Whetstine et al., 2000) have been identified that may partially explain this observation, no studies have identified similar polymorphisms for FMO1 and none have implicated any contribution from regulatory polymorphisms. However, sequence variants have been identified for both the HNF1α and HNF4α genes, some of which clearly represent rare alleles, whereas others represent polymorphisms (for example, Iwasaki et al., 1997; Urhammer et al., 1997). At least three splice variants also have been identified for HNF4α that result in altered function (Sladek et al., 1999). Interest in these variants has been heightened by the linkage of several HNF1α and HNF4α mutations with maturity onset diabetes of the young (Yamagata et al., 1996; Chèvre et al., 1998) and the demonstration that loss of function HNF4α mutations can dramatically alter expression of genes involved in glucose transport and metabolism (Stoffel and Duncan, 1997). Given our results implicating both HNF1α and HNF4α as important in regulating humanFMO1 expression, sequence variants at these loci may also contribute to variability in FMO1 expression. Because humanFMO1 is expressed in the fetal liver, these variants also may be significant in altering the susceptibility of the fetus to toxicants oxidized by FMO1.
In contrast to the hepatocyte nuclear factors, YY1 is ubiquitously expressed. Furthermore, this highly conserved member of the zinc finger family of transcription factors participates in both the positive and negative regulation of a large number of genes (Thomas and Seto, 1999). Most of the known YY1 regulatory elements are distal to the promoter, serving as enhancers or silencers of basal promoter activity. Instances wherein YY1 serves as a basal transcription factor are more limited. YY1 was first shown to bind an Inr within the AAV P5 gene (Shi et al., 1991). It was subsequently shown to participate as part of the basal transcriptional machinery for the phosphotyrosine phosphatase activator gene (Janssens et al., 1999), and cytochrome oxidase VIIc (Seelan and Grossman, 1997), VIA1 (Wong-Riley et al., 2000), and Vb subunit genes (Basu et al., 1997). In the last case, there is evidence that YY1 binding to the basal promoter is stabilized by interactions with transcription factor IIB and also that YY1 interacts directly with the large subunit of RNA polymerase II, directing it to the transcription initiation site (Usheva and Shenk, 1996). In contrast, YY1 binding to the initiator element within the human DNA polymerase β basal promoter is dispensable for basal promoter activity. Rather, a complex containing TATA box binding protein, transcription factor IIB, transcription factor IIF, and RNA polymerase II was shown to form on this promoter in an Inr-dependent but YY1-independent manner (Weis and Reinberg, 1997). These authors failed to determine whether YY1 would have any effect on the ability of more distal, upstream sequences to modify basal promoter activity, or whether it may alter Inr complex stability. The data presented herein would suggest a somewhat analogous situation for the FMO1 P0 promoter, except that in this instance, YY1 acts to either de-stabilize the protein complex forming on the Inr at the major transcription start-site, or more likely, suppresses the positive effect of upstream enhancers. However, the diversity of YY1 functions and its ability to interact with many different coregulatory proteins (Thomas and Seto, 1999) suggest several possible modes of action in regulatingFMO1 expression.
In summary, this study is the first to describe molecular mechanisms responsible for regulating FMO expression. We have demonstrated that both HNF1α and HNF4α are important in enhancing rabbit FMO1 expression and probably are critical for both its tissue- and developmental-specific expression patterns. We also have demonstrated that FMO1 promoter activity can be negatively regulated by YY1, but that this factor is dispensable for basal promoter activity. It is presumed that the Inr at the major transcription start site is necessary for this latter activity, a hypothesis currently under investigation. Finally, the conservation of these regulatory elements between the rabbit and human FMO1genes, as well as the ability of the orthologous sequences to compete for specific nuclear protein binding, is consistent with these same elements being important for regulating human FMO1expression.
Acknowledgments
We thank Drs. D. Gail McCarver and Jennifer L. Morris for their critical evaluation of this study and Dr. Stephen A. Duncan for his helpful discussions regarding the hepatocyte nuclear factor family of transcription factors.
Footnotes
- Received June 21, 2001.
- Accepted August 10, 2001.
-
↵1 The nucleotide sequence for rabbit FMO1 position −2131 through +4838, including exons 0 (major 5′ untranslated region) and 1 (alternative, minor 5′ untranslated region) and exon 2 has been deposited in the GenBank database under accession number AF355464.
-
These studies were supported in part by U.S. Public Health Services Grant CA53106 from the National Cancer Institute and by generous funds from the Children's Hospital Foundation of Wisconsin.
-
↵2 The nucleotide sequence for the human chromosome 1 fragment exhibiting a high degree of identity to rabbit FMO1 exon 0 and immediate 5′ flanking sequence has been deposited in the GenBank database under accession number HS127D3.
Abbreviations
- FMO
- flavin-containing monooxygenase
- YY1
- ying yang 1
- HNF
- hepatic nuclear factor
- COUP/TF
- chicken ovalbumin upstream promoter/transcription factor
- EMSA
- electrophoretic mobility shift assay
- DMS
- dimethylsulfate
- DEPC
- diethylpyrocarbonate
- bp
- base pair(s)
- Inr
- initiator element
- AAV
- adeno-associated virus
- C/EBP
- CCAAT/enhancer-binding protein
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}