


Abstract
Chk2 is a protein kinase involved in the ATM-dependent checkpoint pathway (http://discover.nci.nih.gov/mim). This pathway is activated by genomic instability and DNA damage and results in either cell cycle arrest, to allow DNA repair to occur, or cell death (apoptosis). Chk2 is activated by ATM-mediated phosphorylation and autophosphorylation and in turn phosphorylates its downstream targets (Cdc25A, Cdc25C, BRCA1, p53, Hdmx, E2F1, PP2A, and PML). Inhibition of Chk2 has been proposed to sensitize p53-deficient cells as well as protect normal tissue after exposure to DNA-damaging agents. We have developed a drug-screening program for specific Chk2 inhibitors using a fluorescence polarization assay, immobilized metal ion affinity-based fluorescence polarization (IMAP). This assay detects the degree of phosphorylation of a fluorescently linked substrate by Chk2. From a screen of over 100,000 compounds from the NCI Developmental Therapeutics Program, we identified a bis-guanylhydrazone [4,4′-diacetyldiphenylureabis(guanylhydrazone); NSC 109555] as a lead compound. In vitro data show the specific inhibition of Chk2 kinase activity by NSC 109555 using in vitro kinase assays and kinase-profiling experiments. NSC 109555 was shown to be a competitive inhibitor of Chk2 with respect to ATP, which was supported by docking of NSC 109555 into the ATP binding pocket of the Chk2 catalytic domain. The potency of NSC 109555 was comparable with that of other known Chk2 inhibitors, such as debromohymenialdisine and 2-arylbenzimidazole. These data define a novel chemotype for the development of potent and selective inhibitors of Chk2. This class of drugs may ultimately be useful in combination with current DNA-damaging agents used in the clinic.
In response to genomic instability and DNA damage, cells activate signaling pathways to arrest the cell cycle (to enable DNA repair) or to invoke apoptosis. Two primary pathways are initiated in response to DNA damage, one mediated by the ATM-Chk2 axis, and the other via the ATR-Chk1 axis (Bartek and Lukas, 2003). The ATM/Chk2 pathway responds primarily to DNA double-strand breaks, whereas the ATR-Chk1 pathway responds mainly to replication-associated DNA lesions. However, cross-talk exists between the pathways, such that certain lesions can activate both pathways. For example, in response to ionizing radiation induced DNA double-strand breaks, the ATM-Chk2 pathway is activated first in a transient manner, whereas the ATR-Chk1 pathway is activated secondarily in a sustained way (Jazayeri et al., 2006). Chk2 shares no sequence homology with Chk1, although both Chk2 and Chk1 are capable of phosphorylating similar substrates such as Cdc25C and p53 (Pommier et al., 2005).
Chk2 is activated primarily by ATM and DNA-PK, which phosphorylates Thr68 of Chk2 (Ahn et al., 2000). Phosphorylation of Thr68, Chk2, causes homodimerization, resulting in trans-activating autophosphorylations of Thr383 and Thr387 (Ahn and Prives, 2002) and cis-phosphorylation of Ser516 (Wu and Chen, 2003). Once activated, Chk2 phosphorylates a number of downstream substrates involved in cell cycle arrest (Cdc25A, Cdc25C, BRCA1) and/or apoptosis (p53, PML, E2F1) (Fig. 1). For a comprehensive overview of these interactions, see Pommier et al. (2005, 2006) and http://discover.nci.nih.gov/mim. Chk2 is endogenously activated in precancerous lesions with genomic instability (Bartkova et al., 2005; Gorgoulis et al., 2005) and in cancer cells grown in culture (DiTullio et al., 2002; Bartek and Lukas, 2003). Endogenous Chk2 activation also colocalizes with abnormal replication foci in BLM-deficient cells (Rao et al., 2007).
Selective inhibition of Chk2 is desirable because cells with endogenous activation may succumb to Chk2 inhibition. Second, selective inhibition of Chk2 in p53-deficient tumor cells, which show a defect in the G1 checkpoint and apoptosis, may provide chemo/radiosensitization by primarily targeting the G2 checkpoint (Zhou and Bartek, 2004; Pommier et al., 2005) (Fig. 1). This rationale is supported by the observation that inhibiting Chk2 activity in p53-defective cells enhances the apoptotic response to ionizing radiation (Yu et al., 2001). Chk2 is also activated in tumor cells by a wide range of chemotherapeutic drugs, including topoisomerase inhibitors (Yu et al., 2001; Park and Avraham, 2006; Takemura et al., 2006), DNA synthesis inhibitors (Matsuoka et al., 2000; Liu et al., 2005) and radiomimetic agents (Ahn and Prives, 2002; Bartek and Lukas, 2003). Thus, it is plausible that a Chk2 inhibitor would increase the therapeutic indices of DNA-targeted agents in p53-defective tumors (Fig. 1) (Pommier et al., 2006). Moreover, because of the importance of p53 in the induction of DNA-damage induced apoptosis, inhibition of Chk2 in normal cells may protect normal tissues (Fig. 1). This may reduce side effects associated with chemotherapy or radiotherapy when used in combination with current clinical therapies. In support of this, Chk2-null mice are viable but show increased survival when exposed to ionizing radiation (Hirao et al., 2000; Takai et al., 2002).
