


Abstract
We hypothesized that lacosamide modulates voltage-gated sodium channels (VGSCs) at clinical concentrations (32-100 μM). Lacosamide reduced spiking evoked in cultured rat cortical neurons by 30-s depolarizing ramps but not by 1-s ramps. Carbamazepine and phenytoin reduced spike-firing induced by both ramps. Lacosamide inhibited sustained repetitive firing during a 10-s burst but not within the first second. Tetrodotoxin-sensitive VGSC currents in N1E-115 cells were reduced by 100 μM lacosamide, carbamazepine, lamotrigine, and phenytoin from Vh of -60 mV. Hyperpolarization (500 ms) to -100 mV removed the block by carbamazepine, lamotrigine, and phenytoin but not by lacosamide. The voltage-dependence of activation was not changed by lacosamide. The inactive S-stereoisomer did not inhibit VGSCs. Steady-state fast inactivation curves were shifted in the hyperpolarizing direction by carbamazepine, lamotrigine, and phenytoin but not at all by lacosamide. Lacosamide did not retard recovery from fast inactivation in contrast to carbamazepine. Carbamazepine, lamotrigine, and phenytoin but not lacosamide all produced frequency-dependent facilitation of block of a 3-s, 10-Hz pulse train. Lacosamide shifted the slow inactivation voltage curve in the hyperpolarizing direction and significantly promoted the entry of channels into the slow inactivated state (carbamazepine weakly impaired entry into the slow inactivated state) without altering the rate of recovery. Lacosamide is the only analgesic/anticonvulsant drug that reduces VGSC availability by selective enhancement of slow inactivation but without apparent interaction with fast inactivation gating. The implications of this unique profile are being explored in phase III clinical trials for epilepsy and neuropathic pain.
Lacosamide (LCM, R-2-acetamido-N-benzyl-3-methoxypropionamide, also formerly known as Harkoseride, SPM 927, or ADD 234034; Fig. 1A) is a novel anticonvulsant currently in phase III clinical trials that has shown significant promise in the treatment of partial seizures with or without secondary generalization (Doty et al., 2007) and diabetic neuropathic pain (McCleane et al., 2003). In preclinical screens, the functionalized amino acid molecule was more potent than both phenytoin (DPH) and phenobarbital in preventing tonic hind limb extension in the maximal electroshock seizure (MES) test in vivo in both rats (ED50 = 3.9 mg/kg p.o.) and mice (ED50 = 4.5 mg/kg i.p.) (LeTiran et al., 2001). LCM was also anticonvulsant in two animal models for status epilepticus and showed a high degree of stereoselectivity (the S-stereoisomer SPM 6953 was 10 to 30 times less potent in the MES test) (LeTiran et al., 2001).
The molecular mode of action of LCM is still unknown. Binding studies have revealed that LCM does not displace radioligands from a plethora of recognized anticonvulsant receptor/channel binding sites, including those for NMDA (phencyclidine, MK-801, glycine), AMPA, GABAA, GABAB, 5-hydroxytryptamine, and dopamine (Errington et al., 2006). In electrophysiological studies, LCM (100 μM) did not affect currents evoked by application of exogenous NMDA, AMPA, or GABA in cortical neuronal cultures but did significantly reduce the incidence of both spontaneous inhibitory postsynaptic currents and excitatory postsynaptic currents, as well as producing the inhibition of spontaneously firing action potentials (Errington et al., 2006). These early data suggested that LCM does not interact with known recognition sites for anticonvulsants on ligand-gated ion channels or other synaptic receptors.

A, functionalized amino acid structure of the novel anticonvulsant molecule LCM. The asterisk indicates the chiral center: the R isomer LCM is biologically active, whereas the L isomer (SPM 6953) had no significant effect in preclinical epilepsy models DPH (B), CBZ (C), LTG (D).
As disclosed previously, LCM has shown significant potency in the MES test in vivo and a relative lack of efficacy in the threshold pentylenetetrazol model. This profile is similar to the antiepileptic drugs carbamazepine (CBZ), lamotrigine (LTG), and DPH, all of which are selective for the former experimental seizure model over the latter (Miller et al., 1986; Lang et al., 1993; Meldrum, 2002). It is now generally accepted that CBZ, LTG, and DPH share a common primary mode of action (although postulated novel molecular target sites may contribute to the distinct pharmacological profiles of the drugs in different cellular compartments; Cunningham and Jones, 2000; Poolos et al., 2002; Riddall et al., 2006) in altering fast inactivation gating of voltage-gated sodium channels (Willow et al., 1985; Lang et al., 1993; Ragsdale et al., 1996), producing tonic and use-dependent blockade.
In our earliest mechanistic studies, sustained repetitive firing (SRF; 750 ms) evoked by somatic current injection was weakly but significantly reduced in frequency by LCM without apparent changes (amplitude, duration) in individual spike properties (Errington et al., 2006). The subtle reduction in the number of spikes throughout a 750-ms period of SRF by LCM was markedly different from that produced by acknowledged sodium channel-blocking anticonvulsants. These older drugs typically produce a complete block of regenerative spiking within a few tens of milliseconds of an evoked SRF burst (McLean and Macdonald, 1983; Willow et al., 1985; Lang et al., 1993; Lees and Leach, 1993). Nonetheless, the marginal effect of LCM on electrogenesis in this experiment may suggest that the novel anticonvulsant could be acting, in part, via inhibition of VGSCs. This postulate was reinforced by binding data that showed LCM (10 μM) was capable of producing 25% displacement of [3H]batrachotoxin binding to VGSC site 2 in rat brain homogenates (Errington et al., 2006). Our earlier experiments suggested that the drug could interfere preferentially with seizure spread, that it could reduce synaptic traffic (indiscriminately for excitation and inhibition) and spontaneous action potentials, and that voltage-gated potassium and calcium channels were not targeted (Errington et al., 2006; Lees et al., 2006). On the basis of these leads, in this article, we examined the hypothesis that LCM may be a modulator of VGSC but that it may require different biophysical conditions (or exploit a new target site) to produce inhibition compared with existing anticonvulsant drugs in the pharmacopoeia (CBZ, LTG, and DPH), which have been used for comparison. We report that LCM does not modify fast inactivation of the VGSC like the other VGSC modulating anticonvulsants but that it has a unique inhibitory action in promoting slow inactivation of the VGSC (a new mechanism for blocking a target acknowledged for its importance in the treatment of epilepsy and pain). The implications of this novel mechanism for the pharmacological profile of the drug are unknown but are currently being characterized in ongoing clinical trials.
Materials and Methods
Primary Neocortical Neuron Culture. Neuronal cultures were prepared from cerebral cortices of embryonic day 16 to 18 Sprague-Dawley rats. Donor animals were humanely killed, and embryos were collected by Caesarian section in accordance with Schedule 1 of the Home Office Animals (Scientific Procedures) Act, United Kingdom, 1986, and with the approval of University of Otago Animal Ethics committee. Cortices were minced with a razor blade and dissociated by trituration through Pasteur pipettes (without the addition of proteolytic enzymes). Isolated cells were plated onto poly(d-lysine)-coated glass shards (approximately 40 mm2) at densities between 50,000 and 100,000 cells/ml in Neurobasal medium (Invitrogen, Paisley, UK) containing 2% B27 (Invitrogen), 1% Glutamax-1 (Invitrogen), 100 U/μg per milliliter of penicillin/streptomycin (Sigma, Poole, Dorset, UK) and 25 μM l-glutamate (Sigma) as described previously (Errington et al., 2006). Cells were maintained in a humidified incubator at 37°C and 5% CO2. After 3 days in vitro (DIV), 50% of the media was removed and replaced with twice the volume of fresh media as defined above but without l-glutamate. After 7 DIV, a further feed was given to allow maintenance of viable cultures for several weeks. Cells were used in experiments typically after 21 to 28 DIV.
N1E-115 Mouse Neuroblastoma Cell Culture. N1E-115 mouse neuroblastoma cells were obtained from the European Collection of Animal Cell Cultures (Wiltshire, UK). Confluent cells (70-80%) were subcultured twice weekly and grown on poly(d-lysine)-coated glass shards in Dulbecco's modified Eagle's medium (Invitrogen) containing 10% fetal calf serum (Sigma) and 100 U/μg per milliliter of penicillin/streptomycin (Sigma). Cells were incubated at 37°C in 5% CO2 in triple-vented 35-mm cell culture dishes (Iwaki, Tokyo, Japan). Cells were used for electrophysiological experiments 24 to 48 h after plating.
