


Abstract
UDP glucuronosyltransferase 2B17 is present in the prostate, where it catalyzes the addition of glucuronic acid to testosterone and dihydrotestosterone and their metabolites androsterone and androstane-3α,17β-diol. Hence, changes in UGT2B17 gene expression may affect the capacity of the prostate to inactivate and eliminate male sex hormones. In this work, we identify a prevalent polymorphism, −155G/A, in the proximal promoter of the UGT2B17 gene. This polymorphism modulates UGT2B17 promoter activity, because luciferase-gene reporter constructs containing the −155A allele were 13-fold more active than those containing the −155G allele in prostate cancer LNCaP cells. The −155G/A polymorphism is contained within a putative binding site for the transcription factor Forkhead Box A1 (FOXA1). Using gene reporter, electromobility shift, and chromatin immunoprecipitation analyses, we show that FOXA1 binds to this site and stimulates the UGT2B17 promoter. Furthermore, down-regulation of FOXA1 in LNCaP cells substantially reduces UGT2B17 mRNA levels. The binding of FOXA1 and subsequent stimulation of the UGT2B17 promoter is greatly reduced in the presence of the −155G allele compared with the −155A allele. Consonant with its capacity to be stimulated by FOXA1, the UGT2B17 −155A allele, compared with the −155G allele, is associated with higher levels of circulating androstane-3α,17β-diol glucuronide. Although the initial phases of prostate cancer are androgen-dependent and UGT2B17 inactivates androgens, there was no association of the UGT2B17 −155G/A polymorphism with prostate cancer risk. In summary, this work identifies FOXA1 as an important regulator of UGT2B17 expression in prostate cancer LNCaP cells and identifies a polymorphism that alters this regulation.
Introduction
Androgens have important roles in the development and maintenance of many organs and tissues, including the prostate. They are effectively inactivated and eliminated by glucuronidation, because the addition of the glucuronic acid moiety to the steroid prevents binding to the androgen receptor (AR) and enhances its elimination in the bile or urine (Mackenzie et al., 2005). There are 19 members of the human UDP glucuronosyltransferase superfamily of detoxifying enzymes that use UDP glucuronic acid to glucuronidate lipophilic chemicals. Of these UGTs, UGT2B17, UGT2B15, and UGT2B7 are primarily responsible for glucuronidating the active androgens testosterone and dihydrotestosterone and their metabolites androsterone and androstane-3α,17β-diol (Bélanger et al., 2003).
A major site of androgen glucuronidation is the liver, where UGT2B7 is primarily involved in the glucuronidation of the 3α-hydroxyl group of androgens, and UGT2B17 and UGT2B15 are primarily involved in glucuronidation of the 17β-hydroxy group. In addition to the liver, androgen glucuronides are formed in many other tissues, including the prostate, skin, and other androgen-sensitive tissues. Indeed, it has been suggested that most of the circulating androstane-3α,17β-diol glucuronides are derived from these extrahepatic tissues (Bélanger et al., 2003). Furthermore, because the 17β-glucuronide comprises 80% of the total androstane-3α,17β-diol glucuronide in the blood of both men and women (Bélanger et al., 2003), UGT2B17 and UGT2B15 seem to be the main contributors to these levels of circulating steroid glucuronide.
The important contribution of UGT2B17 to androgen glucuronidation and elimination is especially highlighted by the physiological effects of a deletion in the UGT2B17 gene. First described by Murata et al. (2003), this deletion, in the homozygous state, leads to substantially reduced levels of urinary testosterone glucuronide (Jakobsson et al., 2006; Juul et al., 2009) and, to a lesser extent, serum androstane-3α,17β-diol-17-glucuronide (Swanson et al., 2007). The deletion seems to be a predictor of fat mass and insulin sensitivity in men (Swanson et al., 2007) and is associated with susceptibility for osteoporosis (Yang et al., 2008). The UGT2B17 gene deletion has also been associated with risk of prostate cancer (Park et al., 2006, 2007; Karypidis et al., 2008), although studies showing no association have also been reported (Gallagher et al., 2007; Olsson et al., 2008).
Although the prostate is exposed to circulating testosterone, the local synthesis of the potent androgen receptor agonist dihydrotestosterone from adrenal dehydroepiandrosterone and/or testis-derived testosterone and its subsequent metabolism are important determinants of androgen response in this organ (Bélanger et al., 2003). Dihydrotestosterone is inactivated by glucuronidation, or converted by hydroxysteroid dehydrogenases to androstane-3α,17β-diol, which is subsequently glucuronidated. Because UGT2B7 is not expressed in the prostate, only UGT2B17 and UGT2B15 catalyze these reactions within this organ. Both enzymes glucuronidate androstane-3α,17β-diol with equal efficiencies; however, UGT2B17 is 24-fold more efficient than UGT2B15 in glucuronidating dihydrotestosterone (Turgeon et al., 2001). Hence, UGT2B17 and UGT2B15 levels in the prostate are important factors in regulating androgen concentrations and androgen receptor signaling pathways. To date, the transcriptional mechanisms that modulate UGT2B17 and UGT2B15 levels are not fully understood.
The UGT2B17 and UGT2B15 genes are located in tandem on chromosome 4q13 (Beaulieu et al., 1997; Mackenzie et al., 2005). Studies with gene reporter constructs indicate that basal UGT2B17 gene expression is controlled by interactions between the transcription factors HNF1α and Pbx2 in liver-derived HepG2 cells but not in prostate-derived LNCaP cells (Gregory et al., 2000; Gregory and Mackenzie, 2002). In the prostate, UGT2B17 is present in basal cells, whereas UGT2B15 is present in luminal cells (Bélanger et al., 2003). However, the mechanisms directing this differential expression in the prostate and the mechanisms that modulate constitutive UGT2B17 and UGT2B15 expression, and expression in response to circulating hormones, are not well defined. Androgen receptor agonists (DHT and R1881), calcitrol, and growth factors (epidermal growth factor, interleukin 1α) are known to down-regulate UGT2B17 and UGT2B15 expression in LNCaP cells via ill-defined mechanisms that differ in the two genes (Beaulieu et al., 1997; Lévesque et al., 1998; Kaeding et al., 2008a).