Currently the number of Chk2 inhibitors remains limited (Zhou and Sausville, 2003; Pommier et al., 2005), and there is no specific inhibitor of Chk2 in the clinic. The reported Chk2 inhibitors include VRX0466617 (Carlessi et al., 2007), 2-arylbenzimidazole (Arienti et al., 2005), debromohymenialdisine (DBH) (Curman et al., 2001; Wan et al., 2004), staurosporine (and analogs UCN-01, Go6976, and isogranulatimide) (Yu et al., 2002; Collins and Garrett, 2005) and a series of isothiazole carboxamides (Larson et al., 2007). Among those inhibitors, only 2-arylbenzimidazole and the series of isothiazole carboxamides seem to be specific for Chk2. Recent cocrystal structures of the catalytic domain of Chk2 with either DBH or ADP have provided an insight into the mechanism of competitive inhibition of Chk2 with respect to ATP (Oliver et al., 2006), which should allow the discovery of more potent and specific inhibitors of Chk2.

Rationale for using Chk2 inhibitors as chemotherapeutic agents. The substrates of Chk2 have an impact on both cell cycle checkpoint and apoptosis. A, in a p53-deficient tumor, Chk2 may act primarily as a cell cycle checkpoint inducer. Therefore, Chk2 inhibition would sensitize the tumor to DNA-damaging agents used in chemotherapy. B, in contrast, in normal tissues, Chk2 may act as a proapoptotic effector. Therefore, Chk2 inhibitors would protect healthy tissues but sensitize the tumor to chemotherapy. The conventions used for the interactions (Kohn, 1999) are as follows: the double line with P indicates phosphorylation, the line with an open circle indicates an enzymatic stimulation of the reaction, the line with an arrow indicates general stimulation, and the line with a bar on indicates inhibition. For an overview of the molecular interaction map for Chk2. see Pommier et al. (2006)
Herein, we describe the identification and biochemical characterization of a novel and selective Chk2 inhibitor, NSC 109555. The drug was initially identified from a high-throughput screen of more than 100,000 compounds. We demonstrate the specificity and potency of the drug for Chk2 but not Chk1. Molecular docking of NSC 109555 into the ATP binding site of Chk2 suggests competitive inhibition of Chk2 as a potential mechanism of action for NSC 109555. NSC 109555 also provides a novel biological tool for characterization of the function and implication of Chk2 inhibition.
Materials and Methods
Expression and Purification of Recombinant Proteins. The expression and purification of recombinant Chk2 has been described in detail elsewhere (Yu et al., 2002). In brief, Chk2 was cloned into a pET-15b plasmid (Novagen, Madison, WI) and was expressed in BL-21 (DE3) bacteria after isopropyl β-d-1-thiogalactopyranoside induction. The cell pellet from a 1-liter culture was washed in ice-cold 20 mM Tris-HCl buffer, pH 7.9, sonicated, and then centrifuged at 14,000g for 30 min. The Chk2 polypeptide was purified from the supernatants over nickel-column chromatography (Novagen, Madison, WI) according to the manufacturer's instructions. To generate a Cdc25C fragment as a GST fusion protein, the cDNA fragment encoding amino acids 200 to 256 of Cdc25C was amplified by polymerase chain reaction from a human cDNA library and cloned into the pGEX-2T vector as described previously (Yu et al., 2002). The expressed protein was purified using a GST purifying kit according to manufacturer's instructions (Pierce, Rockford, IL). Recombinant Chk1 was purchased from Upstate (Charlottesville, VA). Histone H1, from calf thymus, was purchased from Roche (Indianapolis, IN).
