


Abstract
G protein-coupled receptors (GPCRs) are key regulators of cell physiology and control processes ranging from glucose homeostasis to contractility of the heart. A major mechanism for the desensitization of activated GPCRs is their phosphorylation by GPCR kinases (GRKs). Overexpression of GRK2 is strongly linked to heart failure, and GRK2 has long been considered a pharmaceutical target for the treatment of cardiovascular disease. Several lead compounds developed by Takeda Pharmaceuticals show high selectivity for GRK2 and therapeutic potential for the treatment of heart failure. To understand how these drugs achieve their selectivity, we determined crystal structures of the bovine GRK2-Gβγ complex in the presence of two of these inhibitors. Comparison with the apoGRK2-Gβγ structure demonstrates that the compounds bind in the kinase active site in a manner similar to that of the AGC kinase inhibitor balanol. Both balanol and the Takeda compounds induce a slight closure of the kinase domain, the degree of which correlates with the potencies of the inhibitors. Based on our crystal structures and homology modeling, we identified five amino acids surrounding the inhibitor binding site that we hypothesized could contribute to inhibitor selectivity. However, our results indicate that these residues are not major determinants of selectivity among GRK subfamilies. Rather, selectivity is achieved by the stabilization of a unique inactive conformation of the GRK2 kinase domain.
Introduction
G protein-coupled receptor kinases (GRKs) catalyze the phosphorylation of serine and threonine residues in the cytoplasmic tails and loops of activated G protein-coupled receptors (GPCRs) (Krupnick and Benovic, 1998). These phosphorylated receptors are then bound by molecules of arrestin, which uncouple the GPCRs from G proteins, target the receptors to clathrin-coated pits for endocytosis, and serve as adaptors for other signaling pathways such as those of mitogen-activated protein kinases. GRKs are found in all metazoans and are classified into three subfamilies based on their gene structure and homology. The GRK1 subfamily is vertebrate-specific and consists of GRK1 (rhodopsin kinase) and GRK7, which are expressed in the rod and cone cells of the retina. The GRK2 subfamily, consisting of GRK2 and GRK3, are ubiquitously expressed. The GRK4 subfamily consists of GRK4, GRK5, and GRK6. GRK5 and GRK6 are ubiquitously expressed, whereas GRK4 is found primarily in testes and kidneys.
The central, catalytic domain of GRKs is a serine/threonine kinase domain ∼32% identical in sequence to the catalytic subunit of protein kinase (PK) A and is thus a member of the PKA, PKG, and PKC (AGC) family of kinases (Manning et al., 2002). The kinase domain consists of two lobes, termed the small (or N) and large (or C) lobes (Fig. 1). ATP binds at the interface of these lobes, adjacent to a shallow canyon formed primarily by the large lobe where polypeptide substrates bind. The ATP-binding site is highly conserved among all protein kinases and is the binding site for most reported inhibitors of GRKs and other kinases (Johnson, 2009). There are several critical structural elements that cluster around the ATP-binding site of protein kinases (Fig. 1), including the phosphate-binding loop (P-loop), the αC-helix, the hinge connecting the large and small lobes, and the activation loop, which is typically a site of phosphorylation (although not in GRKs). Because of the high conservation of the ATP-binding site among of the ∼500 kinases, the majority of small molecule kinase inhibitors target the ATP-binding site in a binding mode similar to that of ATP itself, generally resulting in inhibitors that lack selectivity (Bogoyevitch and Fairlie, 2007). However, with the discovery of imatinib it became clear that the inactive conformation of a given kinase can be quite unique and therefore targeted to produce selective inhibitors (Noble et al., 2004; Breitenlechner et al., 2005a; Rabiller et al., 2010).

Structural features of GRK2. GRK2 is oriented to show the ATP-binding site with the kinase domain colored green and the regulator of G protein signaling homology (RH) and pleckstrin homology (PH) domains colored slate. ATP binds between the small and large lobes (connected via the hinge region) and is modeled on the basis of the GRK1-ATP structure (PDB 3C4W). Most small molecule kinase inhibitors target the ATP-binding site. The structure shown corresponds to that of the GRK2-Gβγ complex (PDB 3PSC). The Gβγ subunits, which bind to the PH domain, are omitted for clarity.
Since the discovery of a linkage between the overexpression of GRK2 and heart failure, GRK2 has been considered a pharmaceutical target for the treatment of cardiovascular disease (Dorn, 2009). The first reported inhibitors of GRK2 were polyanionic compounds, such as heparin and dextran sulfate, that had nanomolar potencies but were considerably nonselective (Benovic et al., 1989). Other GRK2 inhibitors represent poor drug leads because of their lack of potency, selectivity, or non–drug-like properties (Benovic et al., 1990; Palczewski et al., 1990; Iino et al., 2002; Winstel et al., 2005). For example, the natural product balanol is a potent inhibitor of GRK2 but is relatively nonselective among AGC kinases (Setyawan et al., 1999; Tesmer et al., 2010).
Structural analysis of bovine GRK2 has led to several crystal structures including complexes with the heterotrimeric G proteins, Gαq and Gβγ, and balanol (Lodowski et al., 2003b; Tesmer et al., 2005, 2010). Comparison of the apoGRK2 structure with the balanol complex revealed that balanol stabilizes a slightly more closed conformation of the kinase domain (4° closure of the large lobe relative to the small lobe). However, an additional 16° rotation of the large lobe is still required to achieve what is expected to be the fully closed state. Subtle conformational changes were also observed in the P-loop and αB-αC-helices of the small lobe with residues moving up to 1.6 Å away from the active site to accommodate the ligand. Thus, balanol seems to recognize and stabilize a unique inactive conformation of GRK2 (Tesmer et al., 2010).
A class of heterocyclic compounds (Ikeda et al., 2007) that show therapeutic potential and exhibit higher selectively for GRK2 than balanol was discovered by Takeda Pharmaceutical Company Ltd. (Osaka, Japan) (Fig. 2). To determine the mechanism of selectivity for these inhibitors, we cocrystallized two of the compounds (CMPD103A and CMPD101) with the GRK2-Gβγ complex. We then tested whether GRK2 subfamily-specific residues in the P-loop and αB-αC-loop region could contribute to the selectivity of these compounds for GRK2. However, converting these residues to their equivalents in GRK1 had only subtle effects on their potency. Instead, the conformation of the kinase domain of GRK2 in its inactive state relative to other GRKs probably provides the most important contribution to selectivity.