In this work, we identify the Forkhead Box A1 (FOXA1) transcription factor as a regulator of the basal expression of the UGT2B17 gene in LNCaP cells. We also identify a novel polymorphism in the FOXA1 binding site of the UGT2B17 promoter and show that it modulates UGT2B17 promoter activity and is associated with altered levels of circulating androstane-3α,17β-diol glucuronide.
Materials and Methods
Genotyping.
To screen for single-nucleotide polymorphisms (SNPs) or other DNA variations (deletions/insertions, etc.) in the UGT2B17 proximal promoter, a 321-bp region of the UGT2B17 gene (−265/+56) was amplified from genomic DNA (n = 89) by PCR with the primers 5′-GGAGCCTCTCACCTGCCACTG-3′ and 5′-CTAAAGTAACAACTGAGCTGC-3′, and the PCR products were sequenced. To assess the occurrence of the −155G/A SNP, identified in the screen above, a PCR-based assay was established as follows. Using the sequenced genomic DNA samples and a bacterial artificial chromosome vector containing the deleted UGT2B17 allele (Murata et al., 2003) (RP11-185H6; BACPAC Resources, Children's Hospital Oakland Research Institute, Oakland, CA) as controls, two PCR screens were developed to determine the UGT2B17 genotype of unknown DNA samples. To detect the UGT2B17 deletion, a 616-bp fragment was amplified with primers spanning the deleted region (5′-CCTGACAGAATTCTTTTG-3′ and 5′-ATGTCTCTCGTACATAGTGAT-3′) using an annealing temperature of 57.5°C with AmpliTaq Gold DNA polymerase (Applied Biosystems, Foster City, CA) and 2 mM MgCl2. Generation of a PCR product indicated that one or both UGT2B17 alleles contained the deletion. To differentiate between the UGT2B17 −155G/A SNP alleles, a second PCR screen was used. This involved a multiplex PCR in which four primers were used simultaneously to interrogate the UGT2B17 gene at an annealing temperature of 60.4°C in an AmpliTaq Gold reaction with 4 mM MgCl2. The outer primers (5′-GCATCTTCACAGAGCTTTATATTA-3′ and 5′-GGAGATAATACTGATTATTGTAGTGAAA-3′) amplified an 865-bp fragment of the UGT2B17 gene (−473 to +392), regardless of genotype at the −155G/A SNP, serving as an internal positive PCR control. The internal primers (5′- TGTTTGGTGTTCTTTTATATTC-3′ and 5′-TATTGCTTGACTAGAGTAATTGTA-3′) were positioned with their 3′ ends at the −155G/A SNP. Thus, in addition to the control fragment, a 340-bp fragment was produced if the “G” allele was present at the UGT2B17 −155 position and/or a 570-bp fragment was produced for the “A” allele. Genotype was established by separation of the three potential products on a 1.5% agarose gel. All PCR products were sequenced to ensure specificity for UGT2B17 amplification, with no cross-reaction with the highly related UGT2B15 sequences.
DNA samples from the blood of 826 case subjects and 731 control subjects from an Australian population-based case-control study of prostate cancer were genotyped for the −155G/A SNP and the deletion allele, as described above. This case-control study is described in detail elsewhere (Giles et al., 2001; Severi et al., 2003) and consisted of incident cases identified through the Victorian Cancer Registry and the Western Australian Cancer Registry during the period 1994 to 1998. Case subjects were men with histologically verified prostate cancer diagnosed at age less than 70 years. Patients with poorly differentiated tumors (i.e., Gleason score <5) were excluded. Population control subjects were randomly selected from the State Electoral Rolls and frequency matched to the cases according to the age distribution. Participants completed epidemiologic questionnaires and donated a blood sample. Plasma levels of dehydroepiandrosterone sulfate, sex hormone-binding globulin, testosterone, 17β-estradiol, androstenedione, and androstane-3α,17β-diol glucuronides were determined for control subjects (Hayes et al., 2007) using methods described previously (Severi et al., 2006).
Generation of Luciferase Reporter Constructs.
The UGT2B17−694/−2 promoter construct containing −155G was generated as described previously (Gregory et al., 2000) and designated 2B17−694/−2 “G” (Luc) in the present study. In this article, nucleotides are numbered with +1 as the A of the initiation codon, as recommended by the Gene Nomenclature Committee. In our previous article, nucleotides were numbered from the transcription start site (Gregory et al., 2000). Hence the UGT2B17 −557/+43 promoter construct described in the former study has been renamed −694/−2 to follow convention. With this construct as template, a mutation of “G” to “A” at base −155 was prepared to generate the −155A-containing construct 2B17−694/−2“A”(Luc) by site-directed mutagenesis with the primer 2B17−694/−2M“A” (Table 1) and the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA).
Primers used in this study for promoter cloning, mutagenesis, real-time PCR, EMSAs, and ChIP assays
Bold characters represent the putative FOXA1 site in its wild-type or mutated type. Underlined are mutated bases within the FOXA1 site. The –155G/A SNP is italic.
Mutagenesis of the Putative FOXA1 Binding site in Reporter Constructs.
Mutagenesis was performed with the QuikChange kit, and complementary pairs of oligonucleotide primers containing the desired mutation(s). The sense sequences of the primers are given in Table 1. With construct 2B17−694/−2 “A” (Luc) as template, the FOXA1 site (5′-TGTAAATATAAA-3′) was mutated to 5′-TGCCCCCCTAAA-3′ using the primer 2B17-FOXA1-MT1 and to 5′-TGCGCATATAAA-3′ using the primer 2B17-FOXA1-MT2, to generate two mutated constructs 2B17−694/−2/FOXA1-MT1(Luc) and 2B17−694/−2/FOXA1-MT2(Luc), respectively. Mutations are underlined. The promoter sequences of all reporter constructs were confirmed by sequencing.
Transient Transfection and Luciferase Reporter Assay.
The LNCaP and VCaP prostate cancer cell lines and MCF-7 breast cancer cell line were obtained from the American Type Culture Collection (Manassas, VA). The LNCaP and MCF-7 cells were maintained in RPMI 1640 medium (Invitrogen, Carlsbad, CA) supplemented with 5% (v/v) fetal bovine serum (FBS). The VCaP cells were maintained in DMEM supplemented with 10% FBS. All transfections were performed using Lipofectamine 2000 (Invitrogen) according to the protocols of the manufacturer.