High-Throughput Screening. The IMAP Screening Express Kit (Molecular Devices, Sunnyvale, CA) was used for the high-throughput screening experiments. Compounds from the Developmental Therapeutics Program's (DTP) Open Repository were solubilized and diluted in DMSO and initially tested at one concentration in the assay. Active compounds were subsequently titrated at 20 2-fold dilutions. Reactions were performed using recombinant Chk2 or Chk1 (Chemicon International, Temecula, CA) with the indicated drug concentrations in reaction buffer (10 mM Tris-HCl, pH 7.2, 10 mM MgCl2, 0.1% bovine serum albumin, 1 mM dithiothreitol, 10 μM ATP, and 100 nM Chktide substrate) in a total volume of 5 μlin 384-well plates [384-well low-volume black microplate (Greiner Bio-One, Longwood, FL)] for 60 min at room temperature. Substrates used in the assay were 5-carboxyfluorescein-CHK1tide (Molecular Devices) for Chk1 and Fam-CHK2tide (Molecular Devices) for Chk2. Fifteen microliters of IMAP binding reagent were added to each well, plates were incubated for 30 min at room temperature, and fluorescence polarization was measured using a Tecan Ultra plate reader. Each screening plate contained staurosporine as a positive control.
Protein Kinase Assays. NSC 109555 was diluted in water. All other drugs were dissolved in DMSO, in which case the final DMSO concentration in reactions was 10%, and the controls were performed under comparable conditions. Four hundred nanograms of recombinant Chk2 or 100 ng of recombinant Chk1 were incubated with either 0.5 μg of GST-Cdc25C or 1 μg of histone H1 in reaction buffer (50 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 1 mM dithiothreitol, 10 μM ATP, and 10 μCi of [γ-32P]ATP) in a total volume of 10 μl and incubated at 30°C for indicated times. For the drug inhibition experiments, samples were coincubated with drug during the reactions. Reactions were stopped by adding 10 μl of 2× sample buffer, and samples were boiled for 5 min. Reaction products were separated by 4–20% SDS-PAGE. Chk2 protein kinase activity, measured as 32P incorporation into Chk2, GST-Cdc25C, or histone H1, was determined using a PhosphorImager (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK). Densitometry was performed using Image-Quant.

High-throughput screen for Chk2 inhibitors. A, schematic of IMAP assay (Molecular Devices) used for high-throughput screening. B, inhibition of Chk1 and Chk2 by staurosporine (positive control) and NSC 109555 in IMAP assay. Reactions were performed using recombinant Chk1 or Chk2 with increasing drug concentrations in reaction buffer in a total volume of 5 μl in 384-well plates for 60 min at room temperature. IMAP binding reagent (15 μl) was added to each of the wells, and the plates were scanned for fluorescence polarization. NSC 109555 inhibits Chk2 but not Chk1.

Inhibition of Chk2 but not Chk1 by NSC 109555 using an in vitro kinase assay. Reactions containing kinase, substrate, and increasing concentrations of NSC 109555 with reaction buffer in a total volume of 10 μl were incubated at 30°C for 30 min. Reactions were stopped by the addition of SDS-PAGE loading buffer and subjected to SDS-PAGE. Densitometry was performed on gels. A, recombinant Chk2 (400 ng) was incubated with 1 μg of histone H1 and increasing NSC 109555 concentrations. The data represent an average of five experiments. B, representative gel of an in vitro kinase assay containing Chk2 and histone H1 with increasing concentrations of NSC 109555. C, representative gel an in vitro kinase assay containing 400 ng of Chk2 and 0.5 μg of GST-Cdc25C with increasing concentrations of NSC 109555. D, recombinant Chk1 (100 ng) was incubated with 1 μg of histone H1. E, chemical structures and IC50 values against Chk2 in the in vitro kinase assay determined for NSC 109555, staurosporine, 2-arylbenzimidazole, and DBH.
Kinase Profiling. Detection kits from PerkinElmer Life and Analytical Sciences (Waltham, MA) were used for the protein kinase profiling experiments. For the tyrosine kinase family, the AlphaScreen phosphotyrosine detection kit was used; for all other kinases, the AlphaScreen IgG (Protein A) detection kit was used (PerkinElmer Life and Analytical Sciences). The enzymes and their biotinylated substrates were obtained from Upstate, Cell Signaling Technology (Danvers, MA) and/or Invitrogen (Carlsbad, CA). The drugs were sequentially diluted in kinase buffer to obtain 10, 5, 2.5, 1.25, 0.63, 0.32, 0.16, and 0.08 μM concentration in a final volume of 10 μl. The kinases were pretreated with drug for 30 min, and then 5 μl of substrate was added. An ATP concentration of 100 μM was maintained in all kinase reactions. After 30 min of incubation, 5 μl of a mixture of an antibody-conjugated acceptor and streptavidin-conjugated donor beads was added and then incubated for a further hour. Assays were performed in 384-well white plates (PerkinElmer Life and Analytical Sciences) in a final volume of 20 μl. All the reactions were performed at room temperature. The plates were read on a Fusion-α microplate analyzer (PerkinElmer Life and Analytical Sciences) to measure the fluorescence. The data obtained were analyzed using Cricket Graph and IC50 values were calculated.