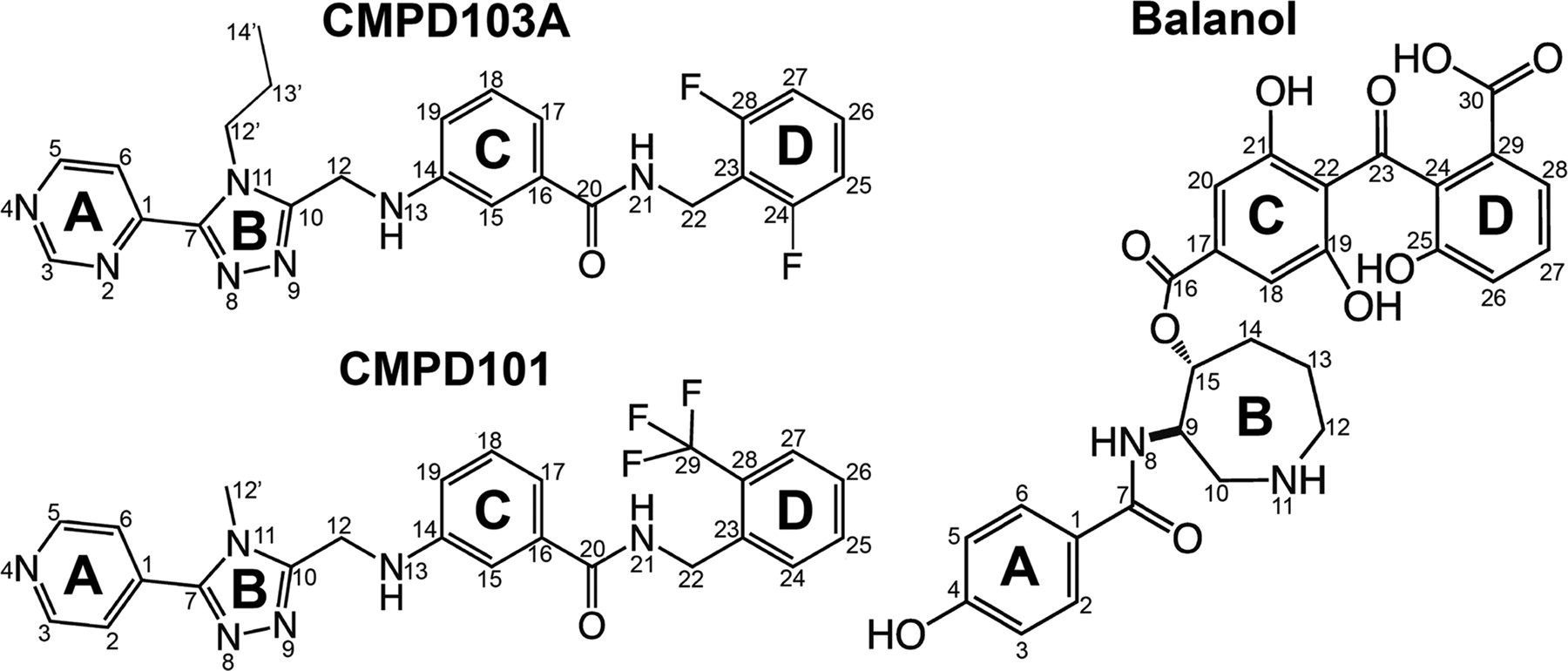
Chemical structures of three potent small molecule inhibitors of GRK2. CMPD103A and CMPD101 are potential cardiotonic drugs discovered by Takeda Pharmaceutical Company, Ltd. (Ikeda et al., 2007). Balanol is a less selective AGC kinase inhibitor isolated from the fungus Verticillium balanoides (Koide et al., 1995). The ring structures are labeled as follows for CMPD103A/CMPD101: A, pyrimidine/pyridine; B, substituted 1,2,4-triazole; C, aminobenzamide; D, substituted benzene; and for balanol: A, p-hydroxybenzamide; B, azepane; C and D, substituted and esterified benzophenones.
Materials and Methods
Reagents.
Balanol was purified from a natural source as described previously (Tesmer et al., 2010). CMPD103A and CMPD101 (Fig. 2) were synthesized as described previously with one modification (Ikeda et al., 2007). For CMPD103A, a pyrimidine on ring A was substituted for a pyridine group. [γ-32P]ATP was purchased from MP Biomedicals (Solon, OH). Dark-adapted bovine retinas were purchased from W. L. Lawson Company (Lincoln, NE). 1-Anilinonaphthalene-8-sulfonic acid (ANS) was purchased from Sigma-Aldrich (St. Louis, MO).
Protein Expression and Purification.
GRK2 was expressed from a baculovirus vector containing the cDNA for bovine GRK2-S670A with an engineered C-terminal hexahistidine tag and purified as described previously (Huang et al., 2009). Bovine GRK1 and GRK5 were expressed as C-terminal truncations (GRK1535 and GRK5561) and were purified as described previously (Huang et al., 2009). Mutagenesis of GRK2 was performed using the QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). Mutations were verified using DNA sequencing. Geranylgeranylated bovine Gβ1γ2 was expressed and purified as described previously (Lodowski et al., 2003a). All proteins were frozen in small aliquots at −80°C for storage, and their concentrations were determined by absorbance at 280 nm.
Crystallization of GRK2-Gβγ Complexes.
The GRK2-Gβγ complex was formed by mixing purified bovine GRK2-S670A (200 μl, 2.14 mg) with purified Gβγ (200 μl, 1.6 mg) and then was supplemented with additional CHAPS and MgCl2 to final concentrations of 10 and 5 mM, respectively. The protein mixture was incubated on ice for 30 to 60 min and filtered with a 0.2-μm Nanosep device (Pall Life Sciences, Ann Arbor, MI) and then loaded onto two tandem S200 gel filtration columns. Formation of the complex was verified by SDS-PAGE, and the GRK2-Gβγ-containing fractions were pooled and concentrated (Amicon Ultra 50 kDa; Millipore Corporation, Billerica, MA).