LNCaP or VCaP cells were plated into 24-well plates in 800 μl of RPMI 1640 medium or DMEM, respectively, and transfections were performed when cells reached 50 to 60% confluence. In brief, 500 μl of medium was aspirated from each well and replaced with 300 μl of serum-free medium transfection mixture containing 0.5 μg of each reporter construct and 25 ng of pRL-null vector, which served as an internal control for transfection efficiency. At 16 h after transfection, 300 μl of medium was aspirated from each well and replaced with 500 μl of fresh RPMI 1640 medium or DMEM supplemented with 5% FBS. At 48 h after transfection, cells were lysed in passive lysis buffer and analyzed for firefly and Renilla reniformis luciferase activity using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI) and a Packard TopCount luminescence and scintillation counter (PerkinElmer Life and Analytical Sciences, Waltham, MA). To transfect MCF-7 cells, cells were seeded into 96-well plates at a density of 3 × 104 cells/well in 200 μl of phenol-red-free RPMI 1640 medium (Invitrogen) supplemented with 5% dextran-coated charcoal-stripped FBS. After 24 h, transfections were carried out in 50 μl of phenol red-free RPMI 1640 medium without serum using 100 ng of each reporter construct and 1 ng of pRL-null vector. At 5 h after transfection, the transfection mix was replaced with 200 μl of fresh phenol red-free RPMI 1640 medium containing 5% dextran-coated charcoal-stripped FBS. At 48 h after transfection, cells were harvested and analyzed for firefly and R. reniformis luciferase activity as described above.
Electrophoretic Mobility Shift and Supershift Assays.
Nuclear extracts were prepared from LNCaP cells as reported previously (Gardner-Stephen et al., 2005). The plasmid expressing FOXA1 was generated by cloning the full-length cDNA into the EcoRI site of the mammalian expression vector pCMX-PL2. Recombinant FOXA1 protein was prepared using this vector and the TnT Quick -Coupled Transcription/Translation kit according to the manufacturer's instructions (Promega). Electrophoretic mobility shift assays (EMSAs) were performed with probes encompassing −162 to −128 bp of the UGT2B17 promoter. These probes had a wild-type FOXA1 site with an “A” (Table 1, 2B17-FOXA1-A) or a “G” at nucleotide −155 (2B17-FOXA1-G) or a mutated FOXA1 site (TGCGCACATAAA; 2B17-FOXA1-MT3, mutated bases underlined). Probe PSA1 from the core enhancer of the prostate-specific antigen (PSA) gene, which is known to bind to FOXA1 (Gao et al., 2003), served as a positive control. The anti-FOXA1 antibody (H-120) was purchased from Santa Cruz Biotechnology (Santa, Cruz, CA). EMSAs were performed as reported elsewhere (Hu and Mackenzie, 2009).
Chromatin Immunoprecipitation Assay and Quantitative Real-Time PCR.
ChIP-qPCR was performed as reported previously (Hu and Mackenzie, 2009). In brief, LNCaP cells were cross-linked by 1% formaldehyde and subsequently quenched by 125 mM glycine solution. Cells were lysed, sonicated, and then subjected to immunoprecipitation with 10 μg of each antibody, including anti-estrogen receptor α antibody and anti-FOXA1 antibody or equivalent amounts of control IgG. All antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The precipitates were then captured on protein A Sepharose CL-4B beads (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK) and washed several times in various buffers. The protein-DNA complexes were eluted from the beads, and cross-linking was reversed by heating the eluates at 65°C overnight. The precipitated DNA was digested with proteinase K, followed by phenol-chloroform extraction and ethanol precipitation. The DNA pellets were resuspended in 50 μl of Tris-EDTA buffer. Quantitative real-time PCR was performed using 2 μl of each of the resultant DNA samples and primers, which were specific to the UGT2B17 promoter, the coding region of the HCG3 gene (GenBank accession number NM_001001394) as a reference gene or two other control loci. The control loci were positioned as reported in the March 2006 version of the human genome (http://genome.ucsc.edu/cgi-bin/hgGateway). Both positive and negative control loci contain a FOXA1 site; however, FOXA1 binds only to the positive control locus in LNCaP cells (Lupien et al., 2008). qPCR primers for UGT2B17 are listed in Table 1, whereas the primers for HCG3 (Hu and Mackenzie, 2009) and the control loci (Lupien et al., 2008) were the same as reported previously. Data obtained from the HCG3 gene were used to normalize the starting amounts of immunoprecipitated DNA added to each PCR.
siRNA Knockdown.
Both On-TARGETplus SMARTpool siRNA against FOXA1 (GenBank accession number NM_004496, designated anti-FOXA1 siRNA) and On-TARGETplus Nontargeting pool siRNA (nontarget siRNA) were purchased from Dharmacon RNAi Technologies (Lafayette, CO). LNCaP cells were maintained in RPMI 1640 medium supplemented with 5% FBS. For transfection assays, 1.5 ml of fresh RPMI 1640 medium with 106 cells was combined with 0.5 ml of serum-free RPMI 1640 medium containing 8 μl of Lipofectamine (Invitrogen) and 400 nM anti-FOXA1 siRNA or nontarget siRNA and subsequently plated into six-well plates. At 48 h after transfection, the medium was replaced by 3 ml of fresh RPMI 1640 medium with 5% FBS. Cells were incubated for a further 24 h and harvested for mRNA. Levels of each target gene mRNA were determined by quantitative real-time RT-PCR with gene-specific primers as described previously (Hu and Mackenzie, 2009). Table 1 lists the RT-PCR primers for FOXA1, PSA gene, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Primers for UGT2B17 and 18S rRNA were reported previously (Congiu et al., 2002). Data from 18S rRNA transcripts were used as a reference to normalize the amount of total cDNA amplified in each reaction.
Statistical Analyses.