Molecular Docking. NSC 109555 was docked into the catalytic domain of Chk2 (residues 208–504). First, the structure of NSC 109555 was drawn in ChemDraw (CambridgeSoft, Cambridge, MA), and energy minimized in Chem3D (CambridgeSoft) using the MOPAC energy minimization routine. NSC 109555 was then docked using the coordinates of the crystal structure of Chk2 kinase (PDB code 2CN5) (Oliver et al., 2006) with the program Molegro (Molegro ApS, Aarhus C, Denmark) and standard procedures as outlined in the manual (Thomsen and Christensen, 2006).
Results
High-Throughput Screening of Chk2 Inhibitors. A modified IMAP assay (Molecular Devices) was employed to screen for compounds capable of inhibiting the kinase activity of Chk2 (Fig. 2A). In brief, a fluorescently labeled peptide is phosphorylated in a kinase reaction. Addition of the IMAP Binding reagent stops the kinase reaction and specifically binds the phosphorylated substrates via a high-affinity interaction of trivalent metal-containing nanoparticles with phosphogroups on the substrate. Phosphorylation and subsequent binding of the substrate to the beads can be detected by fluorescence polarization. The open screening repository from the Developmental Therapeutics Program was used to screen for potential Chk2 inhibitors. NSC 109555 was identified as the sole lead chemotype. The inhibitory effect of NSC 109555 on recombinant Chk2 kinase activity in the IMAP assay is shown in Fig. 2B. The IC50 for NSC 109555 was 0.2 μM. The compound was subsequently counter-screened against Chk1 and found to have no effect on Chk1 kinase activity (Fig. 2B). Staurosporine was used as a positive control because it inhibits both Chk1 and Chk2.

Kinetics of Chk2 inhibition by NSC 109555 and competitive inhibition with ATP. A, Chk2 (400 ng) was incubated with 1 μg of histone H1 and 1 μM NSC 109555 and reaction buffer in a total volume of 10 μl for the indicated times at 30°C in an in vitro kinase assay. Reactions were stopped by the addition of SDS-PAGE loading buffer and subjected to SDS-PAGE. Densitometry was performed on the gels. NSC 109555 slows the phosphorylation of Chk2 and subsequent histone H1 phosphorylation. B, competitive inhibition of Chk2 by NSC 109555 with respect to ATP. Chk2 (400 ng) was incubated with 0.5 μg of GST-Cdc25C in reaction buffer for 10 min at 30°C, and reactions were stopped by the addition of SDS-PAGE loading before being subjected to SDS-PAGE. The ratio of unlabeled ATP and 32P-labeled γ-ATP was kept constant, whereas the concentration was altered (100, 50, 40, and 25 μM). This set of reactions was repeated in the presence of 0.25, 0.5, and 0.75 μM NSC 109555. Densitometry was performed on the gels, and the data were plotted using double reciprocal (Lineweaver-Burk) plots.
In Vitro Kinase Assays using NSC 109555. To further study the activity of NSC 109555 on Chk2 and subsequent substrate phosphorylation, an in vitro kinase assay using radiolabeled γ-ATP was used. Figure 3A shows inhibition of Chk2 autophosphorylation and histone H1 phosphorylation in a concentration-dependent manner by NSC109555. Histone H1 has been previously demonstrated to be phosphorylated by Chk2 in vitro (Yu et al., 2002). The IC50, 0.24 μM, was consistent with the IMAP high-throughput assay results. Figure 3B shows a representative gel demonstrating the concentration-dependent inhibition of Chk2 autophosphorylation and histone H1 phosphorylation by NSC 109555. A recombinant GST-Cdc25C peptide was also used to demonstrate inhibition of an in vivo substrate phosphorylation (Ahn and Prives, 2002). NSC 109555 inhibited the Chk2-mediated phosphorylation of GST-Cdc25C (Fig. 3C). The potency of NSC 109555 in the in vitro kinase assay was also comparable with the selective Chk2 inhibitor 2-arylbenzimidazole and to the Chk1/Chk2 inhibitor DBH (Figure 3E). Staurosporine was around 3.5-fold more potent than NSC 109555; however, it is not a specific inhibitor of Chk2. To confirm selectivity for Chk2 inhibition by NSC 109555, recombinant Chk1 was used in an in vitro kinase assay. Figure 3D shows NSC 109555 was not able to inhibit Chk1 autophosphorylation and subsequent Chk1-mediated histone H1 phosphorylation.