For cocrystallization of compounds with GRK2-Gβγ, CMPD103A and CMPD101 solubilized in 100% DMSO were added to the concentrated GRK2-Gβγ complex and incubated on ice for 30 min at a final concentration of 100 μM each (2% DMSO final). Crystals were grown at 4°C by the hanging drop vapor diffusion method with crystals observable after 1 day. The best diffraction data for a GRK2-CMPD103A-Gβγ crystal was collected from crystals harvested from drops composed of 2 μl of protein (10 mg/ml protein) mixed with 2 μl of well solution (7% PEG3350, 100 mM MES, pH 6.25, and 250 mM NaCl). The best data for GRK2-CMPD101-Gβγ were collected from crystals harvested from drops composed of 2 μl of protein (10 mg/ml protein) mixed with 2 μl of well solution (7% PEG3350, 200 mM NaCl, and 100 mM MES, pH 5.25). For comparison, GRK2-ATP-Gβγ crystals were generated by addition of 1 mM ATP to the initial GRK2-Gβγ complex and then crystallized using 1 μl:1 μl hanging drops with a well solution containing 9% PEG3350, 200 mM NaCl, and 100 mM MES, pH 6.5. All crystals were harvested into a cryoprotectant solution containing: 20 mM HEPES, pH 8.0, 100 mM MES (at the well solution pH), 300 mm NaCl, 10 mM CHAPS, 5 mM MgCl2, 2 mM dithiothreitol, 9% PEG3350, 25% ethylene glycol, and either 100 μM CMPD103A, 100 μM CMPD101 (in 100% DMSO, final concentration 2% DMSO), or 1 mM ATP. Then 2% DMSO was added to the harvesting solution for the GRK2-ATP-Gβγ crystals.
Data Collection and Structure Determination.
The GRK2-Gβγ complexes crystallized in space group C2, and data were collected at Advanced Photon Source beamline 21 ID-G. The diffraction data for all three structures is strongly anisotropic with the highest resolution data extending in a direction bisecting the a* and c* axes of the crystals, as described previously (Lodowski et al., 2003a), thus contributing to the poor crystallographic data and refinement statistics in the higher resolution data shells (Table 1). Data were integrated and scaled using HKL2000 (Otwinowski and Minor, 1997), and the structures were solved using molecular replacement with the original GRK2-Gβγ structure (PDB 1OMW) (Lodowski et al., 2003b) as the starting model (REFMAC5). Models for the ligands were generated using Sketcher (CCP4 6.1) and PRODRG (Schuttelkopf and van Aalten, 2004; http://davapc1.bioch.dundee.ac.uk/prodrg/). The ligand-bound GRK2-Gβγ models were built using Coot (Emsley and Cowtan, 2004) and refined using TLS and restrained refinement in REFMAC5 (Murshudov et al., 1997; Winn et al., 2001). MolProbity (Chen et al., 2010) and PROCHECK (Laskowski et al., 1993) were used for structure validation.
Crystallographic data and refinement statistics
Phosphorylation of Rhodopsin.
Urea-washed bovine rod outer segments (bROS) were purified as described previously (Papermaster, 1982). Steady-state kinetic experiments were conducted using saturating concentrations of ATP (0.5 mM ATP + [γ32P]ATP), 2 to 20 μM bROS, and 50 nM GRK in a buffer containing 20 mM HEPES, pH 8.0, 1 mM CHAPS, 5 mM MgCl2, and 2 mM dithiothreitol. The reactions were performed in a 96-well polymerase chain reaction plate. For inhibition assays, 5 μl of varying concentrations of compound were added to each well, followed by addition of 5 μl of GRK and then 5 μl of bROS (added in the dark). The plate was then allowed to equilibrate for at least 30 min. The reaction was initiated by the addition of 5 μl of ATP and exposure to light at room temperature (∼25°C). The reaction was quenched after 5 to 10 min with the addition of 4 μl of SDS-PAGE loading buffer. Reactions were then analyzed by SDS-PAGE. The gels were dried and exposed to a phosphor-imaging screen, and phosphorylated rhodopsin was quantified using a Typhoon 9410 imager (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK). For determining ATP Km values, the data were fit to the Michaelis-Menten equation using Prism v5.0c. For calculating IC50 values, the data were fit to log (inhibitor) versus response with either a fixed or variable slope.
Thermostability Measurements.
Melting temperatures (Tm) were determined by monitoring the fluorescence change of ANS as it binds to the hydrophobic interior of proteins upon denaturation (Mezzasalma et al., 2007). GRK2 or GRK variants (0.05–0.2 mg/ml) were incubated at saturating ligand concentrations and 100 μM ANS in a total volume of 10 μl in triplicate, using ABgene 384-well polymerase chain reaction microtiter plates (Thermo Fisher Scientific, Waltham, MA). Fluorescence was measured at increasing temperatures (4–85°C) in 1°C intervals using a ThermoFluor 384-well plate reader (Johnson & Johnson, New Brunswick, NJ). The fluorescence data were analyzed using ThermoFluor Acquire 3.0 software (default settings).
Results
Determination of Inhibitor Selectivity for GRKs.
CMPD103A and CMPD101 (Fig. 2) are highly potent and selective inhibitors of GRK2 versus PKA, PKC, and Rho kinase (Ikeda et al., 2007). For example, CMPD101 inhibits GRK2 with an IC50 of 35 nM, but >2 μM for other tested kinases. However, the selectivity for members of the GRK family was not reported. We therefore tested the activity of balanol in phosphorylation assays against bovine GRK1535-H6 (GRK1), bovine GRK2-H6 (GRK2), and bovine GRK5561-H6 (GRK5) using bROS as the receptor substrate and ATP at saturating concentrations (0.5 mM). Balanol had an IC50 of 35 nM for GRK2 (Fig. 3A; Table 2), similar to previously reported values (Tesmer et al., 2010). However, balanol was a less potent inhibitor of GRK5 and GRK1 (IC50 values of 440 nM and 4.1 μM, respectively) than previously reported (Tesmer et al., 2010), most likely because of different assay conditions (e.g., the soluble substrate tubulin and 3 μM ATP were used as substrates). Regardless, the selectivity order is the same (GRK2 > GRK5 > GRK1). We then tested CMPD103A and CMPD101 for their ability to inhibit bROS phosphorylation by GRK2, which yielded IC50 values of 54 and 290 nM, respectively (Fig. 3B; Table 2). The IC50 value for CMPD103A is similar to the reported IC50 value for its parent compound (27 nM); however, CMPD101 is 8-fold less potent than its reported value (Ikeda et al., 2007), with the difference again most likely a result of different assay conditions. Neither CMPD103A nor CMPD101 inhibited GRK1 or GRK5 at any of the concentrations tested (up to 125 μM) (Fig. 3, C and D). However, both did seem to increase the activity of GRK1 by up to 3-fold (see Discussion).