Estimates of allele frequencies and tests of deviation from Hardy-Weinberg equilibrium were carried out using standard procedures based on asymptotic likelihood theory. Fisher's exact test was used to test for association between genotype and tumor stage (stage I-II, III, or IV) and grade (Gleason score 5–7, “moderate,” or 8–10, “high”). Case-control analyses were conducted using unconditional logistic regression to test for association between genotype and prostate cancer risk. The association between genotype and disease-specific survival was tested using Cox regression. Because plasma levels of sex hormones and sex hormone-binding globulin were skewed, linear regression of the transformed levels to test the possible association with genotypes was used. Levels of androstane-3α,17β-diol glucuronides and 17β-estradiol were log-10 transformed, whereas the others were square-root transformed. The linear regression models were adjusted for age and laboratory assay and were fitted using all the controls. Statistical analyses were performed using Stata/SE 8.2 (Stata Corporation, College Station, TX).
Results
A Novel G/A Single-Nucleotide Polymorphism Is Prevalent at Nucleotide −155 of the UGT2B17 Promoter.
A proximal 321-bp UGT2B17 promoter fragment was amplified from genomic DNA samples of 89 unrelated patients by PCR to determine the presence of polymorphisms in this region. Sequencing of the PCR products resulted in the identification of a novel single-nucleotide polymorphism (G to A) at base position −155. Subsequent to our study, this polymorphism appeared in the NCBI SNP database as rs59678213, reported as a C/T change in the reverse strand of the UGT2B17 gene (Fig. 1). A second single-nucleotide polymorphism was also identified at base −199 (A to G) in only one sample, and it probably represents a rare polymorphism rather than a mutation (Fig. 1). Subsequent to our study, this polymorphism appeared in the NCBI SNP database as rs62317003, reported as a C/T change in the reverse strand of the UGT2B17 gene. Polymorphisms were not detected in the functional HNF1 and Pbx binding sites identified previously (Gregory et al., 2000; Gregory and Mackenzie, 2002). The prevalence of the −155G and −155A homozygotes was assessed in 731 control subjects and was found to be 17.4 and 10.5% respectively. The prevalence of the known deletion homozygote was 10.9%; similar to that previously determined (Wilson et al., 2004).

The proximal promoter of the UGT2B17 gene. The SNPs at −155 and −199 are highlighted. HNF1 and Pbx binding sites are boxed. The transcription start site is indicated with an arrow and the initiation codon is in bold italic. Nucleotides are numbered from the A of the initiation codon.
The UGT2B17 −155A Variant Has Higher Promoter Activity Than the −155G Variant in LNCaP Cells.
To determine whether the polymorphism at base −155 of the UGT2B17 promoter influences promoter activity, transient transfection assays were performed in LNCaP cells with luciferase reporter constructs carrying the −694/−2-bp fragment of the UGT2B217 promoter. As shown in Fig. 2, the −155A-containing promoter was approximately 13-fold more active than the −155G-containing promoter (p < 0.001). To confirm this finding in another prostate cancer cell line, we transfected these promoter constructs into VCaP cells. As illustrated in Fig. 2, the promoter with the A allele displayed an approximate 4-fold higher activity than that of the G allele in this cell line (p < 0.001). This difference in promoter activity seemed to be specific for the prostate cell lines, LNCaP and VCaP, as it was not observed in two other cancer cell lines, namely MCF-7 (<2-fold) and HepG2 (<2-fold; data not shown).

The polymorphic −155G/A is contained within a putative FOXA binding site in the UGT2B17 promoter and modulates promoter activity in a cell-specific manner. LNCaP, VCaP, and MCF-7 cells were transfected with the indicated promoter constructs and pRL-null vector. At 48 h after transfection, cells were harvested and assayed for firefly and R. renififormis luciferase activities as described under Materials and Methods. After normalizing transfection efficiency with R. reniformis luciferase activities, the relative luciferase activities of promoter constructs are expressed as the fold induction over that of pGL3-basic vector (set at a value of 1). Transfections were performed at least twice in triplicate. Data shown are from a representative experiment performed in triplicate, the error bar representing 1 S.D. ND, not done.
These results demonstrate that the presence of an adenine at −155 significantly enhances UGT2B17 promoter activity in a cell type-dependent manner. Whether this −155 G/A SNP-dependent change in UGT2B17 promoter activity in LNCaP cells was caused by altered binding of a transcription factor(s) was explored further.
The UGT2B17 −155 G/A SNP Is Contained within a FOXA1 Binding Site in the UGT2B17 Promoter.
Interrogation of the transcription factor database TRANSFAC revealed a potential FOXA binding site (core consensus sequence, C/AAAC/T) (Myatt and Lam, 2007) in the UGT2B17 proximal promoter between nucleotides −158 and −147, which incorporated the −155 G/A SNP. The “A” variant (5′-TGTAAATATAAA-3′) maintains the FOXA core sequence, whereas the “G” variant disrupts this sequence (5′-TGTGAATATAAA-3′).
To demonstrate the functionality of this FOXA site, we mutated the core sequence to generate two constructs, namely 2B17−694/−2/FOXA1-MT1(Luc) and 2B17−694/−2/FOXA1-MT2(Luc) and assessed the effects of these mutations on promoter activities in LNCaP cells by transient transfection assays. These mutations decreased UGT2B17 promoter activity (p < 0.001 in both cases) so effectively that both mutated constructs displayed promoter activities similar to that seen with the −155G-containing promoter (Fig. 2). This decrease in promoter activity was also observed in VCaP cells after transfection of the FOXA1-mutated promoter construct (2B17−694/−2/FOXA1-MT1 (Luc) (p < 0.001). Not surprisingly, these mutations had no significant impact on promoter activity in MCF-7 cells (Fig. 2).
Previous studies have shown that only FOXA1, not FOXA2 or FOXA3, is present in LNCaP cells (Mirosevich et al., 2006). After confirming this finding using quantitative real-time RT-PCR (data not shown), we focused on determining whether FOXA1 binds to the putative site in the UGT2B17 promoter using in vitro synthesized recombinant FOXA1 protein. We included a probe containing the FOXA1-binding site of the PSA gene in these EMSAs as a positive control.
