


Abstract
Conserved features of the sequences of dopamine receptors and of homologous G-protein-coupled receptors point to regions, and amino acid residues within these regions, that contribute to their ligand binding sites. Differences in binding specificities among the catecholamine receptors, however, must stem from their nonconserved residues. Using the substituted-cysteine accessibility method, we have identified the residues that form the surface of the water-accessible binding-site crevice in the dopamine D2 receptor. Of approximately 80 membrane-spanning residues that differ between the D2 and D4 receptors, only 20 were found to be accessible, and 6 of these 20 are conservative aliphatic substitutions. In a D2 receptor background, we mutated the 14 accessible, nonconserved residues, individually or in combinations, to the aligned residues in the D4 receptor. We also made the reciprocal mutations in a D4 receptor background. The combined substitution of four to six of these residues was sufficient to switch the affinity of the receptors for several chemically distinct D4-selective antagonists by three orders of magnitude in both directions (D2- to D4-like and D4- to D2-like). The mutated residues are in the second, third, and seventh membrane-spanning segments (M2, M3, M7) and form a cluster in the binding-site crevice. Mutation of a single residue in this cluster in M2 was sufficient to increase the affinity for clozapine to D4-like levels. We can rationalize the data in terms of a set of chemical moieties in the ligands interacting with a divergent aromatic microdomain in M2-M3-M7 of the D2 and D4 receptors.
The dopamine D2 receptor has been implicated in the mechanism of drugs used in the treatment of disorders such as schizophrenia and Parkinson's disease. The identification of several D2-like receptors (D2, D3, and D4) (Bunzow et al., 1988; Sokoloff et al., 1990; Van Tol et al., 1991) has spurred research into the therapeutic relevance of these receptor types, and several drugs selective for the D2, D3, and D4 receptors have been identified. Whereas the atypical antipsychotic clozapine has nearly 30-fold higher affinity for the D4 receptor than for the D2 or the D3 receptors (Van Tol et al., 1991), other compounds, such as (3-[4-(4-chlorophenyl)piperazin-1-yl]methyl-1H- pyrrolo[2,3-b] pyridine (chlorophenylpiperazinyl methylazaindole; CPPMA), are more than 1000-fold selective (Kulagowski et al., 1996).
The binding sites of the dopamine receptors are formed among their seven, mostly hydrophobic, membrane-spanning segments (Oprian, 1992;Strader et al., 1994) and are accessible to charged, water-soluble agonists, such as dopamine. Thus, for each of these receptors, the binding site is contained within a water-accessible crevice, the binding-site crevice, extending from the extracellular surface of the receptor into its transmembrane domain. The surface of this crevice is formed by residues that can contact specific agonists and/or antagonists and by other residues that may play a structural role and affect binding indirectly.
Conserved features of the sequences of dopamine receptors and of homologous G-protein-coupled receptors, such as the adrenergic receptors, point to regions and amino acid residues within these regions that contribute to the binding sites in the dopamine receptors. Structural features that contribute to ligand affinity include an electrostatic interaction between a protonated amine of the ligand and a conserved Asp in the third membrane-spanning segment (M3) (Strader et al., 1988; Mansour et al., 1992; Javitch et al., 1995b), a hydrogen-bonding group or groups that interact with serines in M5 (Strader et al., 1989; Cox et al., 1992; Mansour et al., 1992), and an aromatic ring that interacts with the aromatic cluster in M6 (Choudhary et al., 1993, 1995; Cho et al., 1995; Roth et al., 1997; Javitch et al., 1998). These contact residues are completely conserved among all catecholamine receptors. The pharmacological differences among the D2-like receptors, however, cannot be found among the conserved features of their sequences but rather must reside in differences in their sequences and folded structures.
We reasoned that the residues that form the surface of the binding-site crevice but are not conserved in the D2 and D4 receptors are the best candidates for determinants of the pharmacological differences between these receptors. We have identified the residues that form the surface of the binding-site crevice in the human D2 receptor, using the substituted-cysteine accessibility method (SCAM) (Akabas et al., 1992,1994; Javitch et al., 1995a,b, 1998, 1999; Fu et al., 1996). We have now taken two parallel approaches to identify the structural determinants of pharmacological specificity: 1) a purely empirical approach in which we systematically substitute all of the residues accessible in the D2 receptor binding-site crevice not conserved in the D4 receptor and 2) use of our current molecular models of the receptors to develop structural hypotheses regarding the likely determinants of specificity.
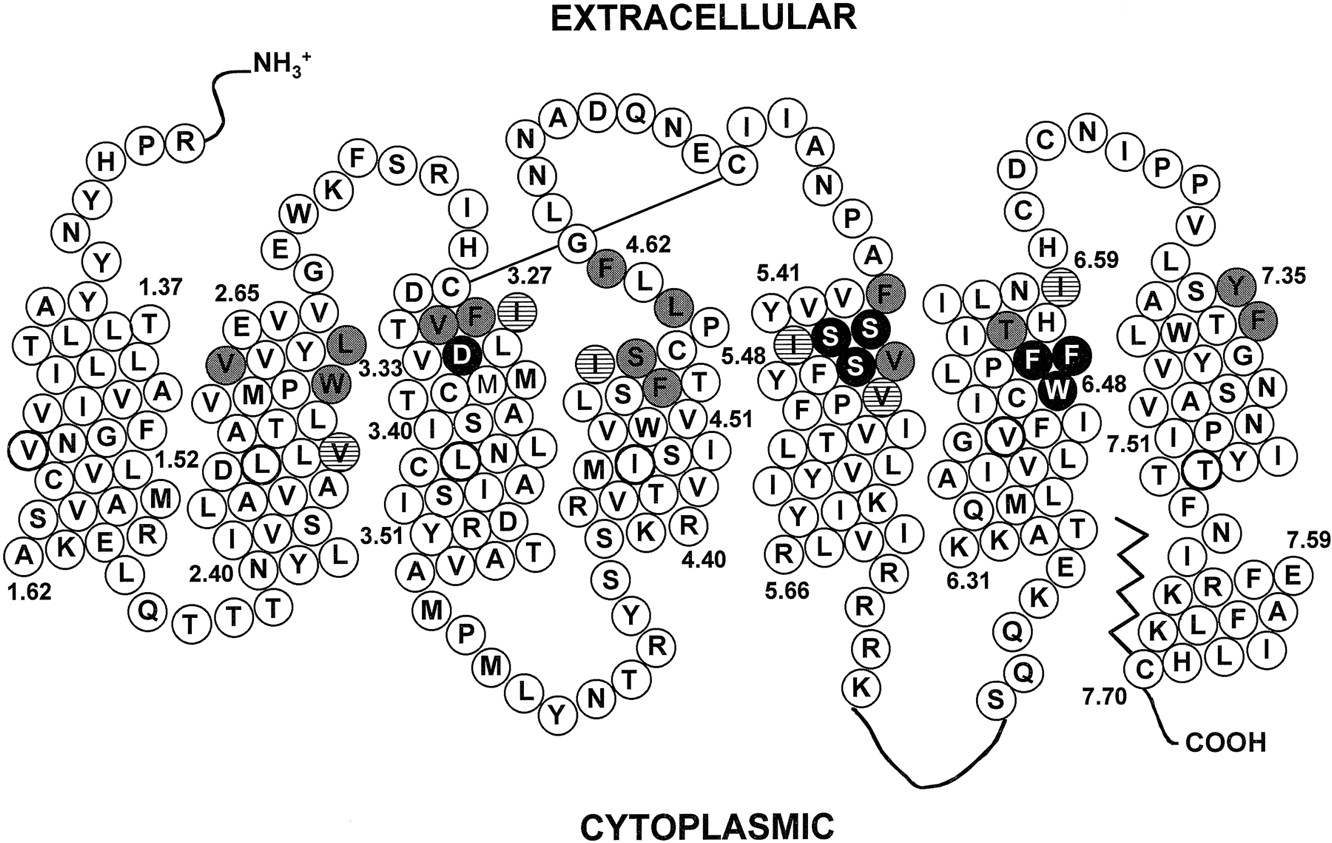