Inhibition of GRK-mediated phosphorylation of bROS by small molecules. A, inhibition of GRK1, GRK2, and GRK5 activity by balanol. Modulation of GRK2 (B), GRK5 (C), and GRK1 (D) activity by CMPD103A, CMPD101, or the negative control DMSO. In A and B, data are normalized to percent GRK2 and GRK5 activity based on a negative control containing no inhibitor and a positive control equivalent to background. The GRK1 data are normalized to the lowest concentration of balanol tested because of the increase in activity versus the control as also seen in D. Data shown are representative of three or more experiments performed in duplicate (Table 2). CMPD, compound.
ATP Km values and inhibitor IC50 values for GRK variants
All experiments were performed with 2 to 20 μM bROS and 50 nM GRK with the reaction quenched at a single time point between 5 and 10 min at ∼25°C. For determining IC50 values, 0.5 mM ATP was used. Values shown represent means ± S.E.M. from three to seven experiments, each run in duplicate.
To determine whether the Takeda compounds are selective for GRK2 or instead for all members of the GRK2 subfamily, we tested CMPD101 against human GRK2 and human GRK3 in phosphorylation assays run under our previously reported assay conditions (Tesmer et al., 2010). Under these conditions, CMPD101 inhibited GRK2 and GRK3 with IC50 values of 54 and 32 nM, respectively (data not shown). Therefore, CMPD103A and CMPD101 are selective for the GRK2 subfamily, which is not surprising given that the primary sequences of their kinase domains are 92% identical.
X-Ray Crystal Structures.
To better understand how CMPD103A and CMPD101 achieve their selectivity, we cocrystallized these inhibitors with the GRK2-Gβγ complex and solved their atomic structures using diffraction data extending to 2.5 Å spacings. As a control, the GRK2-Gβγ complex was cocrystallized with ATP under similar conditions, and the structure was solved using data to 2.7 Å spacings (Table 1). The omit map for ATP revealed only weak electron density in the active site, as in the original GRK2-Gβγ structure (Lodowski et al., 2003b), and hence it is referred to herein as the “ligand-free” structure or apoGRK2.
Both CMPD103A and CMPD101 bind deep in the active site of GRK2 and overlap extensively with the binding site for ATP (Fig. 4, A and B). The primary difference in their conformation is in their “D” rings, which are the most chemically divergent (Figs. 2 and Fig. 4, A and B). The binding of CMPD103A and CMPD101 induce similar conformational changes in the P-loop and αB-αC-loops of the kinase domain relative to the apoGRK2-Gβγ structure, in which individual atoms move up to 0.8 and 0.9 Å, respectively. For example, the side chains of Ile196, Ile197, and Leu235 all adopt different rotamer conformations to accommodate ligand binding. The binding of CMPD103A and CMPD101 also induces a slight closure of the kinase domain, with the large lobe rotating relative to the small lobe by 3.6 and 2.4°, respectively (Fig. 4C).

Crystal structures of inhibitor-bound GRK2. All ligands are shown as a ball and stick model with carbons colored white, nitrogens blue, oxygens red, and fluorines yellow. CMPD103A (A) and CMPD101 (B) bind in the active site of GRK2. Corresponding 3σ |Fo| − |Fc| omit map density is shown as a green cage. C, superposition of the Cα traces of apoGRK2 (blue) and CMPD103A (green). The large lobe, the P-loop, and the αB-αC-loop undergo small conformational changes upon inhibitor binding. The small lobe of GRK2 (residues 181–190, 212–223, and 247–268) was used to superimpose the structures.
The ATP-binding site is composed of several binding pockets including the adenine, ribose, triphosphate, and hydrophobic subsites. The A rings of CMPD103A and CMPD101 bind in the adenine subsite and form a hydrogen bond between the pyridine/pyrimidine N4 atom of the A ring and the backbone amide nitrogen of Met274 in the hinge of the kinase domain (Fig. 5A). The ribose subsite is partially occupied by the B ring 1,2,4-triazole moiety (Fig. 5B), which sits deeper in the binding pocket than ribose and forms a hydrogen bond with the free amine of Lys220 and nonpolar interactions with the side chains of Ile197, Val205, Leu271, and Ser334.

Interactions of the Takeda inhibitors in the four ATP subsites. Carbons in ATP are colored pink, CMPD103A white, and CMPD101 cyan. ATP is modeled from PDB 3C4W. The structure shown is that of the GRK2-CMPD103A complex. Dashed lines indicate hydrogen bonds. A, the adenine subsite. B, the ribose subsite. C, the triphosphate subsite. D, the hydrophobic subsite. The side chains of residues Phe202, Leu235, and Leu338 are shown as spheres and make up one wall of the subsite.
The aryl C rings of CMPD103A and CMPD101 are positioned in the triphosphate subsite with the amino group forming hydrogen bonds with the side chains of Asp335 and Lys220. The carbonyl oxygen of the benzamide ring is positioned under the P-loop and is within hydrogen bond distance to the backbone amide nitrogens of Gly201, Phe202, and Gly203 (Fig. 5C). The position and interactions of the benzamide ring in the active site are analogous to that formed by the structurally similar benzophenone ring of balanol (Tesmer et al., 2010). The D rings of CMPD103A and CMPD101 form nonpolar interactions with Gly201, Phe202, Leu235, Glu239, Gly337, and Leu338 in the hydrophobic subsite (also termed the “allosteric site”) formed by the P-loop, the αB- and αC-helices, and the activation loop (Fig. 5D).
Potential Determinants of Selectivity in the Inhibitor-Binding Site.