Electrophysiology. Cultured neurons adhering to glass shards were placed in a Perspex trench on the stage of a Nikon Diaphot (Nikon, Tokyo, Japan) inverted phase-contrast microscope and superfused (approximately 2 ml/min) with buffered physiological saline containing 142 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, and 30 mM d-glucose, pH adjusted to 7.4 with NaOH. Recordings were made from neurons of pyramidal morphology (unless otherwise stated) using the whole-cell patch-clamp technique with intracellular solution consisting of 142 mM potassium gluconate, 1 mM CaCl2, 2 mM MgCl2, 10 mM HEPES, and 11 mM EGTA, pH 7.4 (KOH). Micropipettes were fabricated on a model P97 Flaming/Brown Micropipette puller (Sutter Instrument Company, Novato, CA) using GC150T-10 borosilicate glass (Harvard Apparatus Ltd, Edenbridge, England, UK) resulting in pipettes with an impedance of typically 4 to 5 MΩ. All patch-clamp recordings were performed at room temperature (∼22 ± 1°C) using an Axopatch 200 integrating amplifier (Molecular Devices, Sunnyvale, CA). For sustained repetitive firing experiments, 2 mM CoCl2 and the AMPA receptor antagonist 6-cyano-7-nitroquinoxaline-2,3-dione (10 μM) were added to the bath to prevent calcium entry and recurrent excitability.
For voltage-clamp experiments on N1E-115 cells, the bath was continuously perfused with a solution containing 140 mM NaCl, 5 mM KCl, 1.8 mM CaCl2, 0.8 mM MgCl2, and 10 mM HEPES, pH 7.3 (NaOH) at a flow rate of approximately 1.5 to 2 ml/min. Patch pipettes (3-5 MΩ) were filled with solution containing 10 mM NaCl, 20 mM tetraethylammonium chloride, 110 mM CsCl, 1 mM CaCl2, 2 mM MgCl2, 11 mM EGTA, and 10 mM HEPES, pH 7.4 (CsOH). To reduce the capacitance of the microelectrode, filled glass pipettes were immersed in Sigmacote (Sigma) or were coated with Sylgard 184 (Dow Corning, Midland, MI). Isolated, spherical, and unclumped neuroblastoma cells were selected for patch-clamp experiments (to minimize space clamp problems), and whole-cell capacitance and series resistance were cancelled using the preamplifier. Series resistance compensation (70-85%) was routinely applied. To allow accurate measurement of I/V properties, 5 min was allowed after membrane rupture to achieve full dialysis of the cell before data were recorded. Series resistance was monitored throughout all experiments, and cells were discarded if it became greater than three times the open pipette resistance (approximately 15 MΩ). Pulse protocols were applied in control solutions and again after 3-min equilibration with drugs unless otherwise indicated.
Data were filtered at 5 kHz and digitized at 15 to 20 kHz using a CED micro1401 (Cambridge Electronic Design, Cambridge, UK), and pulse protocols were generated using Signal 2.10 (Cambridge Electronic Design) software. To isolate pure ionic currents through sodium channels, leak currents and residual capacitance artifacts were deducted offline using Signal software. For activation curves, conductance (g) through Na+ channels was calculated using the equation g = INa+/(V - Er), where INa+ is the peak sodium current, V is the test potential, and Er is the observed reversal potential. Activation and inactivation curves were fitted to a Boltzmann function of the form  where gmax is the peak conductance, Imax is the peak current, V50 is the voltage at which half-maximal current/conductance occurs, k is the slope factor and V is the test potential. Recovery from steady-state fast inactivation was best fit with a biexponential equation of the form
where gmax is the peak conductance, Imax is the peak current, V50 is the voltage at which half-maximal current/conductance occurs, k is the slope factor and V is the test potential. Recovery from steady-state fast inactivation was best fit with a biexponential equation of the form  where t is time, A is the amplitude component of each exponent, and τ is time constant for recovery. Slow inactivation voltage curves were fit using a modified Boltzmann equation (Carr et al., 2003) of the form
where t is time, A is the amplitude component of each exponent, and τ is time constant for recovery. Slow inactivation voltage curves were fit using a modified Boltzmann equation (Carr et al., 2003) of the form  Details of specific pulse protocols are described in the results text or figure legends. All data are depicted as mean ± S.E.M., and statistical analysis was by one-way analysis of variance (Dunnett's post hoc), paired/unpaired t test, Mann-Whitney test, or Kruskal-Wallis (Dunn's post hoc) test where appropriate.
Details of specific pulse protocols are described in the results text or figure legends. All data are depicted as mean ± S.E.M., and statistical analysis was by one-way analysis of variance (Dunnett's post hoc), paired/unpaired t test, Mann-Whitney test, or Kruskal-Wallis (Dunn's post hoc) test where appropriate.
Pharmacology. All reagents were obtained from Sigma unless otherwise indicated. For electrophysiological experiments CBZ and DPH were obtained from Sigma, and LTG was from Tocris Cookson (Bristol, UK). All drugs were formulated daily by dissolution into dimethyl sulfoxide (Sigma). The final concentration of dimethyl sulfoxide in physiological solutions was not greater than 0.1% (v/v), and all drug-free control solutions contained an equal concentration of the solvent. LCM and SPM 6953 were supplied by Schwarz Pharma GmbH (Monheim, Germany).
Results
Electrophysiology: Primary Neocortical Neurons
Sodium Spikes Evoked by Fast or Slow Depolarizing Ramps Were Differentially Sensitive to Anticonvulsants. To study the effects of LCM upon electrogenesis, cultured rat cortical neurons were voltage-clamped in physiological saline using a K+-gluconate-based intracellular solution. From a holding potential of -70 mV, a slow depolarizing (3 mV/s) voltage ramp to +20 mV was applied to the cell soma through the patch pipette. In a subpopulation of neurons tested, as the result of space clamp complications caused by the extensive neuritic projections present in the mature cultured neurons, adequate voltage clamp could not be maintained in response to the depolarizing ramp. The lack of control over the cellular potential consequently led to the firing of high-frequency trains of action currents mediated by TTX-sensitive voltage-gated sodium channels (Fig. 2). From an electrophysiological stand point, this would usually be considered undesirable. From a pharmacological perspective, it allowed us to examine the effects of drugs on membrane excitability in response to slow and sustained depolarization. LCM could profoundly inhibit the firing of action currents that were induced by this slow ramp depolarization protocol. In drug-free control solutions, slow ramps evoked 50.25 ± 10.25 spikes per burst (n = 4), and this was reduced to 13.50 ± 6.74 (76.54 ± 8.91% reduction, Fig. 2, C and D) spikes per burst after 3-min perfusion with LCM (100 μM; P < 0.05, Kruskal-Wallis), an effect that was reversible upon washout (46.75 ± 7.00). Both CBZ (fast-binding; Kuo et al., 1997) and DPH (slowly binding; Kuo and Bean, 1994) could almost completely occlude the firing of spikes in response to the slow ramps with 99.67 ± 0.33% (n = 4) and 98.75 ± 1.08% (n = 4) reduction, respectively (Fig. 2, C and D). However, in a similar suite of experiments in which the rate of potential change was much more rapid (90 mV/s), the resulting trains of evoked spikes were insensitive to LCM. In control solutions, depolarization by such a ramp induced the firing of 10.50 ± 1.99 spikes per burst, whereas in the presence of LCM, this was only very marginally (P > 0.05, n = 10) reduced to 9.30 ± 2.17 (17.91 ± 9.47% reduction). This depolarization rate-dependent block of bursts by LCM seemed to reinforce previous observation of only very marginal inhibition of brief (750 ms) sustained repetitive firing by LCM (Errington et al., 2006). Furthermore, the spikes evoked by the rapid depolarizing ramp were still highly sensitive to the fast inactivation modifying drugs CBZ and DPH (n = 4), which produced 78.76 ± 6.00% and 75.87 ± 14.26% reduction, respectively (Fig. 2, A and B). When the potential was ramped rapidly, LCM did not increase the latency to the first spike; in fact, a small reduction in time to the first spike was observed compared with control (-1.04 ± 1.29%; Fig. 2, A and B). In contrast, both CBZ and DPH produced increased latency to first spike compared with control ramps with 10.53 ± 10.26% and 3.57 ± 2.60% increase, respectively.