Of the more than 80 nonidentical residues within the membrane-spanning segments of the D2 and D4 receptors, only 20 residues in the M2 through M7 segments were determined to be accessible by SCAM in the D2 receptor (Fig. 1) (Javitch et al., 1995a,b, 1998,1999; Fu et al., 1996; M. M. Simpson, J. A. Ballesteros, L. Shi, J. Chen, V. Chiappa, H. Weinstein, and J. A. Javitch, in preparation). Six of these 20 residues represent conservative aliphatic substitutions between the two receptors, whereas the other 14 residues are not conserved. In a D2 receptor background, we mutated these 14 nonconserved accessible residues to the aligned D4 residue, one at a time and/or in combination. We also made the reciprocal substitutions in a D4 receptor background. We found that mutation of a cluster of residues in M2, M3, and M7 of the D2 receptor was sufficient to impart D4 selectivity for several D4-selective compounds.

Helical net representation of the human D2 receptor. Filled circles are amino acids known to be involved in ligand binding. Shaded and hatched circles indicate accessible residues not identical in the aligned D4 receptor sequence. Shaded circles indicate D2 residues replaced by the aligned D4 residues by site-directed mutagenesis. Hatched circles indicate conservatively substituted aliphatic residues.
Experimental Procedures
Materials.
Human dopamine D2L receptor cDNA (Grandy et al., 1989) was provided by O. Civelli (University of California, Irvine, CA), and the bicistronic expression vector pcin4 was provided by S. Rees (Glaxo-Wellcome, Stevenage, UK) (Rees et al., 1996). [3H]N-methylspiperone (84 Ci/mmol) was obtained from DuPont-NEN (Boston, MA). A synthetic human D4 cDNA was used for these studies (Genebank accessionAF119328). Its creation and characterization is described elsewhere (M. A. Kazmi, L. A. Synder, A. M. Cypress, S. G. Graber, and T. P. Sakmar, in preparation). CPPMA was synthesized according to Kulagowski et al. (1996). Ro 61-6270 (C. Riemer, PCT Int. Appl. 1996, WO 9635666), Ro 10-4548 (T. Godel, D. Hartmann, C. Riemer, PCT Int. Appl. 1996, WO 9641630), and Ro 62-4599 (T. Godel, C. Riemer, A. Edenhofer, PCT Int. Appl. 1997, WO 9713759) were synthesized by T. Godel and C. Riemer (Roche, Basel, Switzerland). (+)-Butaclamol, clozapine, andN-methylspiperone were obtained from Research Biochemicals International (Natick, MA).
Numbering of Residues.
Residues are numbered according to their positions in the human dopamine D2Lreceptor sequence. In some cases, we also index residues relative to the most conserved residue in the membrane-spanning segment in which it is located (Ballesteros and Weinstein, 1995). By definition, the most conserved residue within each helix is assigned the position index 50, e.g., Asp80(2.50) in M2, and therefore Ala79(2.49) and Leu81(2.51). This indexing simplifies the identification of corresponding residues in different G-protein-coupled receptors (GPCRs).
Site-Directed Mutagenesis.
Mutations were generated by the Altered Sites Mutagenesis System (Promega) or by the polymerase chain reaction. Mutations were identified by restriction mapping and confirmed by DNA sequencing. Mutants are named as (wild-type residue)(residue number)(substituted aligned residue), where the residues are given in single-letter code.
Stable Transfection.
The cDNA encoding the dopamine D2L receptor or the appropriate mutant, epitope tagged at the NH2-terminus with the cleavable influenza-hemagglutinin signal sequence followed by the FLAG epitope (DYKDDDDA; Sigma, St. Louis, MO) (Javitch et al., 1998) and with the strep-tag sequence AWRHPQFGG at the COOH-terminus, in the bicistronic expression vector pcin4, was used for all transfections. The D4 synthetic gene was identically tagged at its NH2-terminus and subcloned into pcin4.
Human embryonic kidney (HEK) 293 cells in DMEM/F12 (1:1) with 10% bovine calf serum (Hy-Clone Laboratories Inc., Logan, UT) were maintained at 37°C and 5% CO2. Thirty-five-millimeter dishes of 293 cells at 70 to 80% confluence were transfected with 2 μg of the appropriate construct in pcin4 (see above) with 9 μl of lipofectamine and 1 ml of OPTIMEM (Gibco, Grand Island, NY). Five hours after transfection, the solution was removed and fresh media added. Twenty-four hours after transfection, the cells were split to a 100-mm dish and 700 μg/ml of geneticin was added to select for a stably transfected pool of cells, which required approximately 2 weeks.
Harvesting Cells and Membrane Preparation.
Stably transfected HEK 293 cells were washed with PBS (8.1 mM NaH2PO4, 1.5 mM KH2PO4, 138 mM NaCl, 2.7 mM KCl, pH 7.2), briefly treated with PBS containing 1 mM EDTA, and then dissociated in PBS. For membrane preparation, cells were pelleted at 1000g for 5 min at 4°C and resuspended in binding buffer (140 mM NaCl, 5.4 mM KCl, 25 mM HEPES, 1 mM EDTA, pH 7.4). Cells were then disrupted on ice with a Polytron homogenizer at a setting of 6 for 12 s. The membranes were collected by centrifugation at 40,000g for 15 min at 4°C. The pellet was resuspended in binding buffer, disrupted as described above, and used immediately for binding or stored at −80°C until use.