The binding of balanol, CMPD103A, and CMPD101 induces very similar conformational changes in the active site of GRK2. However, our kinetic data indicate that CMPD103A and CMPD101 are much more selective toward GRK2 than balanol (Fig. 3). Although residues that form the ATP-binding site are very well conserved among AGC kinases (Fig. 6), analysis of our crystal structures indicated that five amino acids in the vicinity of the inhibitor-binding site could contribute to selectivity. The P-loop undergoes significant conformational changes in our inhibitor bound structures, and Ile197, one of three nonconserved residues in the P-loop, makes a nonpolar contact with each inhibitor (Fig. 7A). A direct contact is also formed by Leu235 in the hydrophobic subsite, whose side chain adopts a different rotamer than in the apoGRK2 structure to accommodate the D rings of CMPD103A and CMPD101 (Fig. 7B). The equivalent residue is Gly in GRK1 and Met in GRK4, -5, -6, and -7. Finally, the “gatekeeper residue,” which sits at the back of the adenine subsite (Fig. 5A), is known to be a determinant of inhibitor specificity (Zuccotto et al., 2010). In GRK2, the gatekeeper residue is Leu271, whose side chain contacts the A and B rings of the inhibitors and is not conserved in the GRK1 subfamily.
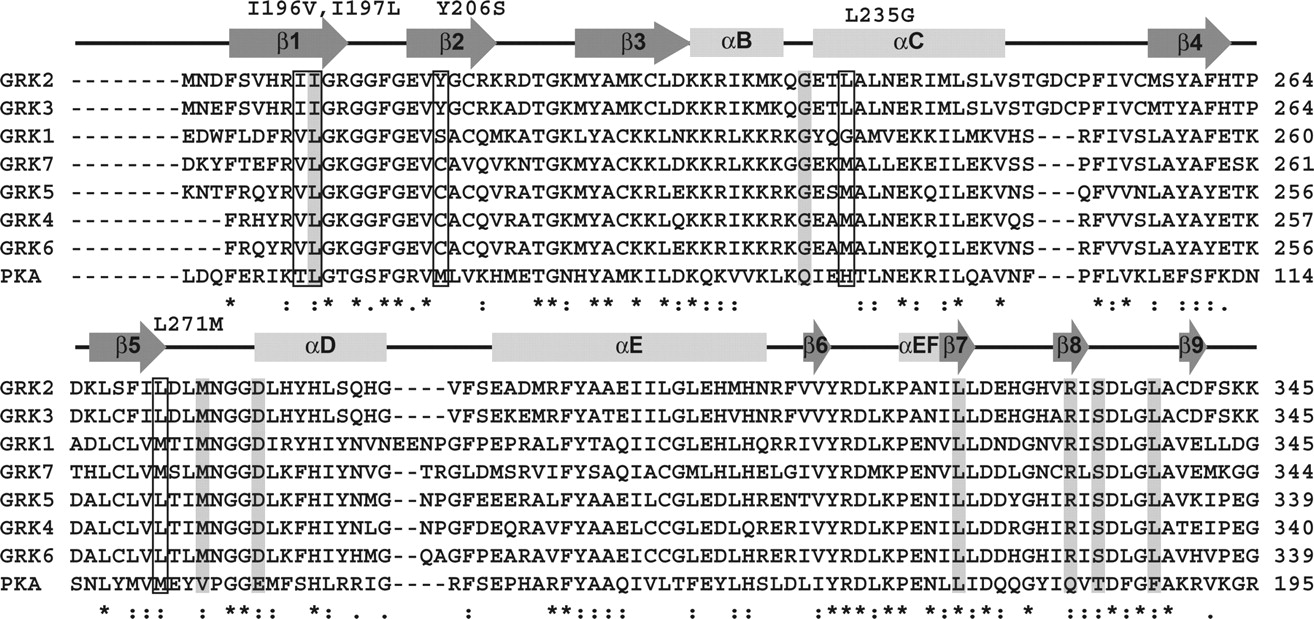
Sequence conservation within the GRK kinase domain. The kinase domain of human GRK1–7 and bovine PKA were aligned using the ClustalW server (bovine GRK2, which was used in this study, has 100% identity in the kinase domain with that of human). Secondary structure is indicated by arrows for β-strands and rectangles for α-helices. Mutations listed along the top correspond to residues in GRK2 (also boxed) that were selected for mutagenesis to the corresponding GRK1 residue. Columns colored gray correspond to positions in either PKA or PKB that have been previously mutated to assess mechanisms of inhibitor selectivity.

Conformational changes in GRK2 upon ligand binding. Electron density is shown for a 3σ |Fo| − |Fc| omit map of the GRK2-CMPD103A-Gβγ structure. Positive and negative density is shown in green and red, respectively. Residues from the apoGRK2-Gβγ structure are colored slate, and residues from the GRK2-CMPD103A-Gβγ structure are colored green. The labeled residues correspond to the mutation from bovine GRK2 to human GRK1 (red). A, nonconserved residues in the P-loop and hinge region undergo small conformational changes upon inhibitor binding. B, displacement of Leu235 by the D ring of CMPD103A.
These five positions were mutated to their equivalents in GRK1 (I196V, I197L, Y206S, L235G, and L271M) to test whether they reduced the affinity of the Takeda compounds. With the exception of the I196V mutant, which did not express, all were purified to homogeneity from baculovirus-infected insect cells. To test whether these mutants produced functional protein, we determined the Km value of ATP for GRK1, 2, 5, and for each GRK2 mutant (Table 2). If the mutants are correctly folded, they should not exhibit greatly different Km values, because all GRK active sites are optimized to bind ATP. GRK1 (Palczewski et al., 1988), GRK2 (Kim et al., 1993), and GRK5 (Kunapuli et al., 1994) exhibited Km values for ATP similar to previously reported values (Table 2), and all GRK2 mutants had Km values for ATP similar to that of wild-type, allowing us to directly compare IC50 values for the various GRK2 mutants.
We then tested the ability of balanol to inhibit the various GRK2 mutants (Fig. 8A; Table 2). The IC50 values for balanol against the I197L, Y206S, and L235G mutations showed little difference from those of the wild type. The gatekeeper mutation L271M, however, increased the potency of balanol by 10-fold, which could reflect a reordering of amino acids around the hinge region that creates more favorable hydrophobic contacts with the A ring of the inhibitor.

Inhibition of GRK2 variants by balanol (A), CMPD103A (B), and CMPD101 (C). Representative curves from three or more separate experiments performed in duplicate are shown for each mutant and inhibitor. Data are normalized to percent GRK2 activity based on a negative control containing no inhibitor and a positive control equivalent to background. Dose-response curves were fit to either a three- or four-parameter model to determine IC50 values (Table 2).