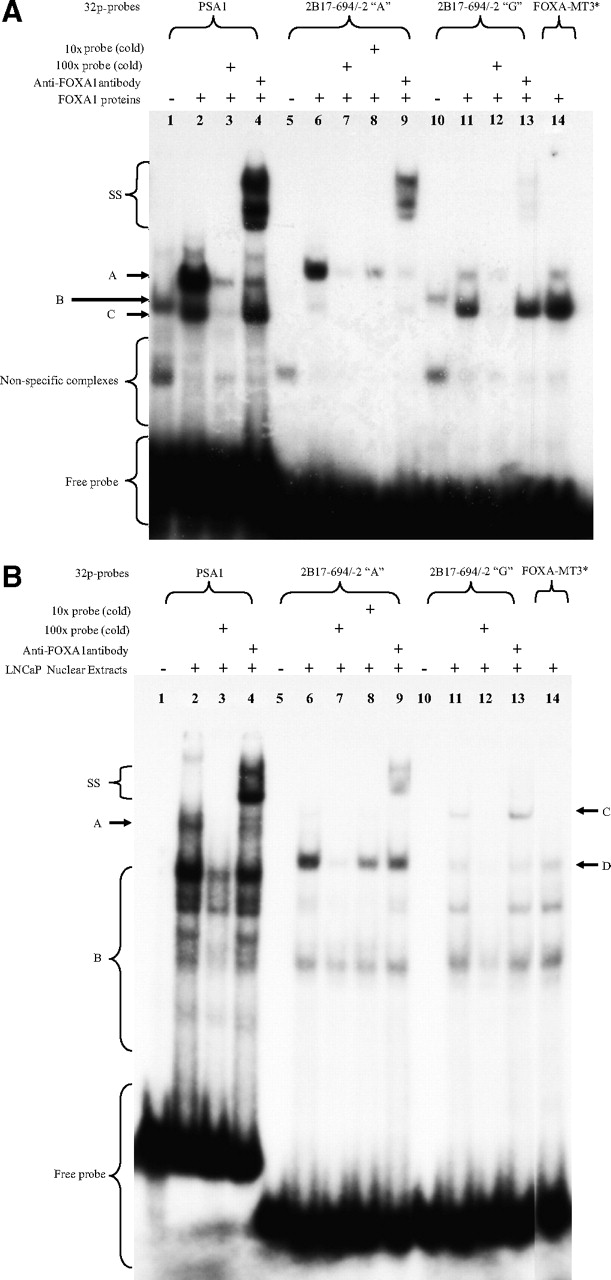
As shown in Fig. 3A, two major complexes (labeled A and C, lane 2) formed on the PSA1 probe after incubation with FOXA1 protein; however, only complex A was supershifted (labeled SS, lane 4) by the anti-FOXA1 antibody, suggesting the presence of FOXA1 in this complex. Incubating probe 2B17-FOXA1-A (Table 1) with FOXA1 protein produced a major complex A and a faint complex C (lane 6). Complex A was significantly reduced (lane 8) or almost completely abolished (lane 7) in the presence of a 10- or 100-fold molar excess of unlabeled probe, and this complex was supershifted (labeled SS, lane 9) by the anti-FOXA1 antibody, thus demonstrating the presence of FOXA1 in this complex. By contrast, a very faint complex A and a major complex C (lane 11) were observed with probe 2B17-FOXA1-G after incubation with FOXA1 protein. Both complexes were probe-specific because their binding was greatly diminished by a 100-fold molar excess of unlabeled probe (lane 12). The addition of the anti-FOXA1 antibody only disrupted complex A and this resulted in the formation of the supershifted complex SS (lane 13), thus indicating the presence of FOXA1 in complex A and its absence in complex C. We further mutated the FOXA1 site to generate probe 2B17-FOXA1-MT3 (Table 1). As expected, complex A that bound to this site (Fig. 3A, lane 14) was much less evident than that binding to the −155A-containing FOXA1 site (lane 6). These results indicate that FOXA1 can bind to the FOXA1 site to form the same complex regardless of the presence of a “A” or “G” at the −155 base position. However, given the difference in the intensities of the FOXA1-containing complex A, FOXA1 seems to have a much higher affinity for the −155A-containing site than its “G” variant.

FOXA1 preferentially binds to the −155A-containing FOXA1 site of UGT2B17 promoter. Using either 1 μl of in vitro transcribed/translated recombinant FOXA1 (A) or 5 μg of LNCaP nuclear extracts (B), EMSAs were performed with 50,000 cpm (∼1 ng) of the indicated 32P-labeled probes as described under Materials and Methods. In supershift assays, 2 μg of anti-FOXA1 antibody (lanes 4, 9, and 13) were added immediately after the addition of labeled probe and incubated for 30 min at room temperature. In competition assays, unlabeled probes (cold) were added at a 10-fold (lane 8) and 100-fold (lanes 7 and 12) molar excess of labeled probe before the addition of labeled probe. Shown in 3A are the FOXA1-containing complexes (A), the FOXA1 antibody supershifted complexes (SS), and the nonspecifically retarded complexes (C). Complexes B in negative controls (lanes 1 and 10) were nonspecific and resulted from unknown proteins present in the rabbit reticulocyte lysate from the TNT kit (Promega). Shown in 3B are the FOXA1-containing complexes with the PSA1 probe (A) and the three UGT2B17 probes (D), the FOXA1 antibody supershifted complexes (SS), and the nonspecifically retarded complexes (B and C). FOXA1-MT3*, 2B17-FOXA1-MT3.
We repeated these experiments with LNCaP nuclear extracts to see whether endogenous FOXA1 proteins could also bind to this UGT2B17 FOXA1 site. As expected, the addition of the anti-FOXA1 antibody decreased the formation of complex A (Fig. 3B, compare lanes 2 and 4) with the PSA1 probe and resulted in a major supershifted complex SS (Fig. 3B, lane 4), thus demonstrating that endogenous FOXA1 proteins could bind to this probe, which carries an authentic FOXA1 site. As shown in Fig. 3B, incubating probe 2B17-FOXA1-A (lane 6) with LNCaP nuclear extracts gave a major complex (labeled D) and nonspecific complexes (labeled B). The formation of complex D was almost abolished (lane 7) and highly inhibited (lane 8) in the presence of a 100- and 10-fold molar excess of unlabeled probe, respectively. Complex D was supershifted (lane 9) by the anti-FOXA1 antibody, indicative of the presence of FOXA1 in this complex. In contrast, the formation of complex D on both the 2B17-FOXA1-G (lane 11) and 2B17-FOXA1-MT3 (lane 14) probes was greatly diminished. The identities of the proteins present in complexes C and B remain to be determined but were probably not FOXA1-related because they were not supershifted by the anti-FOXA1 antibody (lanes 13). By comparing the intensity of complex D in lane 6 to that in lane 11 (Fig. 3B), it is clear that endogenous FOXA1 proteins also bind to the “A” variant FOXA1 site in preference to its “G” counterpart.