Competition Binding Assays.
The IC50values of unlabeled compounds were determined from their inhibition of the binding of [3H]N-methylspiperone (90 pM). In duplicate tubes, we incubated 10 different concentrations of antagonist (100 μl) with 200 μl of membrane suspension diluted in binding buffer (with 0.006% BSA) and 100 μl [3H]N-methylspiperone in a final volume of 1000 μl at room temperature for 60 min. Depending on the level of expression in the various mutants, adjustments in the amount of membrane per assay tube were made as necessary to prevent depletion of ligand in the case of very high expression or to increase the signal in the case of low expression. The mixture was then filtered with a Brandel cell harvester through a Whatman 934AH glass-fiber filter (Brandel). The filter was washed three times with 1 ml of 10 mM Tris-HCl and 120 mM NaCl (pH 7.4) at 4°C. Specific [3H]N-methylspiperone binding was defined as total binding less nonspecific binding in the presence of 1 μM (+)-butaclamol (Research Biochemicals, Inc., Natick, MA).
Calculations of KD andKI.
TheK D values for [3H]N-methylspiperone were determined by fitting the data for N-methylspiperone competition to the following equation for homologous competitive binding with ligand depletion: Y = {B max (cpm) × [free radioligand, nM]/K D + [free radioligand, nM] + [free cold Ligand, nM]} + [free radioligand, cpm] ×NS (nonspecific unitless fraction) (Swillens, 1995) (Prism, Graphpad). Three parameters were set to constant values: specific activity (93 cpm/fmol), volume of assay (1 ml), and radiolabeled cpm added. IC50 values for CPPMA, clozapine, Ro 61-6270, Ro 10-4548, and Ro 62-4599 in competition with [3H]N-methylspiperone were determined by fitting the data to a variable-slope, one-site competition model by nonlinear regression.K I values were calculated with the equation of Cheng and Prusoff (1973) from the IC50 values and the K D for each mutant as determined above.
Modeling.
A computational model of the D2 receptor was developed based on the structure of bovine rhodopsin (Baldwin et al., 1997; Unger et al., 1997) following homology modeling approaches described elsewhere (Ballesteros and Weinstein, 1995). The helical ends (Fig. 1) and orientations for the seven membrane-spanning α-helices were derived from SCAM studies on the D2 receptor (Javitch et al., 1995a,b, 1998, 1999; Fu et al., 1996; M. M. Simpson, J. A. Ballesteros, L. Shi, J. Chen, V. Chiappa, H. Weinstein, and J. A. Javitch, in preparation) and by spin-labeling studies of rhodopsin (Farahbakhsh et al., 1995; Altenbach et al., 1996). The ligands were built and docked manually with the modeling software Quanta (Molecular Simulations Inc., Waltham, MA). Energy minimization was applied to refine the ligand-receptor complexes with the CHARMM force field (Brooks et al., 1983).
Results
Effects on Ligand Binding of Substituting in D2 Receptor the Aligned D4 Residues.
In a D2 receptor background we made 14 amino acid substitutions to the aligned D4 receptor residue (Table1). Twelve mutant receptors were made, 10 with single residue substitutions, and 2 with double residue substitutions in M4. The relatively nonselective ligand [3H]N-methylspiperone bound to all of the mutants stably expressed in HEK 293 cells. The D2 and D4 receptors have KD values of 79 pM and 360 pM, respectively for N-methylspiperone (Table2). The KD values for N-methylspiperone for 9 of the 12 constructs were intermediate between D2 and D4 (Table 2). V91F and F189Y had a 2- to 3-fold increase in affinity for N-methylspiperone compared with D2, whereas V111 M had a 2- to 3-fold lower affinity forN-methylspiperone compared with D4. None of the mutants had dramatic differences in expression level compared with wild-type D2 receptor (data not shown).
Indexing of D2 and D4 receptor residue substitutions
Comparison of affinities of antagonists for wild-type D2 and D4 receptors and mutant D2 receptors
We determined the affinity of the highly D4-selective compound, CPPMA (Fig. 2) for each mutant receptor. Wild type D4 receptor had a 1180-fold higher affinity for CPPMA than did wild type D2 receptor (Table 2). V91F had a nearly 100-fold increase in affinity for CPPMA compared with that of the wild type D2 receptor (Fig. 3). F110L and Y408V had 5- and 3-fold increased affinity, respectively, for CPPMA. The other nine constructs had less than a 3-fold change in affinity for CPPMA compared with D2 receptor (Table 2).
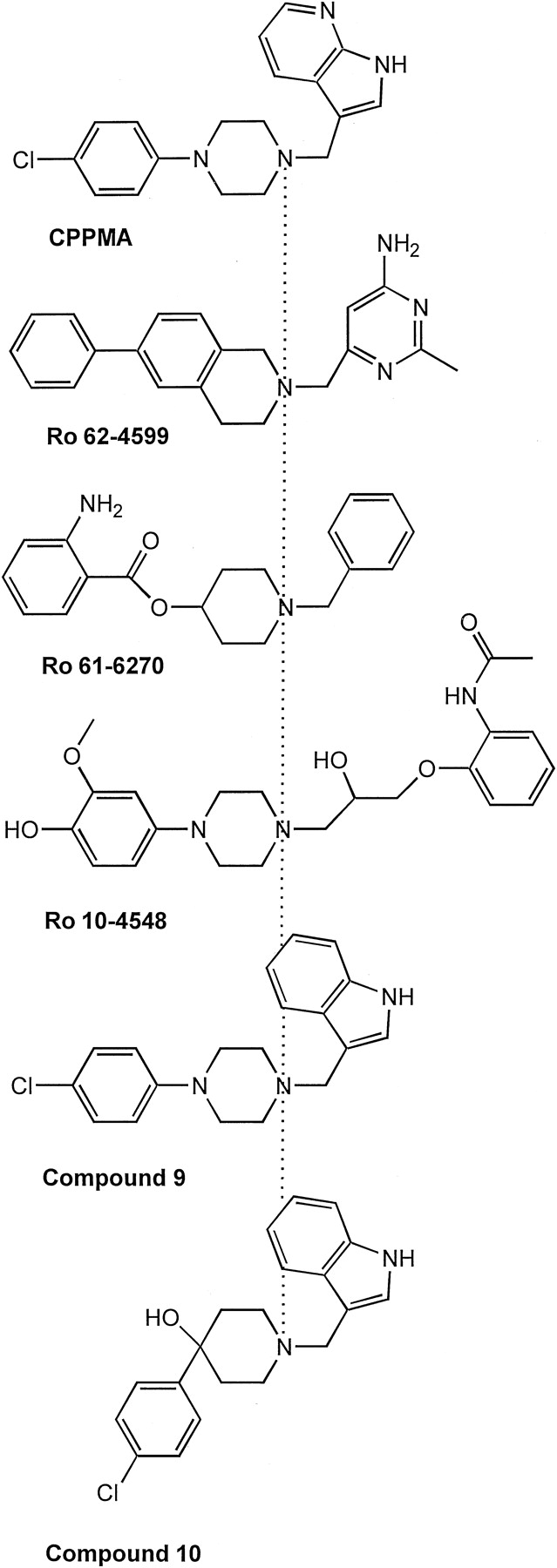
Chemical structures of D4- and D2- receptor-selective antagonists. The compounds are aligned at the protonated amine (dotted line). These chemically distinct compounds all share a protonated amine in a six-atom ring, with an aromatic-containing substituent at each end of the ring (positions 1,4), and thus oriented in opposite directions (see text). Structure-activity relationships suggest that the aromatic moieties oriented toward the left interact with the divergent aromatic microdomain in M2-M3-M7, whereas the aromatic moieties oriented to the right interact with M5 and M6 (see text). All of these compounds are D4 selective, except compound 10, which is D2 selective (Kulagowski et al., 1996). Compounds 9 and 10 are from Kulagowski et al. (1996).