The P-loop mutations I197L and Y206S had no significant effect on the inhibition of GRK2 by CMPD103A or CMPD101 (Fig. 8, B and C; Table 2), whereas the L235G mutation in the αB-αC-loop caused a 2- to 3-fold decrease in the potency of CMPD103A and CMPD101, indicating that the hydrophobic subsite in GRKs makes a small contribution to compound selectivity. The L271M mutation seemed to slightly increase the potency of CMPD103A and decrease the potency of CMPD101 (Fig. 8, B and C; Table 2). Although these latter observations are not statistically significant, they could be explained by the fact that the N2 atom of the A ring of CMPD103A, which is a pyrimidine, could be involved in a favorable electrostatic interaction with the sulfur atom of the substituted methionine. However, taken together, our data demonstrate that the unique residues that compose the inhibitor-binding site in GRK2 do not strongly contribute to the affinity of the compounds, at least not when assessed by competition assays.
Ligand-Induced Protein Stabilization.
Compounds that bind to a native protein will typically cause a stabilization of the protein to an extent that depends on the binding energy of the complex. This can be observed as a rightward shift in thermal denaturation curves. Native GRK2 has a Tm of approximately 36°C, and the addition of 500 μM ATP results in a 5°C increase in its Tm, indicating that ATP binds and stabilizes GRK2 (Table 3). The addition of 10 μM balanol, CMPD103A, or CMPD101 to GRK2 increases its stability by 19, 16.0, and 12°C, respectively (Table 3). These ΔTm values are identical to the rank order of the potency of these compounds for inhibiting GRK2 activity (balanol > CMPD103A > CMPD101), indicating that the thermal stability assay can be used to give valid comparisons of ligand affinity and is thus an orthogonal way to examine the selectivity of inhibitors.
Changes in Tm induced by inhibitor binding
ΔTm is calculated by subtracting the native Tm from the inhibitor-bound Tm. Data represent two to six separate experiments, each run in triplicate. All values listed are means ± S.D. The Tm values shown are for the indicated protein in buffer alone.
GRK1 and GRK5 have melting temperatures of 25 and 29°C, respectively, significantly lower than that of GRK2. Addition of 500 μM ATP stabilizes GRK1 and GRK5 by 17 and 7°C, respectively, consistent with the observation that GRK1 has a significantly lower Km value for ATP than either GRK2 or GRK5, which exhibit smaller ΔTm values upon addition of ATP. Addition of 100 μM balanol (10-fold higher than that added for GRK2) is less effective than ATP in stabilizing GRK1 and GRK5, with Tm shifts of only 10 and 6°C, respectively. CMPD103A and CMPD101, also added at 100 μM, were even less effective with ΔTm values of 6 and 4°C for GRK1 and 4 and 2°C for GRK5, respectively. Thus, CMPD103A and CMPD101 can bind and stabilize both GRK1 and GRK5 under the ThermoFluor assay conditions at high ligand concentrations, yet are ineffective inhibitors of bROS phosphorylation under conditions of saturating ATP.
We next tested the GRK2 active site mutations for thermostabilization upon compound binding (Table 3). The Tm for the P-loop mutation I197L was 37°C, with ΔTm values that are very similar to wild-type ΔTm values for binding the various compounds. The Tm values for Y206S, L235G, and L271M are noticeably lower than those for wild-type GRK2, with values of approximately 30°C, indicating that these substitutions are destabilizing. All three of these mutations exhibit similar ΔTm values for ATP, yet exhibit slight ΔTm differences for inhibitor binding. In particular, the Y206S mutation seems to be more stabilized by inhibitor binding (ΔTm of 21°C for CMPD103A) in comparison to both wild-type GRK2 (16°C) and the other mutants (15°C for I197L, 16°C for L235G, and 17°C for L271M) (Table 3). However, the rank order of the ligands remains the same (balanol > CMPD103A > CMPD101) and correlates well with our IC50 data (Table 2). Thus, our data suggest that the identity of residues in the GRK active site have only modest effects on its affinity for inhibitors and hence cannot explain the exquisite selectivity exhibited by the Takeda compounds for the GRK2 subfamily.
Structural Comparison with Inactive Conformations of GRK1 and GRK6.
If the identity of individual amino acids in the vicinity of the inhibitor-binding site do not strongly contribute to selectivity among GRK subfamilies, then it seems likely that the inhibitors preferentially bind to a unique conformation exhibited by GRK2. A common trend among GRK crystal structures, excluding a GRK6-sangivamycin complex in which the kinase adopts a relatively closed state (Boguth et al., 2010), is that the kinase domains assume relatively open, inactive conformations compared with the nucleotide-bound form of other AGC kinases. Moreover, these observed open states for GRK1, GRK2, and GRK6 are significantly different from each other (Singh et al., 2008). We therefore modeled CMPD103A in the active site of these other GRK structures to assess whether the inhibitors make less optimal interactions.
We first modeled CMPD103A into the apoGRK2 structure (Fig. 9A), which, as expected, generated strong clashes between CMPD103A and the P-loop and αC-helix compared with the GRK2-CMPD103A structure (Fig. 9B). This reflects repositioning of amino acids around the active site that takes place upon inhibitor binding. When docked into the GRK1 structure (Fig. 9C), CMPD103A clashes with multiple residues located in the P-loop, the β3-strand, and the activation loop. In the GRK6 structure (Fig. 9D), CMPD103A clashes with multiple residues in the P-loop, β3-strand, αC-helix, and activation loop. Thus, the GRK1 and GRK6 structures would need to adapt to accommodate the Takeda inhibitors. We ran our initial models/structures through a round of energy minimization using both REFMAC and the YASARA energy minimization server (Krieger et al., 2009) to alleviate the worst contacts; however, in both cases the energy minimized models created worse contacts. Collectively, our biochemical data and modeling studies suggest that CMPD103A and CMPD101 bind to GRK2 in a unique conformation that enzymes in the GRK1 and GRK4 subfamilies cannot readily achieve.