Collectively, these in vitro experiments verified the putative UGT2B17 FOXA site as an authentic FOXA1-binding site and demonstrate that the change from −155A to −155G within this site dramatically decreases its capacity to bind FOXA1. The greater capacity of the site containing −155A to bind FOXA1 correlates well with the higher promoter activity of the UGT2B17 −155A allele.
FOXA1 Binds to the UGT2B17 FOXA1 Site In Vivo in LNCaP Cells.
Having demonstrated the binding of FOXA1 to the UGT2B17 promoter in vitro, we performed chromatin immunoprecipitation assays followed by quantitative real-time PCR to determine whether FOXA1 binds to the endogenous UGT2B17 promoter in its native chromatin context in LNCaP cells.
Because the UGT2B15 and UGT2B17 proximal promoters are 91% identical in sequence, it was necessary to use primers to discriminate between them. In our qPCR experiments, −341 to −232 bp of the UGT2B17 promoter containing the FOXA1-binding site was amplified using a pair of 2B17-specific primers (Table 1), which had a single base mismatch to the UGT2B15 promoter at the 3′-end of both forward and reverse primers. Sequencing confirmed that only UGT2B17 promoter sequences were amplified and revealed that the UGT2B17 deleted allele was absent and that the FOXA1 site contained −155A in LNCaP cells (data not shown).
As shown in Fig. 4, we observed an approximate 6-fold enrichment of the UGT2B17 promoter in samples precipitated with the anti-FOXA1 antibody as opposed to the IgG-precipitated controls (p < 0.001). In contrast, no enrichment was seen in the samples precipitated with the anti-estrogen receptor α antibody, thus confirming the specificity of immunoprecipitation. In agreement with previous findings (Lupien et al., 2008), immunoprecipitation by anti-FOXA1 antibody resulted in an 8-fold enrichment of the positive control locus over the IgG controls (p < 0.001) but no enrichment with the negative control locus, thus demonstrating the reliability of our method. Taken together, these studies provide compelling evidence for in vivo binding of FOXA1 to the UGT2B17 FOXA1 site in LNCaP cells.

FOXA1 binds to the UGT2B17 promoter region harboring the FOXA1 site in LNCaP cells. Cells were cultured in RPMI 1640 medium supplemented with 5% FBS and subjected to ChIP-qPCR to quantify the immunoprecipitated DNA of the indicated target regions as described under Materials and Methods. Both positive and negative loci harbor a FOXA1 site; however, FOXA1 occupies only the positive locus in LNCaP cells. Data are expressed as fold enrichment in DNA samples precipitated with 10 μg of each antibody compared with the control samples (set as a value of 1), which were precipitated from equivalent amounts of IgG. Experiments were performed at least twice. Data shown are from a representative experiment performed in triplicate, the error bars representing 1 S.D. *, p < 0.001.
siRNA-Mediated Knockdown of FOXA1 mRNA Levels in LNCaP Cells Causes a Corresponding Reduction in UGT2B17 Transcript Levels.
Because FOXA1 bound to the UGT2B17 promoter and stimulated its activity, we sought to determine whether siRNA-mediated reduction of FOXA1 gene expression would lead to a significant reduction in UGT2B17 mRNA levels. Transfecting LNCaP cells with anti-FOXA1 siRNA significantly decreased the levels of endogenous FOXA1 transcripts to 42% of that in control cells treated with nontargeting siRNAs (p < 0.01) (Fig. 5). This reduction of FOXA1 mRNA resulted in a decrease of approximately 5-fold in UGT2B17 mRNA levels (p < 0.001), highlighting the importance of FOXA1 in UGT2B17 gene transcription. No significant effect on GAPDH mRNA levels was observed (p > 0.7). GAPDH acted as a negative control for these experiments. In agreement with previous reports (Wang et al., 2007), we found that the mRNA levels of PSA, which is a well known androgen-regulated gene in LNCaP cells, were not affected by down-regulation of FOXA1 expression (p > 0.5). Collectively, these results clearly show that silencing of FOXA1 caused a corresponding reduction of UGT2B17 transcripts, thus reinforcing our hypothesis that FOXA1 is involved in UGT2B17 gene expression.

siRNA against FOXA1 reduces UGT2B17 mRNA levels in LNCaP cells. LNCaP cells were maintained in RPMI 1640 medium supplemented with 5% FBS. Cells were transfected with either anti-FOXA1 siRNA or nontargeting siRNA for 48 h, followed by a further 24 h-incubation in fresh RPMI 1640 medium. Cells were then harvested, and target mRNAs were quantified by real-time RT-PCR as described under Materials and Methods. After normalizing to 18S rRNA, the relative expression levels of target genes in cells transfected with anti-FOXA1 siRNA were expressed as a percentage of that in control cells treated with nontarget siRNA (set as a value of 100%). PSA and GAPDH served as negative controls. Experiments were performed in triplicate and repeated at least twice in independent experiments. Data presented are from representative experiments performed in triplicate with error bars representing 1 S.D. *, p > 0.5; **, p < 0.01.
The UGT2B17 −155 G/A SNP Is Associated with Altered Blood Levels of Androstane-3α,17β-diol Glucuronide but Is Not Associated with Risk of Prostate Cancer.