CPPMA competition of [3H]N-methylspiperone binding to wild-type D2, wild-type D4, and single- (A), double- and triple- (B), and multiple- (C) residue mutant D2 receptors. A, dose-response curves for D2 (●), D2V91F (▴), D2F110L (▪), D2Y408V (♦), and D4 (○). B, dose-response curves for D2 (●), D2V91F/Y408V (▾), D2F110L/Y408V (▵), D2V91F/F110L (■), D2V91F/F110L/Y408V (▿), and D4 (○). C, dose-response curves for D2 (●), D2V91F/F110L/V111M/Y408V (∗), D2W90L/V91F/liter94S/F110L/V111M/Y408V (⋄), and D4 (○). The assays were performed as described in Experimental Procedures. The means ± S.E. (duplicate determinations) are shown from a representative experiment performed three times or more with similar results. The data are fit to a variable-slope, one-site competition model by nonlinear regression.
Clozapine (Fig. 2) is also a moderately D4-selective drug, with a 23-fold higher affinity for D4 receptor than for D2 receptor (Table 2). V91F had a 49-fold higher affinity for clozapine. Thus, this single mutation enabled the D2 receptor to bind clozapine in a D4-like manner. F189Y and Y408V had approximately 5-fold higher affinities for clozapine than did the D2 receptor. The other nine constructs had less than a 3-fold change in affinity for clozapine compared with D2 (Table2).
The 100-fold gain in affinity for CPPMA seen with mutation of Val91 to Phe was substantially less than the approximately 1200-fold higher affinity of the D4 receptor. Because single substitutions of Phe110 to Leu and Tyr408 to Val also had greater than 3-fold effects on the affinity of CPPMA, we brought together the V91F, F110L, and Y408V mutations in various combinations. N-Methylspiperone binding to these constructs was similar to that of the D4 receptor (Table3). Combination of V91F with F110L or with Y408V did not result in a substantial additional gain in affinity for CPPMA. The combination of F110L and Y408V, however, resulted in an additive gain in affinity of 16-fold, and the simultaneous combination of all three mutations led to a receptor with 151-fold higher affinity than D2 receptor (Table 3 and Fig. 3).
Comparison of affinities of antagonists for wild-type D2 and D4 receptors and mutant D2 receptors
In an attempt to construct a D2 receptor mutant with D4-like affinity for CPPMA, we further mutated, in the V91F/F110L/Y408V background, the nonconserved, accessible residues adjacent to Val91 and/or Phe110. Mutation of Val111 to Met, the aligned D4 residue, combined with V91F/F110L/Y408V resulted in a receptor with an affinity for CPPMA 4800-fold higher than the D2 receptor (Table 3 and Fig. 3) and an affinity for N-methylspiperone similar to D4 receptor. W90L/V91F/L94S/F110L/V111M/Y408V also displayed D4-like affinity for CPPMA and N-methylspiperone.
To ascertain whether the set of residues at positions 91, 110, 111, and 408 was essential for the D4 selectivity of other compounds, we examined the binding of Ro 61-6270, Ro 10-4548, and Ro 62-4599 (Fig.2). Ro 61-6270 and Ro 10-4548 have approximately 100-fold higher affinity for the D4 receptor than for the D2 receptor, whereas Ro 62-4599 is highly D4 selective, with a 15,000-fold higher affinity (Table 4). As we observed for CPPMA, mutation of V91F/F110L/Y408V was insufficient to produce full D4-like affinity for these compounds, but Ro 61-6270 bound to V91F/F110L/V111M/ Y408V with D4-like affinity, as did CPPMA. Ro 10-4548 and Ro 62-4599 bound V91F/F110L/V111M/Y408V with intermediate affinity. In contrast, for the six-residue mutant W90L/V91F/L94S/F110L/V111M/Y408V, Ro 10-4548 bound with D4-like affinity and Ro 62-4599 bound with 1600-fold higher affinity than to D2 receptor (Table 4).
Comparison of affinities of Roche compounds for wild-type D2 and D4 receptors and mutant D2 receptors
Effects on Ligand Binding of Substituting the Aligned D2 Residues in the D4 Receptor.
Substitution of critical contact residues in the D4 receptor with the aligned residues from the D2 receptor would be expected to lower affinity for D4-selective ligands. We thus substituted into the D4 receptor the reciprocal 14 D2-divergent residues, which were aligned with the D4 residues substituted into the D2 receptor (see above). As was observed for the mutations in the D2 receptor (see above), the affinity of CPPMA for the mutant D4 receptors was substantially affected by substitution of residues in M2, M3, and M7. Simultaneous substitution of the three accessible, nonconserved residues in M2 of the D4 receptor (L90(2.60), F91(2.61), and S94(2.64)) with the aligned D2 residues reduced the affinity of the receptor for CPPMA by 80-fold (Table 5). Substitution of the two accessible, nonconserved residues in M3 of D4 receptor (L111(3.28) and M112(3.29)) reduced the affinity of the receptor for CPPMA by 15-fold. In contrast, substitution of a single accessible, nonconserved residue in M7 of D4 receptor (V350(7.35)) increased the affinity of CPPMA 7-fold for the receptor. Combining the substitutions of the five residues in M2 and M3 of the D4 receptor nonconserved and accessible in the D2 receptor (L90W/F91V/S94L/L111F/M112V) reduced the affinity of CPPMA 600-fold, nearly to D2-like affinity. Similarly, Ro 61-6270, Ro 10-4548, and Ro 62-4599 bound to L90W/F91V/S94L/L111F/M112V with 40-fold, 13-fold, and more than 15,000-fold lower affinity, respectively, than to D4 receptor (data not shown).