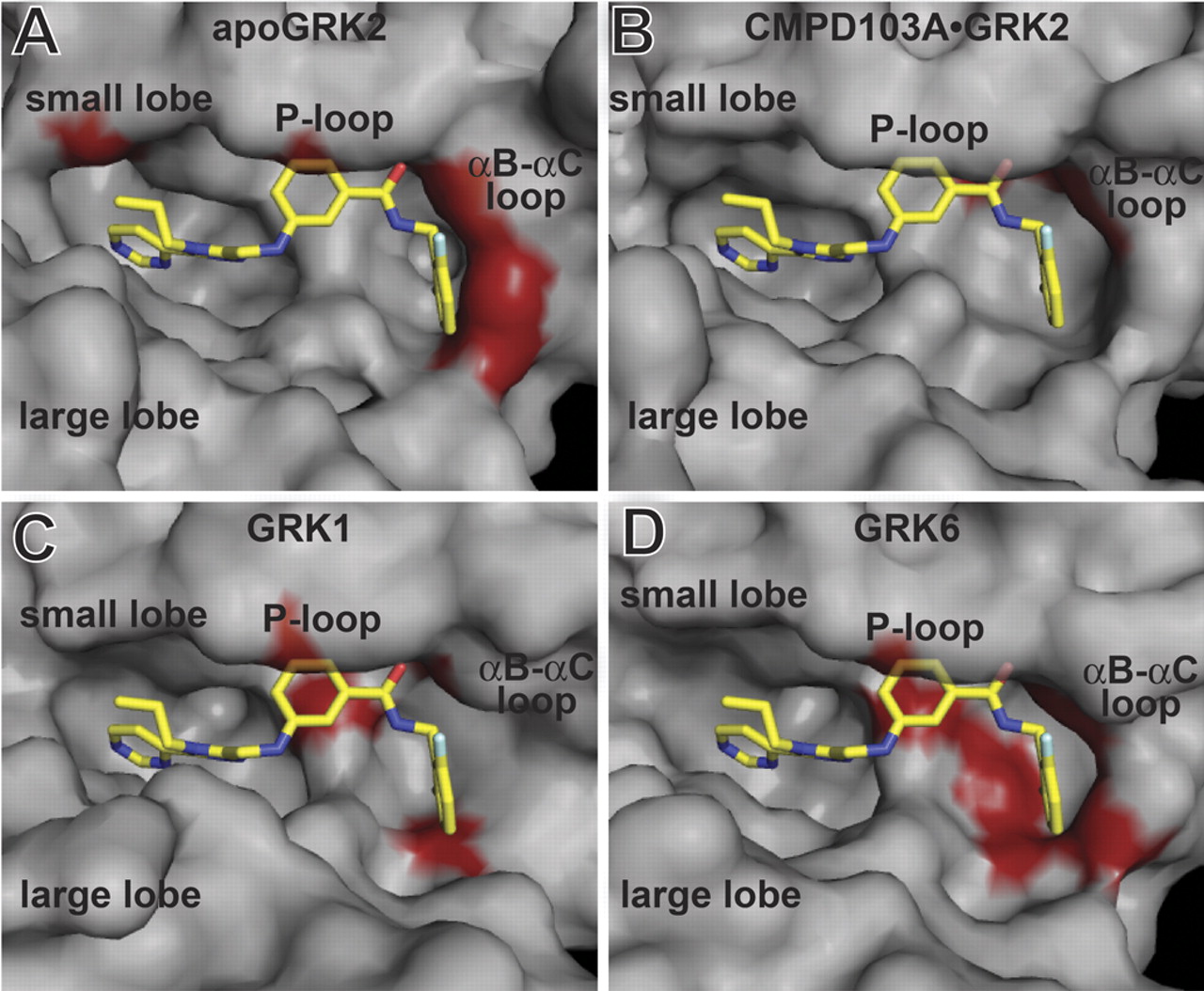
Comparison of various GRK active sites docked with CMPD103A. The molecular surfaces of atomic models for apoGRK2 (A), GRK2-CMPD103A (B), GRK1 (PDB 3C4W) (C), or GRK6 (PDB 2ACX) (D) are shown in gray with CMPD103A modeled in the active site. CMPD103A is represented as a stick model with carbons colored yellow, nitrogens blue, oxygens red, and fluorines white. All structures were superimposed using the kinase small lobe. Red surface coloring represents steric collisions by atoms that are within 3.5 Å of CMPD103A that are not involved in obvious hydrogen bonds.
Discussion
Crystal structures of protein kinases in complex with small molecules have provided valuable insights into how kinase inhibitors can achieve selectivity. Several mechanisms have been discovered, including the stabilization of a unique inactive conformation [e.g., imatinib (Gleevec)] or targeting a less conserved hydrophobic pocket such as the one guarded by the “gatekeeper” residue [e.g., erlotinib (Tarceva)] (Sequist and Lynch, 2008). In this study, we determined crystal structures of two highly selective inhibitors, CMPD103A and CMPD101, in complex with GRK2. These inhibitors bind GRK2, whereas it is in a relatively open, noncatalytic conformation, inducing 3.6 and 2.4° closures, respectively, relative to apoGRK2. CMPD103A and CMPD101 are markedly more selective than balanol, another potent inhibitor of GRK2 (Tesmer et al., 2010). Balanol has a Ki of 4 nM against PKA (Koide et al., 1995) and ∼2 nM against bovine GRK2, whereas CMPD101 has IC50 values of >2000 and 35 nM for PKA and GRK2, respectively (Ikeda et al., 2007). A comparison of the PKA-balanol structure (Narayana et al., 1999) with that of GRK2-CMPD101-Gβγ suggests that the D ring of CMPD101 would collide with Phe187 of PKA in its hydrophobic subsite, thereby precluding binding. In fact, a F187L mutation in PKA, which would convert this residue to its equivalent in GRK2, was previously used to accommodate bulkier substituents to the D ring of a balanol analog (Breitenlechner et al., 2005b). Thus, the identity of the residue at position 187 in PKA is probably a determinant for the selectivity of CMPD103A and CMPD101 versus PKA and related kinases.
CMPD103A and CMPD101 are also remarkably more selective among the GRK subfamilies than balanol, because these compounds can potently inhibit GRK2- and GRK3-mediated phosphorylation of bROS but not that mediated by GRK1 or GRK5 at the concentrations tested (Fig. 3). If the structure of GRK2 in complex with balanol is similar to that of GRK2 in complex with the Takeda compounds, what is the molecular origin of this selectivity among GRKs? In the PKA-balanol structure, it was observed that all polar atoms of balanol are within hydrogen-bonding distance to either enzyme or solvent atoms. As a result, the oxygen-rich substituents on rings C and D are capable of inducing conformational stability and induced-fit binding in a wide variety of protein kinases (Narayana et al., 1999). The C and D rings of the Takeda compounds have five less hydrogen bond donor/acceptor groups than balanol, which render them more reliant on complementary nonpolar interactions. This presumably renders them less able to adapt to the active sites of other GRK subfamilies.