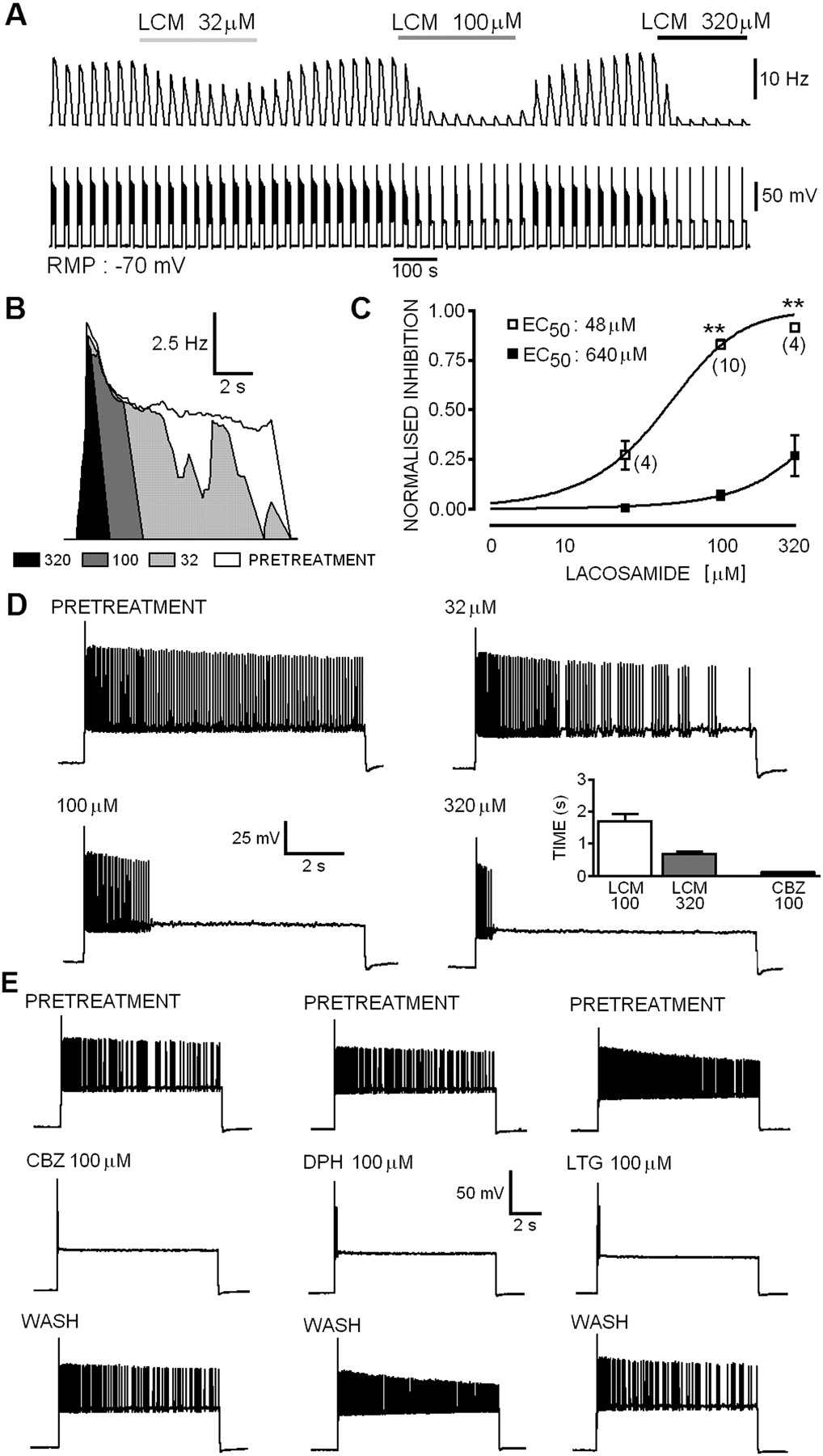
Prolonged Sustained Repetitive Firing Was Markedly Attenuated by Lacosamide but with Slow Kinetics Compared with Other Anticonvulsants. Ictal seizures and epileptiform paroxysmal depolarizing shifts (in vitro and in vivo) often last for tens of seconds to minutes, so we decided to test the ability of LCM to block prolonged SRF bursts (in contrast to brief 750-ms pulses studied previously; Errington et al., 2006). Ten seconds of sustained repetitive firing was evoked in current clamped (Erest -61 ± 1.5 mV, n = 16) cortical neurons. Previous experiments, in a brain slice model of tonic-clonic epileptiform activity induced by 4-AP (Lees et al., 2006), showed that LCM markedly reduced the duration of prolonged tonic firing. The mean firing frequency over the entire 10-s burst (10-s bins) was calculated from the average of four pretreatment or drug-treated (5-min bins) bursts (Fig. 3). In the absence of drugs, neurons produced high-frequency (15.04 ± 0.94 Hz) trains of overshooting (first AP overshoot, 65 ± 3.1 mV, n = 16) action potentials. Neither Erest nor overshoot of the first action potential (LCM 320 μM: -64 ± 9.5 mV; CBZ 100 μM: -65 ± 4.1 mV) were significantly (P > 0.05, Mann-Whitney) affected by LCM (32-320 μM) or CBZ (100 μM). Perfusion of the cellular monolayer with LCM produced a significant concentration-dependent (EC50 value, 48 μM) reduction in the mean firing frequency of neurons compared with pretreatment, an effect that was fully reversible upon washout (Fig. 3, A-C). At 32 μM, LCM produced a noticeable but not significant reduction in the mean firing frequency (control, 12.86 ± 0.84 Hz; LCM, 9.32 ± 0.94 Hz; P > 0.05, n = 4, Wilcoxon matched pairs). When the concentration was increased to 100 μM, the drug significantly inhibited sustained firing (control, 16.25 ± 1.6 Hz; LCM, 2.63 ± 0.43 Hz; P < 0.01, n = 9), and this was enhanced further with an increase in concentration to 320 μM (control, 14.6 ± 1.18 Hz; LCM, 1.28 ± 0.19 Hz; P < 0.01, n = 4). However, the temporal characteristics of the block showed that residual spikes occurring in the presence of the drug are confined to the initial segment of the burst. When the prolonged SRF bursts were analyzed in 1-s bins, the data showed that LCM did not profoundly inhibit the firing rate of action potentials within the early phase of the burst (EC50 value, 640 μM). Even at 320 μM, LCM only produced 27.1 ± 1% reduction in firing frequency and allowed residual high-frequency burst activity of 0.68 ± 0.07-s duration. Unlike the effects of LCM, the fast inactivation modifying anticonvulsants CBZ, LTG, and DPH all produced a marked reduction of firing frequency well within the first second of the burst. CBZ (100 μM; n = 4, Fig. 3, D and E) blocked all spikes except for those occurring in the first 0.11 ± 0.006 s of the SRF burst, and this was accompanied by a dramatic attenuation of the amplitude or full occlusion of the remaining action potentials. Although the data for LTG and DPH were not quantitatively analyzed, they were qualitatively similar to the fast effects of CBZ on prolonged SRF bursts (Fig. 3E). LCM clearly inhibits the firing of repetitive action potentials and reduces cellular excitability, although it is clear that unlike CBZ, LTG, and DPH, relatively prolonged depolarization is required before the block is apparent. The inability of even 320 μM LCM to inhibit the early phase of SRF (Fig. 3D) is strongly indicative that the marked divergence between LCM and CBZ/LTG/DPH on SRF (and bursts evoked by ramps) does not simply reflect a difference in potency at a common target site. The characteristics of the inhibition produced by the novel drug suggest that it may be binding to a different binding site or that the binding rates are significantly slower than for the existing clinically used molecules.

A, current profiles evoked in cultured cortical neurons by a fast ramp depolarization of the holding potential from -70 to 20 mV (90 mV/s). Before drug treatment, high-frequency bursts of TTX-sensitive (data not shown) action currents were observed. CBZ (100 μM) and DPH (100 μM) produced profound and reversible inhibition of spike firing. LCM (100 μM) had only a weak effect on the frequency of spikes induced by fast ramps. B, histograms showing the marked reduction in spike frequency produced by CBZ and DPH (in contrast to the moderate effect of LCM) and the increased latency to first spike in the presence of these drugs. D, spikes evoked by slow depolarizing ramps were weakly sensitive to inhibition by LCM but were completely occluded by CBZ and DPH (all at 100 μM) as summarized in C.