Kinetics of Chk2 Inhibition by NSC 109555. The kinetics of inhibition of Chk2 kinase activity by NSC 109555 was examined. NSC 109555 (1 μM) caused a time-dependent inhibition of Chk2 autophosphorylation and histone H1 phosphorylation (Fig. 4A). However, the kinase activity was not abolished but was delayed over a 2-h period compared with control. Those data suggested that NSC 109555 was acting as a reversible inhibitor of Chk2. To test whether NSC 109555 acted as a competitive inhibitor of ATP, experiments were performed using varying concentrations of ATP while keeping the concentration of recombinant Chk2 and GST-Cdc25C substrate constant. Experiments were repeated using 0.25, 0.5, or 0.75 μM NSC 109555. The data were analyzed using double-reciprocal (Lineweaver-Burk) plots. The convergence of the y-axis intercepts from the four experiments is indicative of competitive inhibition (Fig. 4B). These experiments indicate that NSC 109555 inhibits Chk2 by acting as a competitive inhibitor of ATP.

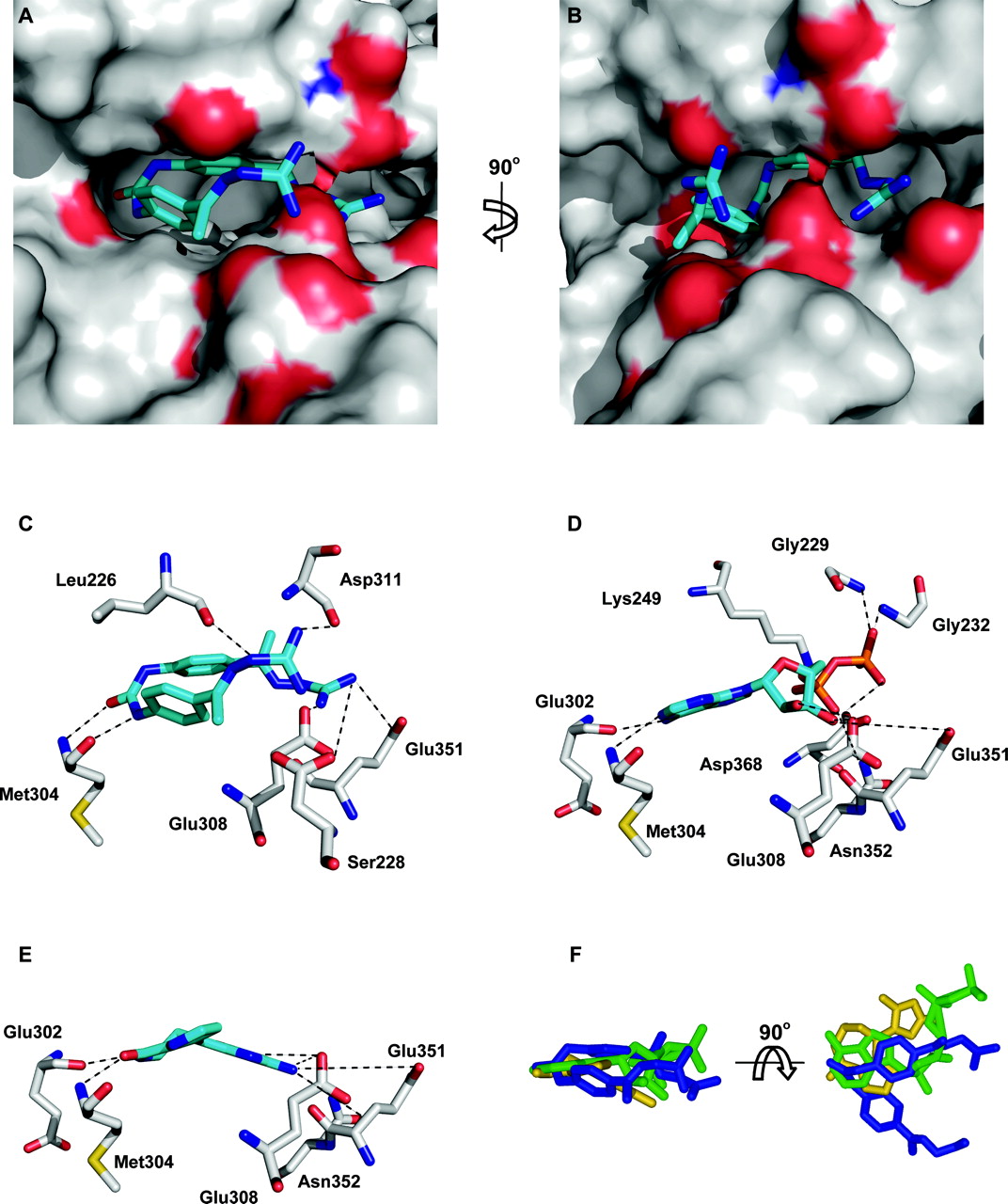
Docking of NSC109555 to the catalytic domain of Chk2. The program MOLEGRO was used to dock NSC 109555 into the catalytic domain of Chk2 using the coordinates of the crystal structure of Chk2 kinase (PDB code 2CN5). Analysis of the structures was performed in PyMol. A, NSC 109555 (blue) is docked into the ATP binding site of the catalytic domain of Chk2. The surface of Chk2 is depicted in gray with amino acids that potentially form hydrogen bonds with NSC 109555 shown in red (oxygen) and blue (nitrogen). B, 90° clockwise rotation of the image shown in A. C, potential hydrogen bonding between NSC 109555 and amino acids of Chk2 around the ATP binding pocket. D, hydrogen bonding between ADP and amino acids of Chk2 around the ATP binding pocket (Oliver et al., 2006). E, hydrogen bonding between DBH and amino acids of Chk2 around the ATP binding pocket proposed by Oliver et al. (2006). F, overlapping of the structures of NSC 109555 (blue), DBH (yellow), and ADP (green) in the ATP binding pocket of Chk2.