UGT2B17 genotypes were determined in 731 control subjects and compared with the levels of dehydroepiandrosterone sulfate, sex hormone-binding globulin, testosterone, 17β-estradiol, androstenedione and androstane-3α,17β-diol glucuronide in blood. The levels of circulating androstane-3α,17β-diol glucuronide were significantly higher in persons homozygous for the UGT2B17 −155A allele compared with those homozygous for the −155G allele (p = 0.004) (Fig. 6). In contrast, those homozygous for the UGT2B17 deletion allele had lower levels of this glucuronide compared with persons homozygous for the −155G allele (p < 0.001). The levels of androstane-3α,17β-diol glucuronide in the blood of persons heterozygous for the three UGT2B17 alleles were in the order −155A/−155G > −155A/del > −155G/del. The circulating levels of the other steroids and proteins measured were not associated with genotype (all p values >0.05). We found no association between UGT2B17 genotype and prostate cancer risk (all p values >0.05). There were also no associations with tumor stage, tumor grade, or survival (not shown).
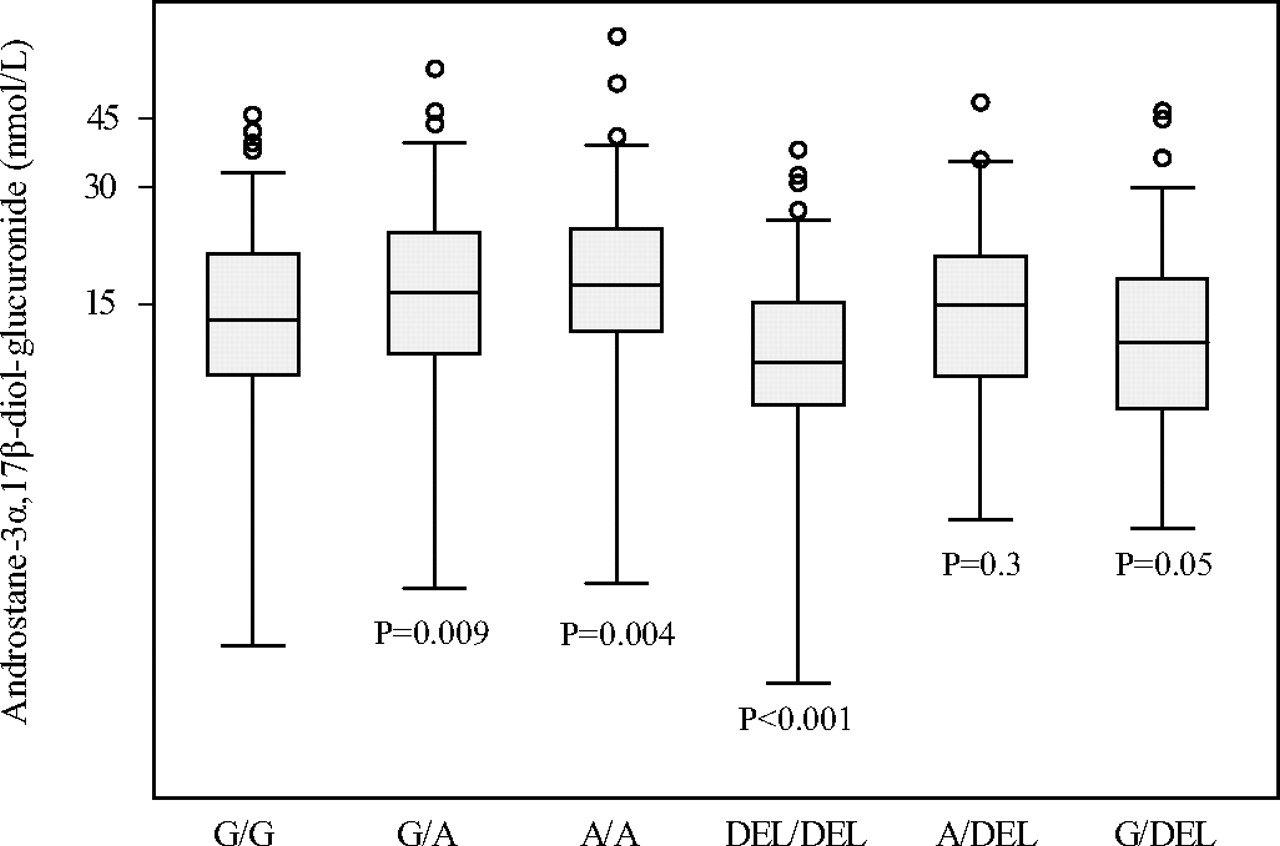
Subjects homozygous for the UGT2B17 −155A allele have higher levels of circulating androstane-3α,17β-diol glucuronide. A whisker plot based on an analysis of the genotypes of the UGT2B17 −155G/A and deletion polymorphisms and serum androstane-3α,17β-diol glucuronide levels, as described under Materials and Methods. The data for each genotype are statistically compared with that for the −155G/G homozygote.
Discussion
UGT2B17 has a major role in the inactivation and elimination of male sex hormones. Its importance in this process is underscored by studies showing that persons who lack UGT2B17 (UGT2B17 del/del homozygotes) have 13-fold less testosterone glucuronide in their urine (Schulze et al., 2008). Hence, factors that regulate UGT2B17 expression in the prostate and in other tissues are likely to be important determinants of androgen effects and homeostasis. In this work, we show that the transcription factor FOXA1 is essential for regulating basal UGT2B17 expression in prostate LNCaP and VCaP cells. We also show that the stimulatory effect of FOXA1 on UGT2B17 expression seems to be cell-type-specific, because it was not observed in breast-derived MCF-7 cells and liver-derived HepG2 cells. This may reflect differences in coregulator profiles in the different cell lines.
Expression of the UGT2B17 gene in the prostate and AR-positive prostate cancer cell lines including LNCaP, VCaP, and LNCaP-abl, and lack of expression in AR-negative prostate cancer cell lines, such as PC3 and Du145, has been well documented (Barbier et al., 2000; Turgeon et al., 2001; Chouinard et al., 2004; Bao et al., 2008). An increasing number of modulatory factors have recently been shown to negatively regulate UGT2B17 expression in LNCaP cells, including natural and synthetic androgens (DHT and R1881), interleukin-1α, epidermal growth factor, calcitrol (1α,25-dihydroxylvitamin D3), and activators for the farnesoid X receptor (FXR) such as chenodeoxylcholic acid and androsterone (Chouinard et al., 2006; Bao et al., 2008; Kaeding et al., 2008a,b). However, although these studies suggest that AR (Chouinard et al., 2006; Bao et al., 2008) or FXR (Kaeding et al., 2008b) are mediating this down-regulation, the relevant cis-regulatory element(s) with which AR or FXR interacts on the UGT2B17 promoter has not yet been characterized.