A, representative experiment showing concentration-dependent inhibition of action potential firing in 10-s bursts of evoked SRF in a single cell pyramidal neuron with a resting potential of -70 mV (bottom trace). Mean firing frequency plot in 10-s bins. B, top, overlaid mean frequency plots (1-s bins) showing the inhibition of firing by LCM. The open portion reflects the control frequency. Light gray shows the conversion of the firing pattern to that of interrupted repetitive firing (or bursting) by 32 μM LCM that can also be seen in the expanded tracing shown in D. The black portion shows the profound effect of 320 μM LCM. Note here, however, that although the duration of the repetitive firing is markedly attenuated, the initial frequency (in the first second) is virtually unchanged. C, concentration-response curves for the data as analyzed in A (10-s bins, □) and in B (1-s bins, ▪). Note the profound shift in potency for prolonged evoked trains. D, expanded representative tracings of the 10-s SRF showing the inhibitory effects of LCM. The inset histogram displays the residual duration of firing at equilibrium. E, in marked contrast to LCM, the other antiepileptic drugs CBZ, LTG, and DPH inhibit all spikes except for those occurring within approximately 100 ms of the burst initiation.
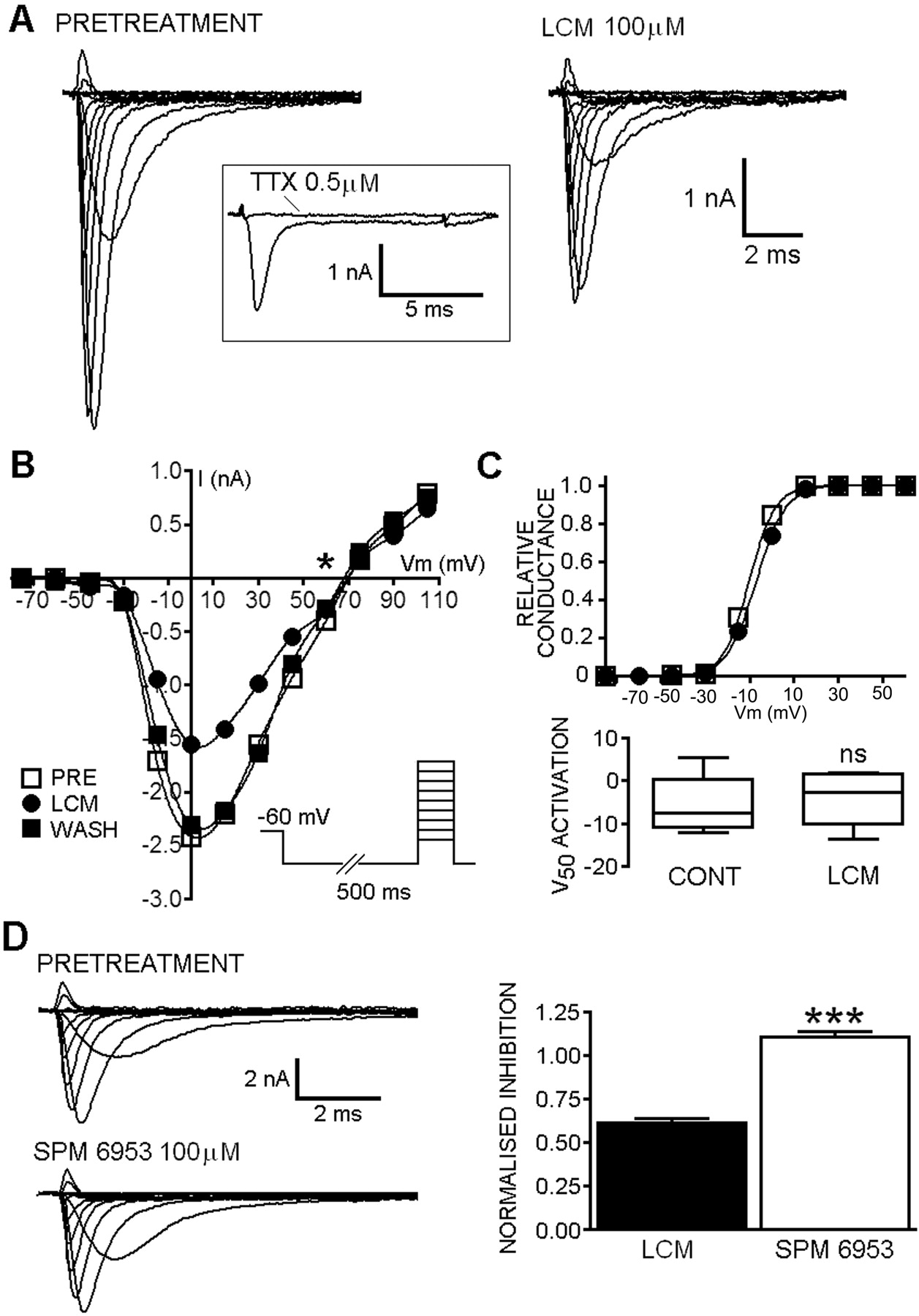
A, families of current responses to a 10-ms depolarizing step (-60 to +110 mV) from a representative cell before and after application of 100 μM LCM. Inset, complete block of the evoked inward current by tetrodotoxin (0.5 μM). Note the presence of a TTX-sensitive persistent sodium current in these cells. B, current-voltage plot taken from cell different from that shown in A demonstrates the reversal potential (*) is close to the predicted Nernstian Na+ equilibrium potential and that LCM reversibly blocked both inward and outward sodium currents. After removal of the drug, the curve was almost coincident with the pretreatment curve in this cell. Inset, the step depolarization protocol used to evoke currents. C, representative normalized conductance-voltage plot from a single cell (different from those used in A and B) showing that curves were almost superimposable in the presence of LCM and in control. The box and whisker plot shows that the V50 value for activation was not significantly shifted (P > 0.05, Mann-Whitney) in replicated experiments (n = 4). This confirms that the block by LCM did not reflect shifts in the voltage-dependence of activation gating. D, the target for LCM is stereoselective. Families of evoked currents were not altered by SPM 6953 (100 μM) compared with pretreatment currents. The maximal current evoked using the inset pulse protocol was reduced by LCM but not by SPM 6953 (which is not anticonvulsant in vivo). The effect of 100 μM LCM in reducing sodium currents was significantly (P > 0.05, n = 3) different to an equal concentration of the S-stereoisomer.
Electrophysiology: Mouse N1E-115 Neuroblastoma Cells
Lacosamide Inhibited Voltage-Activated Sodium Currents. Mouse neuroblastoma cells were patch-clamped in the whole-cell configuration. All cells showed fast, rapidly inactivating inward currents in response to step depolarization. Peak currents were evoked by 10-ms depolarizing steps to varying test potentials (-70 to 100 mV) from a holding potential (Vh) of -60 mV preceded by a 500-ms hyperpolarizing step to -100 mV to remove the influence of fast inactivation (Fig. 4B). The evoked currents were fully blocked by 500 nM TTX (Fig. 4A, inset) but were insensitive to 500 nM Cd2+ (data not shown), and the kinetics, activation, and reversal potentials (Fig. 4) were consistent with fast channels selective for Na+. Application of 100 μM LCM to the clamped cells resulted in a reduction in the peak sodium current observed at all test potentials (Fig. 4, A and B). The maximal current evoked in response to a 10-ms test pulse to 0 mV was 0.61 ± 0.02 (n = 4) that of control in the presence of LCM, and the effects were fully reversible after removal of the drug (Fig. 4B) Inhibition of sodium currents by LCM was not accompanied by shifts in the voltage-dependence of activation gating for the channels (V50: control, -5.3 ± 4.0; k, 8.1 ± 1.5, n = 4; LCM: -4.2 ± 3.7; k, 8.8 ± 1.8, n = 4; P > 0.05; Fig. 4C) and did not alter the reversal potential (although this was not analyzed in detail). In contrast to this, the S-stereoisomer, SPM 6953 at the same concentration marginally increased the fraction of available current (1.11 ± 0.03, n = 4, P > 0.05; Fig. 4D).