Molecular Docking of NSC10955 into the Catalytic Site of Chk2. To further elucidate the mechanism of Chk2 inhibition by NSC 109555, molecular docking studies were performed. The previous kinetic studies suggested that NSC 109555 is a competitive inhibitor of ATP, and thus probably occupies the ATP binding pocket of Chk2. Using recently solved Chk2 cocrystal structures of Chk2 with either ADP (PDB code 2CN5) or the Chk2 inhibitor DBH (PDB code 2CN8), NSC 109555 was docked into the ATP binding pocket of Chk2. Figure 5, A and B, shows NSC 109555 occupying the ATP binding pocket of the catalytic domain of Chk2. NSC 109555 seems to form a “horseshoe” in the ATP binding pocket. A 90° clockwise rotation shows this more clearly (Fig. 5B). The proposed hydrogen bonding between NSC 109555 and amino acid residues of the ATP binding pocket of Chk2 are shown in Fig. 5C. NSC 109555 forms seven possible hydrogen bonds with surrounding amino acids (≤3.2 Å). Figure 5, D and E, show the hydrogen bonding between ADP (5D) and DBH (5E) and the amino acids of the ATP binding pocket of Chk2. These represent predicted hydrogen bonding from the solved cocrystal structures previously published (Oliver et al., 2006). Hydrogen bonding between Met304, Glu308, and Glu351 is common for NSC109555, ADP, and DBH. Figure 5F depicts a representation of NSC 109555 (blue), DBH (yellow), and ADP (green) structures overlapped within the ATP binding pocket of Chk2. A 90° vertical rotation confirms the overlapping of the structures within the ATP binding pocket. These results are consistent with the competitive binding of NSC 109555 with ATP in the Chk2 catalytic site.
Kinase Profiling of NSC 109555 Compared with DBH and 2-Arylbenzimidazole. To determine the selectivity of NSC 109555 for Chk2 inhibition, kinase-profiling experiments were performed. The profile of NSC 109555 was compared with the known Chk2 inhibitors DBH and 2-arlybenzimidazole. Table 1 shows the selectivity of NSC 109555 for Chk2 over a panel of other cellular kinases. Although NSC109555 did exhibit some weak activity against a few other kinases, the IC50 values are more than 6.5-fold higher than that for Chk2, which is comparable with DBH and 2-arylbenzimidazole (Table 1). Thus, NSC109555 is a potent and selective Chk2 inhibitor.
Kinase profiling of NSC 109555, DBH, and 2-arylbenzimidazole.