FOXA1 has been reported to play a critical role in androgen signaling in LNCaP cells and its binding motifs are significantly enriched in the vicinity of functional AR binding sites in androgen target genes (Wang et al., 2007; Lupien et al., 2008). The binding of FOXA1 to its cognate DNA elements within highly compacted chromatin is proposed to facilitate the subsequent recruitment of transcription factors, including the AR in response to androgens (Lupien et al., 2008). In this sense, our findings of FOXA1 binding to the UGT2B17 promoter combined with a recent description of DHT-enhanced recruitment of AR to the same region (Bao et al., 2008) would suggest the possible involvement of FOXA1 in mediating DHT-induced down-regulation of UGT2B17 gene expression in LNCaP and VCaP cells. Hence FOXA1 may be necessary for both the constitutive activation of the UGT2B17 gene, as demonstrated in this work, as well as for facilitating the actions of other transcriptional regulators such as AR. These possibilities are currently under investigation in our laboratory.
The −155A>G change in the FOXA1 binding site reduces the capacity for FOXA1 to bind to and stimulate the UGT2B17 promoter in LNCaP and VCaP cells. In a preliminary study on samples of normal prostate, we found that the −155A allele was also associated with higher levels of UGT2B17 mRNA (D. G. Hui and P. I. Mackenzie, unpublished data). Of the nine polymorphisms in the 5′-flanking region of the UGT2B17 gene that have been detected to date (http://www.pharmacogenomics.pha.ulaval.ca/sgc/ugt_alleles/), the −155G/A SNP is the only polymorphism shown to modulate UGT2B17 expression. This fact and its prevalence in men suggests that it could be relevant to assessments of steroid abuse in athletes and to the etiology of diseases and syndromes that involve androgens.
Testosterone abuse is generally assessed by measuring the urinary testosterone glucuronide/epitestosterone glucuronide (T/E) ratio. These ratios range from 0.9 to 1.6 in healthy men. Ratios above 4 are normally considered to be indicative of testosterone dosing. However, in one study, persons that do not express UGT2B17 (homozygous for the UGT2B17 deletion allele) have on average 20-fold lower T/E ratios than those containing two functional copies of the UGT2B17 gene after a single dose of 500 mg of testosterone (Schulze et al., 2008). Only 40% of the former reached a T/E ratio of 4 after testosterone dosing. Hence the lack of UGT2B17 expression has a profound effect on the capacity to detect testosterone dosing, based on T/E ratios. In a similar manner, persons that overexpress UGT2B17 (homozygous for the UGT2B17 −155A allele) are likely to have naturally elevated T/E ratios, which may be of sufficient magnitude to raise suspicion of testosterone doping. Because consistently elevated urinary T/E ratios have been noted in a small population of persons who have not taken testosterone (Bowers, 2008), the genotyping of persons for the −155G/A SNP may be an important addition to doping tests.
Because the progression of prostate cancer is initially androgen-dependent, higher expression of the androgen-inactivating enzyme UGT2B17 might lower prostate androgen concentrations and retard tumor progression. Indeed, antagonists of the androgen receptor (e.g., flutamide) are used to treat this disease. Hence, one could postulate that persons with the transcriptionally more active UGT2B17 −155A allele might have a lower risk of prostate cancer, and those with the transcriptionally less active −155G allele or with a deleted UGT2B17 gene might be at greater risk. This supposition was not supported by our genotyping and analyses of samples from a large Australian population-based case-control study of prostate cancer, and adds to the evidence in other published studies that do not find a link between UGT2B17 expression and prostate cancer risk (Gallagher et al., 2007; Olsson et al., 2008). UGT2B17 has been identified a susceptibility gene for osteoporosis (Yang et al., 2008). The basic findings of this study were that persons with two alleles of UGT2B17 were at higher risk for osteoporosis than those homozygous for the deletion of this gene. Because we found that the −155A allele is 4 to 13 times more active than the −155G allele, we speculate that the genotype of the −155G/A SNP might influence osteoporosis risk, possibly in the order −155A/A > −155A/G > −155G/G. Because this SNP acts in a strictly cell-specific manner, this assumption seems unlikely but awaits further investigation. It would also be interesting to test the association of the UGT2B17 −155G/A SNP to risk for other disorders linked to androgen involvement.
In summary, we found that FOXA1 is a positive regulator of the UGT2B17 gene in prostate cells, and that the prevalent polymorphism −155G/A in its binding site on the UGT2B17 promoter greatly reduces transcriptional activation by FOXA1. Because UGT2B17 inactivates and eliminates androgens by glucuronidation, this finding may have important implications in diagnostic tests for steroid abuse and in assessing disorders of the prostate and other androgen-dependent tissues and organs.
Footnotes
The work was supported by funding from the National Health and Medical Research Council of Australia [Grant 426705].
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.110.065953.
-
ABBREVIATIONS:
- AR
- androgen receptor
- UGT
- UDP-glucuronosyltransferase
- LNCaP
- lymph node carcinoma of the prostate
- DHT
- dihydrotestosterone
- R1881
- methyltrienolone
- FOXA1
- Forkhead Box A1
- SNP
- single-nucleotide polymorphisms
- PCR
- polymerase chain reaction
- VCaP
- vertebral cancer of the prostate
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- bp
- base pair(s)
- EMSA
- electrophoretic mobility shift assay
- PSA
- prostate-specific antigen
- ChIP
- chromatin immunoprecipitation
- qPCR
- quantitative real-time PCR
- siRNA
- small interfering RNA
- RT
- reverse transcription
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- T/E
- testosterone glucuronide/epitestosterone glucuronide
- HNF
- hepatocyte nuclear factor
- Pbx
- pre-B-cell homeobox
- FXR
- farnesoid X receptor.
- Received April 28, 2010.
- Accepted July 13, 2010.
- Copyright © 2010 The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}