Comparison of affinities of antagonists for wild-type D4 and D2 receptors and mutant D4 receptors
Role of the M2-M3-M7 Aromatic Microdomain in Ligand Specificity.
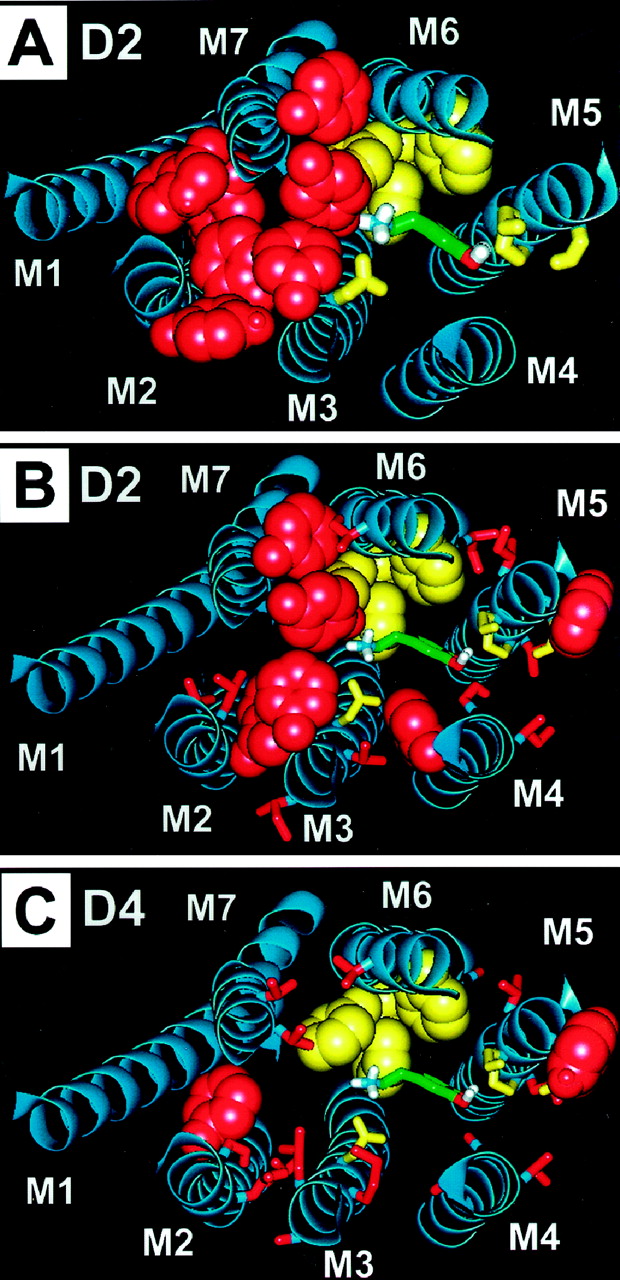
We have recently identified a cluster of aromatic residues in M2, M3, and M7 at the surface of the binding-site crevice of the D2 receptor (Javitch et al., 1999) adjacent to the protonated amine of bound dopamine (Fig. 4A). Analysis of the divergent aligned residues, found to be accessible by SCAM in the D2 receptor (Javitch et al., 1995a,b, 1998, 1999; Fu et al., 1996) revealed that this aromatic cluster is strikingly different in the D2 and D4 receptors, the latter of which lacks Trp90, Phe110, Tyr408, and Phe411 and instead has Phe substituted for Val91 (Fig. 4, B and C). Remarkably, four of these five residues contained in the M2-M3-M7 divergent aromatic cluster plus an additional two residues also belonging to this cluster were found to be among the molecular determinants of selective D2-D4 ligand recognition (Table 3).

An aromatic microdomain in the binding-site crevice of the D2 receptor. Dopamine (green, liquorize) is shown binding between M3, M5, and M6. Residues in these helices previously proposed to contact dopamine (Asp114 in M3; Ser193, Ser194, and Ser197 in M5; and Trp386, Phe389, and Phe390 in M6) are shown in yellow. A, a dense cluster of accessible aromatic residues contained within M2, M3, and M7 is shown in Van der Waals representation in red. The three-dimensional motif defined by this aromatic microdomain is positioned next to the dopamine-binding site at the level of the protonated amine. B, the accessible residues in the D2 receptor not conserved in the D4 receptor are shown in red. C, the equivalent residues present in the D4 receptor at these positions are shown in red on a D2 receptor template.
Because the aromatic microdomain between M2-M3-M7 of the D2 receptor is predicted to be adjacent to the protonated amine of dopamine, which interacts with Asp114(3.32) in M3 (see Fig. 4A), we would expect that aromatic, bulky substituents stemming from the protonated amine would clash with the dense cluster of aromatic residues in the D2 receptor (Fig. 4, A and B) but fit in the D4 receptor pocket (Fig. 4C). The several D4-selective and chemically distinct ligands studied here could be decomposed into three chemical moieties schematically illustrated in Fig. 2: a centrally positioned protonated amine, which we assume interacts with Asp114(3.32) in M3, and two aromatic-containing substituents. In the cases of CPPMA and Ro 62-4599, one of the aromatic-containing moieties (shown to the right of the protonated amine in Fig. 2) contains H-bonding groups. According to structure-activity relationships for catecholamine receptor ligands, this moiety would be expected to interact with a cluster of aromatic residues and with Ser and other polar residues in M5 and M6 (Choudhary et al., 1993, 1995; Cho et al., 1995; Roth et al., 1997; Javitch et al., 1998) (Fig. 5).