A, LCM (top) produced inhibition of transient sodium currents evoked by test pulses to 0 mV from a holding potential of -60 mV with (left) or without (right) a prior 500-ms hyperpolarizing prepulse. In contrast, the blocking effect of DPH (bottom right) could be completely occluded (traces overlaid, bottom left) by the hyperpolarizing pulses. B, bar chart summarizing the profound voltage-dependent blocking action of the typical sodium channel acting anticonvulsants CBZ, LTG, and DPH in the same experiment. All three anticonvulsants produced marked inhibition of currents evoked from -60 mV that was considerably reduced when a 500-ms hyperpolarizing pulse was delivered before the test pulse. LCM, on the other hand, produced approximately 30% reduction in current under both conditions. C, the frequency-dependence of block was examined from a relatively hyperpolarized holding potential and currents were evoked at 10 Hz by 20-ms test pulses to -10 mV (protocol shown inset, bottom). Representative overlaid current traces are shown of the 1st, 10th, 20th, and 30th pulses under control conditions and in the presence (right) of DPH and LCM. D, plot summarizing the frequency-dependent decrement in peak current amplitude (mean value from replicated experiments) produced by CBZ, LTG, and DPH but not LCM (for clarity error, bars are not shown, values are quoted in text). A small decrement (∼10%) in peak current amplitude was observed under control conditions (□). After only 10 pulses, both CBZ and DPH produced significantly more attenuation of current than pretreatment (LTG followed the trend). At the end of the 30-pulse train, all three anticonvulsants produced significant inhibition compared with pretreatment. In contrast, 100 μM LCM exerted no significant (P > 0.05) frequency-dependent facilitation of block even after 30 pulses, although a slow reduction in the peak current was observed beginning after approximately 12 to 13 pulses (arrow).
Inhibition of Sodium Currents by Lacosamide Showed Markedly Different Voltage-Dependent Properties Compared with Other Antiepileptic Drugs. To test the voltage-dependence of block, neuroblastoma cells were maintained at a holding potential of -60 mV and depolarized by a 10-ms test pulse to 0 mV at 0.5 Hz. The protocol was repeated in each cell with a 500-ms hyperpolarizing pulse to -100 mV before the depolarizing test pulse (Fig. 5A). In all cells tested (n ≥ 4), all four of the anticonvulsant drugs (100 μM) produced a reduction in current when Vh was -60 mV (residual current after drug equilibration was for CBZ: 0.29 ± 0.17, n = 4, Fig. 5B; LTG: 0.50 ± 0.08, n = 5, Fig. 5B; DPH: 0.52 ± 0.10, n = 6, Fig. 5B; LCM: 0.68 ± 0.05, n = 7, Fig. 5, A and B). For CBZ, LTG, and DPH, application of a 500-ms hyperpolarizing pulse to -100 mV significantly (two-tailed unpaired t test) reduced the blocking action on the VGSCs, with the fraction available being 0.94 ± 0.19 (P < 0.05), 0.88 ± 0.06 (P < 0.01), and 0.99 ± 0.05 (P < 0.01), respectively, compared with control values. The inhibition produced by LCM was not (P > 0.05) altered by the hyperpolarizing prepulse. When 500-ms prepulses to -100 mV were applied in the presence of LCM, the peak evoked current was still reduced to 0.71 ± 0.06 of the pretreatment maximum (Fig. 5, A and B).
Rapid Frequency-Dependent Facilitation of Block Was Not Observed with Lacosamide. A series of 30 test pulses (20 ms to 0 mV) were delivered from a holding potential of -80 mV at 10 Hz. The available current in control and in the presence of drugs was calculated by dividing the peak current at any given pulse (pulsen) by the peak current in response to the initial pulse (pulse1). CBZ (0.80 ± 0.01, P < 0.01, n = 6), LTG (0.84 ± 0.01, P > 0.05, n = 7), and DPH (0.78 ± 0.01, P < 0.001, n = 7) markedly reduced current amplitude compared with controls (0.90 ± 0.01, n = 10) by the tenth pulse in the train, but the LCM currents were almost superimposable with controls (Fig. 5, C and D). It is interesting that LCM began to show some degree of use-dependent block, with a distinct latency, only after approximately 13 to 14 test pulses (Fig. 5D, arrow). However, even by the last of the 30 pulses delivered, the peak current available was still not significantly different from control (control, 0.89 ± 0.01; LCM, 0.83 ± 0.01; P > 0.05). In contrast, by the 30th test pulse in the train in the presence of CBZ (0.78 ± 0.01, P < 0.01), LTG (0.79 ± 0.01, P < 0.05), or DPH (0.71 ± 0.02, P < 0.001), the available peak current was significantly reduced compared with control.
Steady-State Fast Inactivation Voltage Curves Were Not Shifted in the Hyperpolarizing Direction by Lacosamide. Steady-state fast inactivation curves (Fig. 6A) were fitted to a single Boltzmann function of the form described previously (eq. 1). The protocol (Fig. 6A, inset) was designed with the intention of recruiting predominantly fast sodium channel inactivation and minimizing the development of slowly inactivated conformations. The V50 value for inactivation under control conditions was -66 ± 0.9 mV (n = 21, pooled from all replicates). Significant hyperpolarizing shifts in the inactivation curves (summary statistics in Fig. 6C) were produced by CBZ (V50: - 79 ± 2.6 mV; n = 5, P < 0.001; Fig. 6, A and B), LTG (V50: - 72 ± 1.7 mV, n = 7, P < 0.05; Fig. 6B), and DPH (V50: -77 ± 2.3 mV, n = 7, P < 0.05; Fig. 6B). LCM (100 μM) did not produce a significant shift in the V50 value for inactivation of sodium currents in the neuro-blastoma cells. The voltage for half-maximal inactivation after equilibration with LCM (-65 ± 1.7 mV, n = 7; Fig. 6D) was not significantly different (P > 0.05) from the observed V50 value in control solutions. It is noteworthy that in contrast to the fast inactivation modifiers, the entire curve for the experiments in the presence of LCM displayed a very marginal depolarizing shift, although this was not significant. Furthermore, the slopes (k) of the inactivation curves were decreased marginally by CBZ (k: 7.1 ± 0.6 mV, P > 0.05), DPH (k: 7.3 ± 0.2 mV, P > 0.05), and significantly by LTG (k: 7.5 ± 0.3 mV, P < 0.05), whereas the slope in the presence of LCM was almost identical with the control value (k: 6.6 ± 0.3 mV; Fig. 6B).

A, steady-state inactivation curves before and after equilibration with anticonvulsant drugs. Prepulses (500 ms) between -120 and -20 mV were given followed immediately by a test pulse (30 ms) to -10 mV (A, inset). Tracings showing voltage-dependent steady-state fast inactivation of sodium currents and the effects of CBZ and LCM. B, steady-state fast inactivation curves (Boltzmann fit) were shifted in the hyperpolarizing direction by CBZ, LTG, and DPH (note that the LTG and DPH curves and data points are almost superimposed) but not by LCM compared with pooled control data (n = 21). C, box and whisker plot showing the V50 value for inactivation was significantly (P < 0.05, Kruskal-Wallis with Dunn's multiple comparison test post hoc) shifted in the hyperpolarizing direction for CBZ (P < 0.001, n = 5), LTG (P < 0.05, n = 7), and DPH (P < 0.05, n = 7) but not LCM (P > 0.05, n = 7) compared with control data. LCM seemed to produce a marginal depolarizing shift in steady-state inactivation. V50 value for inactivation in the presence of LCM was not statistically altered compared with controls (P > 0.05), but the slope of the curve was identical, and all points were displaced to the right across the replicated experiments. D, currents depicting recovery from fast inactivation were measured using the pulse protocol illustrated in the absence and presence of drugs (Δt is indicated in milliseconds for each trace). E, plot of the recovery in presence and absence of drugs. Note that recovery was 95% complete after 100 ms in control saline. F, CBZ (100 μM;  ) significantly prolonged the time for recovery from steady-state fast inactivation (P < 0.001). LCM (100 μM; ▪) did not alter the time for recovery significantly (P > 0.05) or displace the control curve (□) compared with the obvious retardation of recovery with CBZ.
) significantly prolonged the time for recovery from steady-state fast inactivation (P < 0.001). LCM (100 μM; ▪) did not alter the time for recovery significantly (P > 0.05) or displace the control curve (□) compared with the obvious retardation of recovery with CBZ.