Structure-Activity Relationship for NSC 109555. Once the lead compound NSC 109555 had been identified and characterized for selectivity, we compared the activity of this compound with available analogs using the modified IMAP assay (Table 2). The meta-substituted analog (NSC 177944), a urea to thiourea substitution (NSC 177941), a symmetrical molecule missing diphenyl urea central core (NSC 67931), an aliphatic analog (NSC 69432, MGBG), a half-parent molecule with thiourea substitution (NSC 177940), and a half parent molecule with a modified guanidinium terminus (NSC 377844) of NSC 109555 were inactive. From the structure activity relationship, it seems that the linker region between the guanidylhydrazone groups is important for activity. This is demonstrated with NSC 177944 and NSC 177941, where orientation of the guanidylhydrazone groups render the molecules inactive. The presence of a different linker group, either a phenyl (NSC 67931) or aliphatic (NSC 69432) moiety, also leads to an inactive molecule. This would indicate that the presence of the diphenylurea linker is important for activity. The monomer (with a thiourea substitution) is also inactive, suggesting that the presence of both guanidylhydrazone groups is crucial for activity. It is not possible to draw a conclusion from NSC 377844 as it represents a half-parent molecule and has a modification of the guanidinium terminus. Nevertheless, NSC 377844 is still inactive. Further structural analogs will be required to determine the importance of the guanidylhydrazone group as well as the urea group for the bioactivity of the molecule. Because NSC 109555 and various congeners and related structures can be readily assembled in two or three steps from commercially available building blocks (Korytnyk et al., 1978), early stage medicinal chemistry exploration of this new lead should be rather easily accomplished.
Structure activity relationship of NSC 109555
Discussion
p53 is a critical regulator of both the G1 and G2 cellular checkpoints (Bunz et al., 1998). Given that mutations of p53 are observed in approximately 50% of tumors (Hollstein et al., 1999), which are therefore defective in the G1 checkpoint, selective inhibition of the G2 checkpoint may be useful in cancer therapy by enhancing the effects of DNA-damaging agents (Nurse, 1997; Roberge et al., 1998). G2 inhibitors, including caffeine, staurosporine, and UCN-01, and selective Chk1 inhibitors have been shown to enhance the cytotoxicity of DNA-damaging agents (Bunch and Eastman, 1996; Bunz et al., 1998; Roberge et al., 1998; Hollstein et al., 1999; Tse et al., 2007). The observation that G2 inhibitors enhance the effects of DNA-damaging agents supports the rationale for using a specific inhibitor of Chk2 in cells defective in the G1 checkpoint. Moreover, specific Chk2 inhibitors may reduce the side effects commonly associated with chemo/radiotherapy as they may inhibit the p53-induced apoptotic response in healthy tissues after exposure to ionizing radiation or chemotherapeutic agents. In contrast, Chk1 inhibition is unlikely to protect healthy tissue from p53-mediated apoptotic effects induced by the DNA-damaging agent, because Chk1 is not implicated in p53-mediated apoptosis. In addition, Chk2 has been shown to induce the release of the antiapoptotic protein survivin from the mitochondria in response to DNA-damaging agents (Ghosh et al., 2006). Inhibition or genetic targeting of Chk2 prevents this release and thus enhances the DNA damage-induced tumor cell apoptosis.
Only a few specific inhibitors of Chk2 have so far been reported (Arienti et al., 2005). Herein, we have identified and biochemically characterized a specific Chk2 inhibitor that represents a novel chemotype, NSC 109555. Our data show that the drug inhibits the kinase activity of Chk2 in a concentration-dependent manner at nanomolar concentrations in two independent in vitro assays. The current study also demonstrates the selectivity of Chk2 compared with Chk1 (Figs. 2B and 3). Kinase-profiling experiments confirmed Chk2 selectivity for NSC 109555 in a panel of kinases, which was comparable with that of two other Chk2 inhibitors, DBH and 2-arylbenzimidazole (Table 1). The potency of NSC 109555 (0.24 μM) is also comparable with DBH (0.4 μM) and 2-arylbenzimidazole (0.1 μM) in the in vitro kinase assay. We have also demonstrated that NSC 109555-mediated inhibition of Chk2 is competitive with respect to ATP, which is consistent with other proposed specific Chk2 inhibitors, 2-arylbenzimidazole (Arienti et al., 2005) and isothiazole carboxamidines (Larson et al., 2007). The disclosure of cocrystal structures of the catalytic domain of Chk2 with either ADP or DBH (Oliver et al., 2006) allowed us to investigate the potential mechanism of action of NSC 109555. Docking of NSC 109555 into the ATP binding site of the catalytic domain of Chk2 enabled us to visualize where NSC 109555 may inhibit the catalytic function of Chk2. Figure 5 demonstrates the similarity of space occupied by DBH and ADP compared with NSC 109555 within the ATP binding pocket of Chk2. Potential hydrogen bonding suggests that this binding is plausible. Finally, the structure activity relationship of NSC 109555 was examined (Table 2). The presence of two aromatic rings in the structure of NSC 109555 seem important in its mechanism of action, in that the aliphatic analog MGBG was unable to inhibit the kinase activity of Chk2. The positions of the guanidylhydrazone groups on NSC 109555 are also critical for its activity, because meta-substitution of these renders the molecule inactive (NSC 177944).