Ligand docking to molecular models of the wild-type D2 receptor (left) and D4-like mutant D2 receptors (right). Contact sites for dopamine (see Fig. 4) are shown in yellow (A–D). The D2-D4 divergent residues sufficient to switch D2-D4-selective recognition of CPPMA (A and B) and of Ro 62-4599 (C and D) are shown in red. Note that these residues cluster together in space and belong to the divergent aromatic microdomain in M2-M3-M7 (see Fig. 4). These residues and their interactions with CPPMA (yellow), Ro 62-4599 (green), and compound 10 (orange) are shown in greater detail in a side view looking from the perspective of M7 (E and F) and in an extracellular view of the M2-M3-M7 microdomain (G and H). The chlorophenyl moiety of CPPMA clashes with F3.28 and Y7.35 in the D2 receptor (A, E, and G) but fits between L3.28 and V7.35 in the D4 receptor (B, F, and H). Moreover, an additional stabilizing interaction of the chlorophenyl moiety of CPPMA with F2.61 occurs in the D4 receptor (B, F, and H) but not in the D2 receptor (A, E, and G). Ro 62-4599 shares the interactions described above for CPPMA (C, D), but because its phenyl ring is rotated 90 degrees relative to that of CPPMA, it also clashes with Trp90 and Leu94 in the D2 receptor (E and G) but not in the D4-like mutant D2 receptor (F and H). The D2-selective compound 10 (orange) is included to illustrate how subtle chemical modifications can alter the geometry and thus the resulting interactions of ligands containing otherwise analogous chemical moieties. Thus, because the chlorophenyl substituent of compound 10 is in a tetrahedral orientation [rather than the planar orientation of the substituent in CPPMA (yellow)], it is oriented away from M2-M3 and toward M7 (G and H), avoiding the clash with M2-M3 in the D2 receptor (E and G) and losing the interaction with Phe2.61 in the D4 receptor (F and H).
The third chemical moiety present in these ligands is an aromatic-containing substituent of the protonated amine (shown to the left of the protonated amine in Fig. 2). In CPPMA and Ro 62-4599, this aromatic moiety is devoid of H-bonding groups. Because the substituents on the six-atom nitrogen-containing ring in the relatively rigid CPPMA or Ro 62-4599 are on opposite ends of the ring (1,4 positions), the aromatic-hydrophobic moiety is expected to be oriented in a direction opposite in space from the other aromatic/H-bonding moiety. Thus, as shown schematically in Fig. 2 and in the molecular models in Fig. 5, because the aromatic/H-bonding moiety is oriented toward M5-M6, the aromatic-hydrophobic moiety would be oriented toward M2-M7 at the level of the aromatic microdomain between M2-M3-M7. This rationale was used to guide the docking of CPPMA and Ro 62-4599 in the D2 receptor and mutant D2 receptor models (Fig. 5).
Although structure-activity data supported the orientation of Ro 61-6270 and Ro 10-4548 relative to our docked Ro 62-4599 shown in Fig.2 (T. Godel, unpublished observations), the altered positioning of the H-bonding groups made us less confident of the orientation of these more complex molecules, and we did not attempt to dock these ligands. Nonetheless, all these ligands share the basic chemical scaffold described above of a protonated amine contained within a six-member ring with two aromatic-containing substituents on opposite ends. Clozapine could not be decomposed into these three chemical moieties, and, thus, we did not attempt to orient or dock it.
Figure 5 shows the experimentally identified critical D2/D4-selective ligand-receptor interactions for CPPMA and Ro 62-4599 that result from our docking procedure onto the D2 (Fig. 5, A, C, E, and G, left) and D4-like mutant D2 receptor (Fig. 5, B, D, F, and H, right) models. Figure 5A shows an extracellular view of CPPMA docked into the D2 receptor. The previously identified dopamine interaction sites, D3.32 in M3, the aromatic cluster in M5-M6, and a serine residue or residues in M5 described in Fig. 4, are shown in yellow for reference. The protonated tertiary amine of CPPMA has an ionic interaction with the aspartic acid D3.32 in M3, in analogy to dopamine and all catecholamine ligands studied here. The six-atom ring containing the protonated amine positions the two aromatic substituents of CPPMA in opposite directions. Structure-activity relationships suggests that one of the aromatic substituents is oriented toward M5-M6, as seen in Fig. 5A, in analogy to dopamine (see Fig. 4A). The second aromatic substituent of the ring containing the protonated amine of CPPMA would thus face toward M2-M3-M7. Panel A shows the detailed docking of CPPMA, with the chlorophenyl substituent oriented toward M2-M3-M7 at the level of the divergent aromatic microdomain shown in Fig. 4. The four residues in the D2 receptor, the substitution of which by their D4 homologs switches the affinity of CPPMA to a D4-like affinity, are shown in red in Fig. 5A. Note that the chlorophenyl substituent of CPPMA clashes with F3.28 in M3 and Y7.38 in M7 but does not interact directly with V2.61 in M2 or V3.29 in M3.
Figure 5B shows the equivalent interactions of CPPMA described above for A in a model of the D4-like mutant D2 receptor. We have labeled these panels D4 for clarity, because the model and ligand-receptor interactions shown would correspond to a D4 receptor if these two receptors were to share a common backbone fold. The steric clash of the chlorophenyl moiety of CPPMA with F3.28 and Y7.35 shown in A for the D2 receptor is absent in the L3.28 and V7.35 D4-like mutant shown in Fig. 5B. Note the added stabilizing interaction between the chlorophenyl moiety of CPPMA and F2.61 of the D4-like D2 mutant receptor. The fourth substituted residue necessary to switch CPPMA affinity to D4-like levels, M3.29, is now in direct proximity to the ligand, participating in favorable Van deer Waals interactions. However, because Met residues are long and flexible, this is one of multiple possible conformations of M3.29; therefore, modeling by itself cannot establish this residue as a significant CPPMA-receptor interaction.
Figure 5, C and D, shows the interaction of the D4-selective ligand Ro 62-4599 with D2 and D4-like mutant D2 receptors, respectively. C and D follow the same color schemes and overall orientation as the interaction of CPPMA shown in A and B, with the addition of two mutated residues, W2.60L and L2.64S in M2. These two additional mutations were necessary to switch the affinity of Ro 62-4599 from D2 to D4-like levels. Docking Ro 62-4599 into these models (Fig. 5, C and D) follows a similar rationale and demonstrates the interactions described for CPPMA. The aromatic substituent of the six-atom ring containing the protonated amine of Ro 62-4599 is oriented toward the divergent aromatic microdomain within M2-M3-M7. Although Ro 62-4599 shares the same interactions as CPPMA with the four-mutant V2.61F/F3.28L/V3.29M/Y7.35V, its distinct aromatic substituent (see Fig. 2) clashes with W2.60 and L2.64 in the D2 receptor (Fig. 5C) but does not clash in the 6 residue D4-like mutant D2 receptor (W2.60L/V2.61F/L2.64S/F3.28L/V3.29M/Y7.35V) (Fig. 5D).
Figure 5, E to H, shows in greater detail the aromatic substituents of CPPMA and Ro 62-4599 and their proposed interactions with the experimentally identified critical D2/D4 selective residues in M2-M3-M7 of the D2 and D4-like receptors. E shows a side view of the D2 receptor from the perspective of the M7 backbone, which has been removed for clarity, leaving only the M3 and M2 backbones shown in blue. The six residues that switch completely the affinity of the D2 receptor to D4-like levels for both CPPMA and for Ro 62-4599 are shown in red in Van deer Waals representations to highlight possible steric clashes with the ligands. The aromatic substituents stemming off the six-atom ring containing the protonated amine of these two ligands and oriented toward M2-M3-M7 are shown superimposed for comparison, with CPPMA in yellow and Ro 62-4599 in green. The equivalent chemical moiety of a third compound, the D2-selective compound 10 (see Fig. 2), is included (orange) to highlight its different orientation within the binding site.