Lacosamide Did Not Significantly Retard the Recovery of Sodium Channels from Steady-State Fast Inactivation. Neuroblastoma cells were held at -90 mV, and a test pulse to -10 mV (20 ms) was given before a conditioning pulse of 500 ms to the same potential. After the conditioning pulse, varying time for recovery (Δt) was allowed before delivery of a second test pulse (Fig. 6D). The holding potential and relatively brief conditioning pulses were designed to remove the potential influence of sodium channel slow inactivation and isolate the fast inactivation gating process. The fraction of current available after each recovery period (depicted Fig. 6E) is the result of the second test pulse divided by the first. In control conditions, a relatively large proportion of the sodium current (0.41 ± 0.03, n = 7; Fig. 6, D and F) was reavailable after only a brief recovery period (3-ms, 50% maximal recovery, ∼4 ms). When 100 μM CBZ was applied to the bath for three min before running the pulse protocol, the fraction available after 3 ms was significantly reduced (P > 0.001, n = 5; Fig. 6, D-F) to 0.18 ± 0.03 (50% maximal recovery, ∼37.5 ms). LCM, on the other hand, did not produce any significant (P > 0.05, n = 5) retardation in recovery of channels from steady-state fast inactivation with the fraction available at 3 ms being 0.40 ± 0.04 (50% maximal recovery, ∼4.3 ms). In response to the 500-ms conditioning potential used, CBZ but not LCM was able to inhibit the recovery of sodium channels from fast inactivation for up to 300 ms after repolarization (as summarized in Fig. 6F).

LCM enhances the entry of sodium channels into the slow inactivated state. A, under drug-free physiological conditions, the pulse protocol used (inset, 10 s) resulted in reduced channel availability (P2/P1) of approximately 30% in voltage-clamped N1E 115 cells. The extent of physiological slow inactivation was consistent across three different groups of cells. LCM produced a concentration-dependent enhancement in the reduction of sodium channel availability attributable to slow inactivation. B, plot of the fraction of current unavailable as a result of physiological slow inactivation (○, n indicated) for each group of cells compared with the fraction unavailable in the presence of increasing concentration of LCM (32-320 μM, •). To account for the tonic blocking action of LCM at the holding potential used, the enhancement of slow inactivation was measured as the fraction of current made unavailable after the conditioning pulse (P2LCM) compared with that available before conditioning (P1LCM) in the presence of the drug. C, concentration-response curves for the inhibition of sodium currents by LCM at the holding potential of -80 mV (P1LCM/P1CONT; □) and after slow inactivation was induced by the conditioning pulse to -20 mV (P2LCM/P2CONT; ▪). LCM produced a greater (but not significant: P > 0.05, n = 5-7, Mann Whitney) reduction in the peak current amplitude in cells in which slow inactivation was prevalent. D, LCM enhances the rate of entry of sodium channels into the unavailable slow inactivated state with time constants for entry as shown. The first data points were taken from the recovery experiments presented in Fig. 6, D and E, and represents the fraction of current available after a 1-s recovery period from a 500-ms conditioning pulse.
Lacosamide Enhanced the Entry of Sodium Channels into the Unavailable Slow-Inactivated State. In light of the ability of LCM to inhibit sodium currents despite any apparent effect on fast inactivation gating, we decided to assess the effect of the novel drug upon voltage-gated sodium channel slow inactivation. In line with the findings of many authors in a varied range of preparations, VGSCs in the mouse N1E-115 cells undergo the physiological process of slow inactivation when exposed to periods of prolonged depolarization. To assess the entry of sodium channels into the slowly inactivated state, cells were held at -80 mV and depolarized for either 10 or 30 s to -10 mV followed by a recovery interval of 1.5 s before a test pulse to measure the peak available current (Fig. 7A, inset). The recovery interval of 1.5 s was used to completely allow the recovery of fast inactivation, making occupancy of the slow inactivated state the sole determinant of the second test pulse amplitude. When neuroblastoma cells were depolarized for 10 s, the peak available current was reduced to 0.73 ± 0.01 (n = 18, pooled from all experiments described below) of that of the preconditioning test pulse, and when the conditioning pulse was increased to 30-s duration, the current available was reduced further to 0.61 ± 0.02 (n = 4).
LCM enhanced the entry of sodium channels into the slow inactivated state in a concentration-dependent manner. Under control conditions, the degree of physiological slow inactivation induced by the conditioning pulse was consistent (P > 0.05; Fig. 7B, ○) across three different groups of cells. LCM (32 μM) produced a slight but not significant (n = 5, P > 0.05, paired t test) increase in the fraction of channels that became unavailable as a result of slow inactivation with 0.63 ± 0.05 available compared with pretreatment value of 0.69 ± 0.05 (Fig. 7, A and B). However, when the concentration of LCM was increased to 100 μM, a significant reduction in the fraction of available channels was noted after the conditioning prepulse. The fraction of channels available was reduced to 0.50 ± 0.02 compared with 0.73 ± 0.02 in the same cells before LCM application (n = 6, P < 0.01; Fig. 7, A and B). LCM (320 μM) significantly (P < 0.001, n = 7) reduced the fraction of available channels to 0.38 ± 0.01 compared with 0.71 ± 0.02 pretreatment. Plots of normalized peak current (P2/P1; Fig. 7D) against conditioning pulse duration were fit with a monoexponential function and yielded a time constant for the physiological entry into slow inactivation (for the pulse protocol used) of τctrl = 11.95 s (Fig. 7D). LCM (100 μM) more than doubled the reduction of sodium channel availability that resulted from entry into the slow inactivated state. In the presence of the drug using the pulse protocol shown, channels entered the slow inactivated state with a time constant for entry of τLCM = 4.79 s. The highly significant, concentration-dependent and reversible (data not shown) LCM-induced changes were measured over a limited number of data points, which prevented a deeper kinetic analysis of the LCM-induced acceleration of entry into the slowly inactivated state.
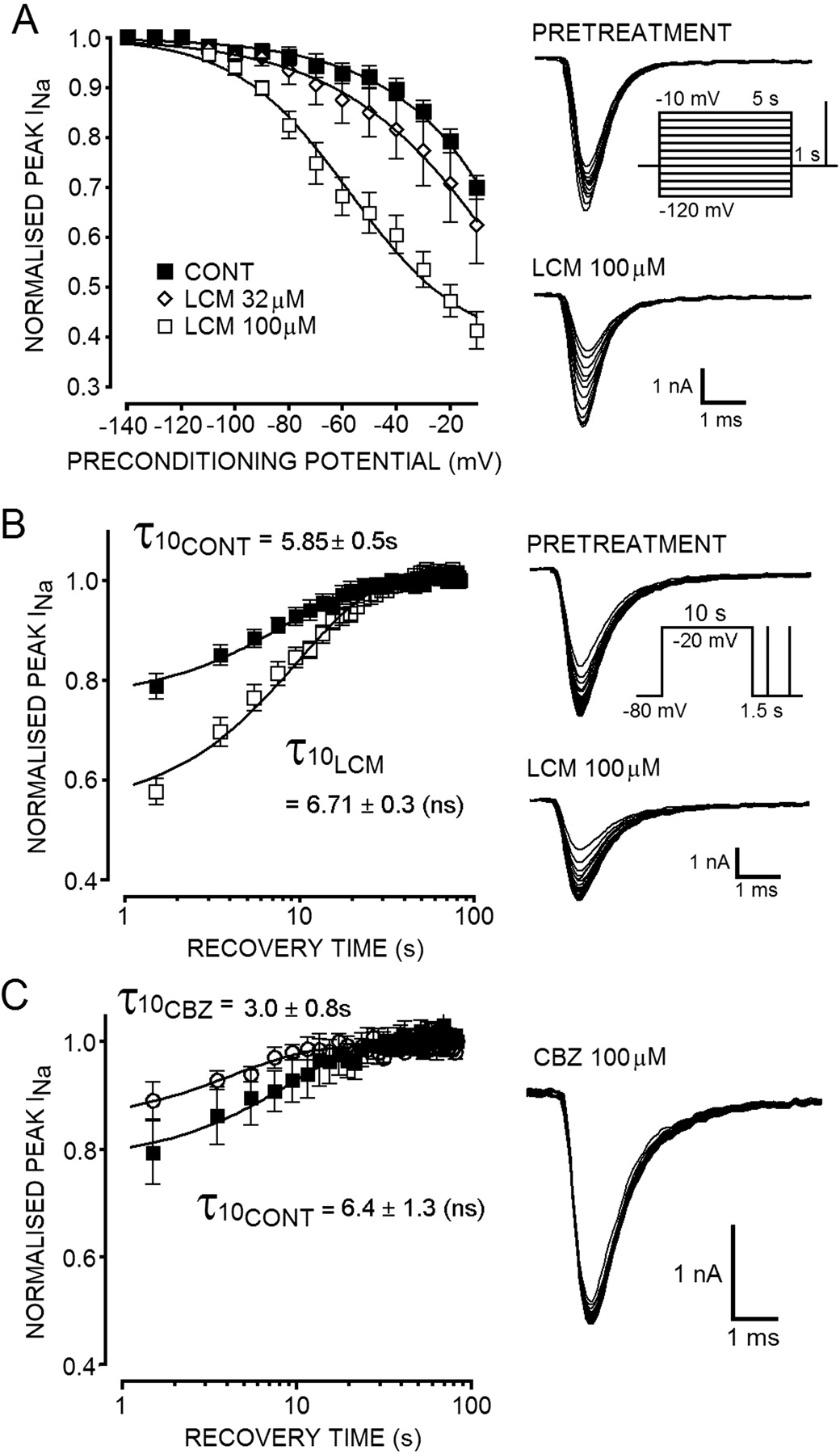
A, in a concentration-dependent manner, LCM shifted the voltage-dependence of slow inactivation of sodium currents. Tracings show the voltage-dependence of slow inactivation under pretreatment (top) and LCM-treated conditions (bottom). The fraction of peak current available in response to a test pulse to -10 mV was significantly reduced by 100 μM LCM across the range of potentials from -80 to -10 mV. In the presence of LCM 100 μM (□), the slow inactivation voltage curve was significantly shifted (P < 0.001, n = 4 -8, Friedman with Dunn's multiple comparison) to more hyperpolarized potentials. The normalized peak amplitude of whole-cell currents evoked after depolarizing conditioning pulses were clearly reduced in the presence of the drug compared with the currents evoked after the same depolarizing conditioning pulse pretreatment (▪). B, tracings show progressive recovery of sodium current from slow inactivation in control (top) and LCM (100 μM, bottom)-treated conditions. The fraction of channels made unavailable by slow inactivation was significantly enhanced by LCM, but the half-life for channel recovery (τ) was not significantly changed (τ values derived from monoexponential curve fits are shown; ns indicates P > 0.05). C, tracing shows recovery of sodium currents in the presence of 100 mM CBZ (n = 3, ○) from slow inactivation induced using pulse protocol shown in B. Unlike the effects of LCM, CBZ did not enhance the fraction of channels made unavailable by running the slow inactivation protocol. The peak current available after the conditioning pulse was in fact slightly greater in the presence of CBZ than control conditions (▪).