NSC109555 belongs to a family of compounds known as the bis(guanylhydrazones), including the aliphatic derivative methylglyoxal-bis(guanylhydrazone) (MGBG). NSC 109555 has previously been shown to demonstrate antiproliferative activity in a number of leukemias, most notably murine L1210 (Mihich, 1975). The molecular mechanism attributed to the antiproliferative effect of NSC 109555 remains unclear, although it is likely to involve interference with polyamine function (Marcus et al., 1987). NSC 109555 at high concentrations has also been shown to inhibit DNA polymerase (Dave et al., 1973), bind to the minor groove of DNA (Dave et al., 1977), and interfere with the structure and function of mitochondria (Byczkowski et al., 1981). However, the data presented here showing NSC 109555 docked into the ATP binding site of Chk2 and specificity to Chk2 from the kinase profiling experiments suggest that Chk2 is also a potential target of NSC 109555. Cellular studies have been performed with NSC 109555 in combination with topotecan. We first examined the effect of NSC 109555 on the topotecan-induced HDMX degradation (Carlessi et al., 2007) in MCF7 cells (p53 wild type). Concentrations up to 100 μM of NSC 109555 did not abrogate the topotecan-induced HDMX degradation (data not shown). We also examined the effect of NSC 109555 in HT29 cells (p53-mutant) to investigate downstream substrates of activated Chk2. We observed no inhibition of the topotecan-induced autophosphorylation of Chk2 at residue Ser516 (which is required for Chk2 activation) after preincubation of the cells with NSC 109555 (up to 100 μM). In addition, NSC 109555 did not abrogate the degradation of Cdc25A after topotecan treatment (data not shown). Furthermore, NSC 109555 was unable to abrogate the G2/M block induced by camptothecin in HT29 cells (data not shown). It is likely that the lack of detectable Chk2 inhibition by NSC 109555 in a cellular environment is due to the other actions of NSC 109555, including DNA binding and interference with polyamine function, that have been demonstrated previously (Dave et al., 1973; Marcus et al., 1987). Furthermore, we cannot exclude the possibility that the pharmacokinetics and drug uptake of 109555 limit the amount of drug able to reach Chk2. We are currently developing chemically modified NSC 109555 structures that will hopefully demonstrate clear in vivo Chk2 inhibition. Promising structure-activity relationship studies are ongoing. Thus, the data we have shown demonstrating the potency and specificity of NSC 109555 for Chk2 inhibition suggest that this molecule represents a chemotype for development of a newer generation of agents that target Chk2 in vivo.
Acknowledgments
We thank Dr. Paula Krosky and Karen Hite for optimizing conditions for expression and purification of Chk2, and Tim Stevenson and Russ Reinhart for technical assistance in optimization and operation of the high-throughput screen. Dr. Michael Currens provided support for screening logistics, chemoinformatic analysis of the primary screening data, and assistance with ATP-competition studies. We thank Dr. George Lountos for help with the computational docking studies and Dr. Christophe Marchand for molecular structure visualization. We thank Dr. Elena A. Semenova for constructive discussions. We would also like to thank Drs. Olivier Sordet, Jennifer Seiler, and Gabrielle Grundy for critical reading of the manuscript.
Footnotes
-
This project was funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract N01-CO12400. This research was supported in part by the Developmental Therapeutics Program in the Division of Cancer Treatment and Cancer Diagnosis and by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research.
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.107.035832.
-
ABBREVIATIONS: DBH, debromohymenialdisine; NSC 109555, 4,4′-diacetyldiphenylurea-bis(guanylhydrazone); UCN-01, 7-hydroxystaurosporine; DMSO, dimethyl sulfoxide; PDB, Protein Data Bank; NSC 69432/MGBG, 2-[[(1E)-1-(diaminomethylidenehydrazinylidene)propan-2-ylidene] amino]guanidine; PAGE, polyacrylamide gel electrophoresis; VRX0466617, 3-hydroxy-N-isopropyl-5-(4-phenoxy-phenylamino)isothiazole-4-carboximidamine; Go6976, 12-(2-cyanoethyl)-6,7,12,13-tetrahydro-13-methyl-5-oxo-5H-indolo(2,3-a)pyrrolo(3,4-c)-carbazole.
- Received March 7, 2007.
- Accepted July 6, 2007.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}