The ligand-receptor interactions shown in Fig. 5E for the D2 receptor can be compared with an analogous representation for the six-residue D4-like D2 mutant receptor shown in Fig. 5F. In E, Ro 62-4599 (green) clashes with W2.60 and L2.64, but the ligand no longer clashes with L2.60 and S2.64 in the D4-like mutant (F). In contrast, CPPMA, because of the different orientation of its aromatic ring, does not clash with W2.60 or L2.64 in the D2 receptor (E). Indeed, the affinity of CPPMA for the four-residue D2 mutant is severalfold higher than for the six-residue mutant or the wild-type D4 receptor (Table 3), consistent with the favorable interaction of CPPMA with W2.60 when F2.61, L3.28, M3.29, and V7.35 are also present.
To complement the side view in Fig. 5, E and F, Fig. 5, G and H show a close-up extracellular view of the proposed ligand-receptor interactions for D2 and D4-like mutant D2 receptors, respectively, following similar conventions and color schemes. Note that the backbone of M7 is now shown, and the six critical D2 and D4 residues are shown in liquorize to facilitate a view of the ligand moieties. The aromatic moieties of both CPPMA and Ro 62-4599 clash with F3.28 and Y7.35 (E and G), and this clash is relieved by mutation to L3.28 and V7.35 (F and H). The D2-selective compound 10 (orange) does not clash with F3.28, F2.60, or L2.64 but is instead oriented toward M7, where it has a favorable interaction with Y7.35.
Discussion
Based on our identification by SCAM of the residues forming the surface of the binding-site crevice of the dopamine D2 receptor, we targeted accessible residues not conserved in the homologous dopamine D4 receptor as candidates for the structural determinants of pharmacological specificity. Excluding six conservative aliphatic substitutions (Fig. 1), this reduced the number of candidate residues approximately 6-fold and left 14 residues. We reasoned that mutation of these candidate residues in the D2 receptor to the aligned residues in the D4 receptor would generally be well tolerated, based on our previous experience in mutating them to cysteine. Indeed, we found that all of the resulting mutants expressed at the cell surface and boundN-methylspiperone with near wild-type affinity.
The combination of the four mutations V91F/F110L/V111M/ Y408V in the D2 receptor increased the binding affinity of CPPMA by three orders of magnitude to D4-like affinity (Fig. 3), suggesting that these four residues encode the molecular determinants for CPPMA D2/D4 binding specificity. The four residues arise from three different membrane-spanning segments (M2, M3, and M7) but are predicted to be in spatial proximity to one another (Fig. 5) and to form part of an M2-M3-M7 microdomain characterized in the D2 receptor by a high density of aromatic residues (Fig. 4A) (Javitch et al., 1999). Notably, this aromatic microdomain is highly divergent between the D2 and the D4 receptors, with the D4 receptor missing four accessible aromatic residues present in the D2 receptor (Fig. 4, B and C). Because this dense and divergent cluster of aromatic residues is adjacent to the protonated amine moiety of the ligand (Fig. 4A), we rationalized that aromatic, bulky substituents oriented toward this M2-M3-M7 microdomain, such as in CPPMA (Fig. 2), may clash in the D2 receptor and yet fit in the D4 receptor (Fig. 5, A and B). This hypothesis is schematically illustrated in Fig. 6 in a simplified illustration.

Schematic model of the proposed interaction of the studied D4-selective compounds with the D2 and D4 receptors. The seven membrane-spanning segments (M1–M7) of the D2 (A) and D4 (B) receptors are shown in an extracellular view and are arranged based on the projection map of rhodopsin. A, we have identified the presence of a dense cluster of aromatic residues within M2-M3-M7 of the D2 receptor (black “bumpers”). This aromatic microdomain in the D2 receptor could act as a barrier, preventing binding of aromatic, bulky substituents stemming from the protonated amine of D4-selective ligands toward M2-M3-M7. B, the D4 receptor lacks several of the aromatic residues present in the aromatic microdomain of the D2 receptor. Thus, ligands having aromatic, bulky substituents stemming from the protonated amine of D4-selective ligands fit in the binding-site crevice of the D4 receptor as shown here schematically. Moreover, these D4-selective ligands gain an additional stabilizing interaction with F2.61 in M2 of the D4 receptor (see text). Compounds having these chemical moieties, properly distributed in space as shown at the molecular level in Fig. 5, are likely to be D4-selective ligands.
Our docking results suggested two major factors in the D4 selectivity of CPPMA: the gain of an interaction of the chlorophenyl substituent with Phe91 and the loss of steric clash of the ligand with Phe110 and Tyr408 (Fig. 5, A, B, and E–H). Although the solitary mutation of Val111(3.29) to Met did not affect CPPMA affinity, combination of this mutation with V91F/F110L/Y408V led to a greater than 30-fold increase in CPPMA affinity to D4-like levels. The mechanism of this increased affinity is not clear and may reflect indirect effects of the Val to Met mutation on the conformation of the residues at positions 91, 110, and 408 or a direct ligand interaction of Met111 that requires the presence of the D4 residues at positions 91, 110, and 408.
To test the generality of this structural motif to the selectivity of other D4 ligands, we studied three other D4-selective ligands that also contained aromatic substituents at the protonated amine. Ro 61-6270 had properties similar to those of CPPMA, in that mutation of the four residues (V91F/F110L/V111M/Y408V) was sufficient to convert the receptor fully to D4-like affinity. In contrast, these four mutations did not fully convert the affinity of the receptor for Ro 10-4548 and R0 62-4599 but left the receptor with intermediate affinity between that of the D2 and the D4 receptors. In our docking, the 90-degree rotation of the phenyl ring of Ro 62-4599 relative to that of CPPMA produced a clash with Trp90 and Leu94 in the D2 receptor (Fig. 5, E and G), which was relieved by the substitution of these residues with the smaller aligned D4 residues (Fig. 5, F and H). Consistent with this, the mutation of W90L and L94S combined with V91F/F110L/V111M/Y408V more fully converted the affinity of Ro 62-4599 and Ro 10-4548 toward D4-like values.