Moreover, LCM was able to produce tonic inhibition of transient sodium currents at -80 mV, but the affinity of the channel for the drug was low with an EC50 value of 569 ± 1.2 μM (n = 5-7; Fig. 7C). When the conditioning pulse to -20 mV for 10 s was applied, the affinity of the drug for the channel was markedly increased with an EC50 value for inhibition of 164 ± 1.2 μM. These values are likely to underestimate the true dissociation constant of channels in both the resting and slowly inactivated states. If cells were held at very negative potentials (-120 mV) for periods exceeding the time constant for entry into slow inactivation (several tens of seconds, which was not possible for technical reasons) it is likely that the EC50 value for LCM would be even greater than that quoted (at -80 mV).
The Slow Inactivation Voltage Curve Was Shifted to More Hyperpolarized Potentials by Lacosamide. To test the voltage dependence of the slow inactivation process in N1E-115 cells, the pulse protocol shown in the inset in Fig. 8A was used. Using this protocol, physiological slow inactivation became prevalent at potentials more depolarized than -80 mV but was only 30% complete at the maximum conditioning pulse of -10 mV (n = 7; Fig. 8, A and B). The inactivation voltage curve was fit using a modified Boltzmann function (eq. 3), but the relatively small fraction of slow inactivation induced meant that the estimated potential for V50 under control conditions was ∼+64 mV. LCM significantly (P < 0.01, Friedman test, n = 7) shifted the slow inactivation voltage curves to more hyperpolarized membrane potentials in a concentration-dependent manner (Fig. 8A). In the presence of 100 μM LCM, the curve was shifted significantly with half-maximal reduction in channel availability at -57 ± 4.5 mV (n = 4). The first significant change in channel availability in the presence of 100 μM LCM was noted at a conditioning potential of -80 mV, which was more hyperpolarized than the typical resting potential of many neurons. LCM application also significantly increased the maximal fraction of current made unavailable by depolarization (-10 mV; control, 0.70 ± 0.02, n = 7; LCM, 0.41 ± 0.04, n = 4; P < 0.01, Mann-Whitney test).
Lacosamide Does Not Alter the Rate of Recovery of Channels from the Unavailable Slow Inactivated State. Although LCM enhanced entry to slow inactivation and reduced the fraction of channels available after a long conditioning pulse, the half-life for recovery of the channels from the slow inactivated state was not significantly altered by the drug. The rate of recovery of channels from slow inactivation was measured using the protocol shown in Fig. 8B (inset). After a 10-s conditioning pulse to -20 mV, a fraction of the current was made unavailable for activation by a second test pulse as described previously. Under control conditions, the half-life for recovery of the current back to steady-state maximal was 5.8 ± 0.5s (monoexponential fit, n = 6). When LCM (100 μM) was applied for 3 min, a proportion of the current was unavailable (before conditioning pulse: Vh =-80 mV), and the fraction made unavailable by the conditioning pulse was enhanced. The rate at which the unavailable channels recovered and became available for activation was not significantly altered by LCM (P = 0.17) with the half-life for recovery from slow being 6.7 ± 0.3 s (Fig. 8B). When the conditioning pulse was extended to 30-s duration, the fraction of current made unavailable by physiological slow inactivation was greater than that resulting from a 10-s pulse. The kinetics of physiological channel recovery was marginally slower for the longer conditioning pulse with a half-life of 7.8 ± 0.6s(n = 4; data not shown). As with the shorter conditioning pulse, LCM produced tonic inhibition of the evoked sodium current and increased the fraction made unavailable by conditioning. Again, however, the half-life for recovery of the channels was not significantly altered in the presence of the anticonvulsant (9.1 ± 0.3 s, P = 0.09; data not shown). In marked contrast to the effects of LCM upon slow inactivation observed using this protocol, in three of three neuroblastoma cells tested, CBZ did not enhance the fraction of channels made unavailable by a prolonged depolarizing pulse. In contrast, a slight but not significant (P > 0.05, Wilcoxon matched pairs) reduction in the fraction of channels made unavailable by slow inactivation was noted in the presence of 100 μM CBZ (0.89 ± 0.03, n = 3) compared with control measurements (0.79 ± 0.06, n = 3; Fig. 8C). CBZ did not significantly alter the kinetics of channel recovery from slow, but the half-life for this recovery process was reduced by the drug (control half-life, 6.4 ± 1.3s; CBZ, 3.0 ± 0.8).
Discussion
We have shown that currents through VGSCs of central nervous system neurons are sensitive to inhibition by the novel anticonvulsant LCM. The inhibitory actions produced by LCM are mechanistically distinct from those seen with VGSC fast inactivation modifiers. Reduction of sodium currents through binding to a high-affinity receptor site that becomes available during depolarization (McPhee et al., 1995; Ragsdale et al., 1996; Kuo, 1998; Ragsdale and Avoli, 1998) plays a major part in the anticonvulsant actions of CBZ, DPH, and LTG. This high-affinity binding site, or “modulated receptor,” is located between the outer pore lining regions of IIIS6 and IVS6, in particular the amino acid residues Phe1764 and Tyr1771. In line with the modulated receptor hypothesis, when neuroblastoma cells were held at a potential of -60 mV (where steady-state fast inactivation is prevalent) CBZ, LTG, DPH, and LCM all produced significant impairment of the fast transient sodium current.
Because inhibition of INa by CBZ, LTG, and DPH follows the voltage-dependence of steady-state inactivation gating, it was impeded, to a large extent, when 500-ms hyperpolarizing pulses to -100 mV were applied before test pulses. This is unsurprising in light of the fact that recovery from steady-state fast inactivation was approximately 90% complete after only 300 ms in the presence of CBZ. Inhibition of INa by LCM was not eroded by a hyperpolarizing prepulse delivered in advance of the test pulse. This suggested that the inhibition produced by the novel drug was not dependent on the exposure of the so-called modulated receptor, as is the case for CBZ, DPH, and LTG. Further evidence supporting this postulate was the lack of any hyperpolarizing shift in the steady-state inactivation curve under conditions in which the peak sodium current was significantly inhibited by LCM. This was in marked contrast to the significant shifts (in V50 values) produced by acknowledged fast inactivation modifiers. We saw a greater shift in the V50 value for steady-state inactivation produced by CBZ over DPH and LTG, reflecting the more rapid binding rate of the former over the latter to the inactivated channel. Despite using relatively short conditioning pulses (500 ms), DPH and LTG, whose on-binding rates to the inactivated channel are low (Kuo and Bean, 1994; Kuo and Lu, 1997), still produced significant hyperpolarizing shifts. In our experiments, inhibition of sodium currents by LCM seemed largely dependent on tonic holding potential (-70 mV) and was not greatly accentuated or reversed by brief (500 ms) depolarizing or hyperpolarizing pulses.