Our data do provide evidence that CMPD103A and CMPD101 bind to members of other GRK subfamilies, albeit weakly and at much lower affinity than ATP, explaining why they are very poor competitive inhibitors. Both GRK1 and GRK5 exhibit 4–6°C higher thermostability upon addition of 100 μM CMPD103A or CMPD101 (Table 3). It was also observed that balanol, CMPD103A, and CMPD101 increase the catalytic activity of GRK1 in our assays, although higher concentrations of inhibitor negate this effect (Fig. 3, A and D). This can potentially be explained by the low thermostability of GRK1 (Tm = 25°C), which is near room temperature, suggesting that GRK1 could aggregate and/or lose function during the course of our phosphorylation assays. The association of CMPD103A or CMPD101, although of low affinity, could stabilize GRK1 versus the controls and result in an apparent increase in catalytic activity.
Previous studies of other AGC kinases have indicated that residues in the active site can significantly influence inhibitor selectivity. Conversion of amino acids in the active site of PKA to their equivalents in PKB conferred greater selectivity for balanol-like derivatives that preferentially inhibited PKB (Breitenlechner et al., 2005b). In another study, residues in the P-loop, hinge region, and activation loop of PKA were converted to their equivalents in Rho kinase, and two mutants, T183A and L49I (corresponding to Ser334 and Ile197 in GRK2), were shown to increase the affinity of Rho kinase-selective inhibitors for PKA (Bonn et al., 2006). Sequence alignment between the various GRK isoforms likewise reveals nonconserved residues in close proximity to bound CMPD103A and CMPD101 (Fig. 6), with 6 of the 28 contacting residues differing. Of these six residues, we hypothesized that three (Ile197, Leu235, and Leu271) could influence selectivity, and the other three residues were left unexamined because they had only backbone-mediated interactions with the inhibitors. In addition, we proposed that Ile196 and Try206 in the P-loop could indirectly play a role in selectivity as these residues undergo large conformational changes upon ligand binding (Fig. 7A). However, when tested in phosphorylation assays against bROS, none of the mutations greatly altered the affinity for inhibitors, suggesting that the identity of the amino acids surrounding the binding site do not strongly contribute to selectivity among GRKs. This is in stark contrast to a previous studies with PKA, in which the L49I mutation (corresponding to GRK2-I197L) decreased the affinity of the staurosporine-like inhibitor (9S,10S,12R)-2,3,9,10,12-hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]-benzo-diazocine-10-carboxylic acid hexyl ester (KT5720) by ∼180 fold (Bonn et al., 2006).
Although it remains possible that multiple substitutions in GRK2, such as I197L in combination with I196V, are required to dictate significant changes in binding affinity for the Takeda inhibitors, it seems more likely that selectivity is dictated by the inactive conformation unique to GRK2. This mechanism is similar to that of the inhibitor imatinib, which is strongly selective for the inactive state of Abl over that of the closely related Src tyrosine kinase (Schindler et al., 2000). The inactive states of these enzymes are dictated, at least in part, by distinct interactions between the kinase domain and the preceding SH2 and SH3 domains. Among GRK subfamilies, the N-terminal regulator of G protein signaling homology domain likewise interacts differentially with both the small and large lobes of the kinase domain, serving as a scaffold that probably helps to confer a unique kinase domain conformation. Thus, the inactive states of GRK1 and GRK6 are not compatible with the Takeda compounds because of steric clashes in the active site (Fig. 9, C and D). For example, in the triphosphate subsite, substituents on the C rings of CMPD103A and CMPD101 are involved in a network of key hydrogen bonds between the activation loop (Asp335), β3-strand (Lys220), αC-helix (Glu239), and P-loop (Fig. 5C) that cannot be replicated in the crystal structures of GRK1 and GRK6.
In conclusion, our studies support the idea that small molecules selective for a particular GRK subfamily can be identified or designed, even when they bind in the active site of the kinase domain. This selectivity seems to be due primarily to the unique conformations of these enzymes in their inactive states. Other families of compounds could have similar properties and likewise serve as future therapeutic leads to treat GRK-related diseases such as heart failure.
Authorship Contributions
Participated in research design: Thal, Yeow, and Tesmer.
Conducted experiments: Thal and Yeow.
Contributed new reagents or analytic tools: Schoenau and Huber.
Performed data analysis: Thal.
Wrote or contributed to the writing of the manuscript: Thal and Tesmer.
Acknowledgments
We thank Dr. Chih-chin Huang for the GRK1535-H6 and GRK2-H6 baculovirus construct and for providing purified GRK5561-H6.
Footnotes
This work was supported by an American Heart Association Predoctoral Fellowship from the Midwest Affiliate (to D.M.T.) and the National Institutes of Health National Heart Lung and Blood Institute [Grants HL071818, HL086865] (to J.J.G.T.). This research used the Cell and Molecular Biology Core of the Michigan Diabetes Research and Training Center supported by the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grant DK20572]. Use of LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor [Grant 085P1000817]. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences [Contract DE-AC02-06CH11357].
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.111.071522.
-
ABBREVIATIONS:
- GRK
- G protein-coupled receptor kinase
- GPCR
- G protein-coupled receptor
- CMPD103A
- N-(2,6-difluorobenzyl)-3-(((4-propyl-5-(pyrimidin-4-yl)-4H-1,2,4-triazol-3-yl)methyl)amino)benzamide
- CMPD101
- 3-(((4-methyl-5-(pyridin-4-yl)-4H-1,2,4-triazol-3-yl)methyl)amino)-N-(2-(trifluoromethyl)benzyl)benzamide
- ANS
- anilinonaphthalene-8-sulfonic acid
- PK
- protein kinase
- P-loop
- phosphate-binding loop
- CHAPS
- 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid
- PAGE
- polyacrylamide gel electrophoresis
- DMSO
- dimethyl sulfoxide
- MES
- 4-morpholineethanesulfonic acid
- PDB
- Protein Data Bank
- bROS
- bovine rod outer segment(s)
- KT5720
- (9S,10S,12R)-2,3,9,10,12-hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]-benzo-diazocine-10-carboxylic acid hexyl ester.
- Received February 1, 2011.
- Accepted May 19, 2011.
- Copyright © 2011 The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}