Because simultaneous substitution of the identified residues in the D4 receptor with the aligned residues from the D2 receptor lowered the affinity of the receptor for CPPMA, Ro 61-6270, Ro 10-4548, and 62-4599 to near D2-like levels, this set of six residues that cluster between M2-M3-M7 acts as a functional motif that can be interchanged between the D2 and D4 receptor. In contrast to the loss of affinity seen by substitution of the identified residues in M2 and M3, however, solo substitution of Val350(7.35) with Tyr did not decrease the affinity for CPPMA, suggesting that this residue may not directly clash with CPPMA but rather may exert an indirect effect on binding. It is also possible that, in the absence of the other critical D4 residues, CPPMA can rearrange within the binding pocket to counteract the detrimental effect of the mutation.
These findings support the importance of the M2-M3-M7 microdomain in the D4 selectivity of multiple chemically distinct ligands and suggest a set of structural properties that confer D4 selectivity: the presence of a protonated amine in a ring and an aromatic substituent stemming from each side of the ring, one interacting with the M2-M3-M7 divergent aromatic cluster (Fig. 2, left) and the other interacting with the M5-M6 aromatic cluster (Fig. 2, right). This hypothesis is, of course, an oversimplified model for the complex interactions of these chemical compounds with D2 and D4 receptors. Nonetheless, it is useful in providing clear guidelines that can be readily applied to drug design. It must be emphasized, however, that small differences in the geometry and orientation of these chemical groups can dramatically alter their functional role. For example, compounds 9 and 10 described inKulagowski et al. (1996) contain the identical chlorophenyl substituents on a nitrogen-containing ring as does CPPMA (Fig. 2). Nonetheless, compound 9 is 44-fold D4 selective, whereas compound 10 is 92-fold D2 selective (Kulagowski et al., 1996). Note in Fig. 5, E through H, the conformational effects of the different geometry of a single chemical bond in compound 10, linking the chlorophenyl substituent to the nitrogen-containing six-atom ring in a tetrahedral orientation (Sp3 hybridization) rather than the planar orientation (Sp2 hybridization) of the identical substituent in CPPMA (yellow). As a result, the chlorophenyl substituent in compound 10 is oriented away from M2-M3 and toward M7 (G and H), avoiding the clash with M2-M3 and gaining a favorable interaction with Tyr408 in the D2 receptor (E and G) while losing the favorable interaction with Phe2.61 in the D4 receptor (F, H). This alternate interaction pattern can explain the reversed D2/D4 selectivity of compound 10 relative to that of CPPMA and compound 9.
Whereas several chimeric studies demonstrate the contribution of large domains to creating high-affinity binding sites (Kozell and Neve, 1997;Kobilka et al., 1988), there are fewer cases where individual residues not adjacent in primary sequence have been shown to have combined effects on specificity (Wu et al., 1994; Ji et al., 1995). In these cases, the residues found to be important for selectivity were identified through exhaustive screening of different combinations of substitutions. We were able to use the accessibility pattern determined by SCAM to narrow our initial screen and to identify a cluster of residues within a three-dimensional structural context from three different membrane-spanning segments that contribute to creating a high-affinity antagonist binding site for D4-selective ligands. Thus, our findings emphasize the importance of considering the role of three-dimensional structural microdomains in receptor function (Ballesteros et al., 1998) as opposed to simply considering the structural and functional roles of single residues or stretches of residues in the primary structure.
Although the exact degree of structural similarity between related GPCRs is unknown, the surface of the binding-site crevice identified in the D2 receptor may represent a close approximation of a universal potential surface for the interaction of rhodopsin-like GPCRs with ligands. In other related GPCRs, mutation of residues at positions 2.60, 2.61, 2.64, 3.28, 3.29, and 7.35 have been reported to affect ligand binding (Kristiansen et al., 1996; van Rhee and Jacobson, 1996). Thus, the residues on the surface of this crevice are potential targets for novel drugs, even if the residues are not contacts for currently existing compounds. As we found in this study, we would expect that different positions and residues play a more or less critical role in different receptors and with different ligands.
Although we found the multiple and variant ligand-receptor interactions to be complex at the atomic level, a molecular level of understanding could be accomplished by considering ligand-receptor interactions in terms of chemical moieties of the ligands (e.g., an aromatic moiety stemming from the protonated amine) interacting with specific receptor motifs (e.g., a divergent aromatic cluster in M2-M3-M7). Notably, understanding the key ligand-receptor interactions at this level may be sufficient to critically inform the drug discovery process and may be especially suited for guiding the design of combinatorial chemistry libraries. Although the residues we identified in this article are critical in determining the pharmacological specificity of the particular D4 receptor antagonists we studied, it is possible, or even likely, that other D4-selective compounds contact an alternate or overlapping set of residues accessible in the binding-site crevice. Indeed, substitutions in the aromatic/H-bonding moiety that is expected to interact with the M5-M6 aromatic cluster also affect the D4 selectivity of analogs of CPPMA (Kulagowski et al., 1996), suggesting the presence of additional structural determinants of specificity in this domain as well. Because none of the substitutions of these divergent residues in M5 and M6 of the D2 receptor substantially affected the selectivity of CPPMA, it is possible that differences in the backbone and/or helical packing of M5 and M6, rather than simple differences in side-chain identity, may contribute toward the specificity of other compounds and may in fact account for our inability to completely switch the specificity of Ro 62-4599 to D4-like levels.
Acknowledgments
We thank Olivier Civelli and Steve Rees for the human D2 receptor cDNA and the pcin4 vector, respectively. We thank Thomas Livelli for the HEK 293 cells and for valuable advice. We thank Myles Akabas, Arthur Karlin, George Liapakis, Irache Visiers, and Harel Weinstein for valuable discussion and for comments on the manuscript.
Footnotes
- Received July 19, 1999.
- Accepted September 20, 1999.
-
Send reprint requests to: Dr. Jonathan A. Javitch, Center for Molecular Recognition, Columbia University, P & S 11-401, 630 West 168th St., New York, NY 10032. E-mail: jaj2{at}columbia.edu
-
↵1 Current affiliation: Department of Lead Discovery, Astra Arcus USA, Inc., Worcester, MA 01605.
-
↵2 Current affiliation: Linguagen Corp., Clifton, NJ 07015.
-
This work was supported in part by National Institutes of Health Grants MH-57324 and MH-54137, the G. Harold & Leila Y. Mathers Charitable Trust, the Lebovitz Trust, and the Aaron Diamond Foundation.
Abbreviations
- CPPMA (chlorophenylpiperazinyl methylazaindole)
- 3-[4-(4-chlorophenyl)piperazin-1-yl]methyl-1H-pyrrolo[2,3-b]pyridine
- Mn
- nth membrane-spanning segment
- SCAM
- substituted-cysteine accessibility method
- GPCRs
- G-protein-coupled receptors
- HEK
- human embryonic kidney
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}