Drugs that bind to voltage-dependent conformations of VG-SCs generally demonstrate frequency-dependent facilitation of block (Willow et al., 1985; Lang et al., 1993; Vedantham and Cannon, 1999). In response to a train of depolarizing pulses (10 Hz) from a holding potential of -80 mV, the probability of channels occupying the inactivated state is dramatically increased and rapid cumulative block was observed with CBZ, LTG, and DPH. We did not see the characteristic response observed with fast inactivation modifying anticonvulsants when we tested LCM using this protocol. In contrast, the inhibition of sodium currents by LCM showed a slowly developing facilitation of block that was only apparent after several pulses (1-1.5 s). Furthermore, during prolonged episodes of sustained repetitive firing, in complete contrast to CBZ, LTG, and DPH, LCM was only able to facilitate spike failure after a similar temporal lag. Two plausible mechanisms may explain these findings. We cannot discount the possibility of a high-affinity, ultra-slow binding interaction of the novel drug with a target site on the channel protein that becomes exposed for liganding rapidly during depolarization. For example, it is known that CBZ binds to the fast inactivated channel with 3-fold lower affinity than DPH but that the rate of binding is five times faster (Kuo and Bean, 1994; Kuo et al., 1997). The slow onset of inhibition by LCM may ultimately be the result of slow association (binding) to the fast inactivated (gating transition) channel or to transition states along the activation pathway, which underpin the frequency of dependent block by local anesthetics (Vedantham and Cannon, 1999). If this is the case, however, our results demonstrate a rate of binding that is dramatically slower than existing anticonvulsant drugs. A second and perhaps a more likely explanation (based on our findings) is that LCM may be reducing the availability of sodium channels by enhancing the intrinsic inhibitory physiological mechanism of slow inactivation via a novel binding site.
Slow inactivation of VGSCs was first discovered in squid axon (Rudy, 1978) and subsequently in mammalian preparations, including hippocampal neurons (Jung et al., 1997; Mickus et al., 1999) and neuroblastoma cells (Quandt, 1988). It involves a presumptive structural channel rearrangement that develops over several hundreds of milliseconds to seconds (roughly 100- to 1000-fold greater than fast inactivation) of sustained depolarization (Toib et al., 1998; Carr et al., 2003) or in response to prolonged high-frequency trains of repetitive firing (Jung et al., 1997). Slow inactivation of sodium channels may be pivotal in regulating firing properties of a range of neurons by increasing spike threshold, curtailing prolonged action potential bursts, and limiting active back propagation of action potentials into dendritic regions (Jung et al., 1997; Maurice et al., 2004). The last of these regulatory mechanisms may serve to dampen the excitability of dendrites by regulating NMDA receptor or voltage-gated calcium channel-mediated dendritic calcium spikes and hence could potentially affect processes such as spike timing-dependent synaptic plasticity (Carr et al., 2003). Paroxysmal depolarizing shifts associated with epileptiform cellular activity would present ideal conditions for the recruitment of this intrinsic inhibitory mechanism, and pharmacological manipulation of this process would most likely have profound anticonvulsant effects. In support of this (Lees et al., 2006), we have shown previously that LCM exerts depressant effects on ictal-like events in rodent brain slices at concentrations (EC50 values ∼40-60 μM) similar to those that significantly reduce sodium channel availability in this study.
Recently published studies suggest that certain opiate analgesics (Haeseler et al., 2006), carbamazepine (Cardenas et al., 2006), and a preclinical congener of DPH (Lenkowski et al., 2007) to some extent promote slow inactivation of VGSCs in dorsal root ganglion cells or central nervous system neurons. However, upon examination of the voltage-clamp experiments performed using these ligands, it is apparent that they all concurrently interact with fast inactivation (in contrast to LCM). For example, the DPH analog produced shifts in fast inactivation curves with prepulses that were as brief as 3 ms. LCM is the only anticonvulsant thus far that seems to selectively promote slow sodium channel inactivation. The preclinical δ opiate agonist SNC80 has been elegantly demonstrated to exert effects on VGSC slow inactivation in acutely isolated hippocampal cells (Remy et al., 2004), but unlike with LCM, the rate of recovery from fast inactivation was also notably impaired.
Several studies have proposed potential structural correlates for slow inactivation, including changes in the configuration of the outer channel mouth (Struyk and Cannon, 2002; Xiong et al., 2003) and reduced efficiency of bending at the putative glycine gating hinge residue (Zhao et al., 2004), but the submolecular mechanism(s) of slow inactivation is still poorly understood. LCM as a selective modifier of slow channel inactivation may be a useful tool to understand key domains on the VGSC that regulate availability by slow inactivation and to determine the pharmacophore for this novel modulatory mechanism.
The implications of selective promotion of slow inactivation for the pharmacological profile of such drugs in vivo have been investigated in animal experiments. The classic sodium channel-modulating anticonvulsants are relatively inactive in the 6-Hz psychomotor model of treatment-resistant seizures (Barton et al., 2001), whereas LCM has shown full efficacy in this test (Beyreuther et al., 2007). Moreover, LCM has been compared with LTG in the streptozotocin model for diabetic neuropathic pain (Beyreuther et al., 2006). Although LCM showed full efficacy against mechanical hyperalgesia and thermal allodynia, LTG was completely inactive. These results imply that selective enhancement of VGSC slow inactivation reported here and/or the novel interaction of LCM with cytoskeletal collapsin response mediator protein 2 (Beyreuther et al., 2007) can result in a distinct pharmacological profile compared with drugs affecting fast inactivation of VGSCs as their primary mode of action.
In summary, our results suggest that LCM does not share any of the mechanistic hallmarks of VGSC fast inactivation modifiers that have been used in treating epilepsy for some years. Instead, LCM alters the voltage-dependence of the channel rearrangement underpinning slow inactivation and accelerating the process of entry into the slow inactivated state. Doses of LCM used in clinical trials yield plasma concentrations of 10 to 60 μM (Beyreuther et al., 2007; Doty et al., 2007), and we show that at these concentrations, the drug produces sufficient modulation of sodium channel slow inactivation as to significantly constrain the ability of pyramidal neurons to sustain prolonged bursts (the hallmark of neurons in epileptic foci). This novel mechanism probably underpins the short-term anticonvulsant and analgesic effects of the drug that are currently being profiled in clinical trials.
Acknowledgments
We acknowledge the contributions of Gareth Bruce, Marc Smith, and Liping Huang who helped in the production of neuronal cell cultures for this study and Kevin Markham for technical support.
Footnotes
-
The Otago project (consumables and electrophysiological apparatus) and A.C.E.'s studentship was funded by Schwarz BioSciences GmbH, Monheim, Germany. LCM and SPM 6953 were supplied by Schwarz Pharma GmbH (Monheim, Germany).
-
Some of the data presented here have been published previously in abstracts form: Epilepsia47 (Suppl 3):84-85, 2006.
-
These findings were presented at the 37th Annual Meeting of the Society for Neuroscience, San Diego, CA, November 2007.
-
ABBREVIATIONS: LCM, lacosamide, R-2-acetamido-N-benzyl-3-methoxypropionamide; CBZ, carbamazepine; DPH, phenytoin; LTG, lamotrigine; SRF, sustained repetitive firing; VGSC, voltage-gated sodium channel; MES, maximal electroshock seizure; NMDA, N-methyl-d-aspartate; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; DIV, days in vitro; TTX, tetrodotoxin; AP, action potential; SNC80, (+)-4-[(αR)-α-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenzyl]-N,N-diethylbenzamide; MK-801, 5H-dibenzo[a,d]cyclohepten-5,10-imine (dizo-cilpine maleate); 4-AP, 4-aminopyridine; SPM 6953, (S)-2-acetamido-N-benzyl-3-methoxypropionamide.
- Received July 10, 2007.
- Revision received October 15, 2007.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}