


Abstract
1,3-Bis-(2-hydroxy-5-trifluoromethyl-phenyl)-urea (NS1643) is a newly discovered activator of human ether-a-go-go-related gene (hERG) K+ channels. Here, we characterize the effects of this compound on cloned hERG channels heterologously expressed in Xenopus laevis oocytes. When assessed with 2-s depolarizations, NS1643 enhanced the magnitude of wild-type hERG current in a concentration- and voltage-dependent manner with an EC50 of 10.4 μM at –10 mV. The fully activated current-voltage relationship revealed that the drug increased outward but not inward currents, consistent with altered inactivation gating. NS1643 shifted the voltage dependence of inactivation by +21 mV at 10 μM and +35 mV at 30 μM, but it did not alter the voltage dependence of activation of hERG channels. The effects of the drug on three inactivation-deficient hERG mutant channels (S620T, S631A, and G628C/S631C) were determined. In the absence of channel inactivation, NS1643 did not enhance hERG current magnitude. The agonist activity of NS1643 was facilitated by mutations (F656 to Val, Met, or Thr) that are known to greatly attenuate channel inhibition by hERG blockers. We conclude that NS1643 is a partial agonist of hERG channels and that the mechanism of activation is reduced channel inactivation.
Long QT syndrome (LQTS) is a disorder of ventricular repolarization that predisposes affected individuals to cardiac arrhythmia and sudden death. Although inherited LQTS can result from mutations in Na+ and Ca2+ channels, it is most often caused by loss of function mutations in HERG or KCNQ1 K+ channel genes (Keating and Sanguinetti, 2001). Acquired LQTS is more common and can be induced as an unintended side effect of treatment with class III antiarrhythmic drugs (Waldo et al., 1996). LQTS is associated with torsade de pointes, a peculiar tachyarrhythmia characterized by sinusoidal twisting of the QRS axis around the isoelectric line of the electrocardiogram. This arrhythmia usually reverts to normal sinus rhythm but can degenerate into ventricular fibrillation, the cause of sudden death (Schwartz et al., 2000, 2001). More recently, many noncardiovascular drugs have been shown to cause torsade de pointes and sudden death, including such common medications as terfenadine, cisapride, and erythromycin (Haverkamp et al., 2000). These drugs were developed and approved for human use without any knowledge of their untoward effects on ventricular repolarization. Only after they had been used by large numbers of patients, oftentimes when coadministered with other drugs that inhibit specific cytochrome P450 isoenzymes, were these drugs discovered to possess significant proarrhythmic risk. In the past decade, several commonly used drugs, including terfenadine, cisapride, sertindole, thioridazine, and grepafloxacin, were withdrawn from the market, or their approved use was severely restricted when it was discovered, albeit very infrequently, that they caused arrhythmia or were associated with unexplained sudden death (Pearlstein et al., 2003). In most clinically relevant cases, drug-induced torsade de pointes seem to result from block of human ether-a-go-go related gene (hERG) K+ channels. These channels conduct the rapid delayed rectifier K+ current (Sanguinetti et al., 1995; Trudeau et al., 1995), and their function is vital for normal repolarization of the human ventricular myocardium.
Inherited LQTS is usually treated by administration of β-adrenergic receptor blockers (Schwartz et al., 1991; Napolitano et al., 1994). For serious cases or when β-blocker therapy is inadequate, patients can opt to receive an implantable defibrillator or pacemaker. Treatment of acute drug-induced LQTS is inadequate and consists of magnesium sulfate and discontinuing use of the offending drug. In theory, drugs that activate cardiac K+ channels could be used to enhance net repolarizing current reduced by gene mutations or block of hERG channels. KATP channel activators are not used for this purpose because of side effects such as postural hypotension (Lawson, 2000). Drugs that activate KCNQ1 or hERG channels would seem preferable because these channels are highly expressed in the heart, and their activation might be associated with less severe side effects. We previously characterized the benzodiazepine derivative R-L3, the only known KCNQ1-specific activator (Salata et al., 1998; Seebohm et al., 2003). Novel and specific activators of hERG channels have been reported recently. These compounds, RPR260243 (Kang et al., 2005), PD-118057 (Zhou et al., 2005), and NS1643 (Hansen et al., 2006), seem to increase hERG current by different mechanisms. RPR260243 slows hERG deactivation, whereas PD-11807 and NS1643 enhanced current magnitude by an unknown mechanism without measurable effects on current kinetics. As expected for hERG channel activators, all three compounds shorten the action potential duration of ventricular myocytes (Kang et al., 2005; Zhou et al., 2005; Hansen et al., 2006).
Here, we have studied the mechanism of action of NS1643 on hERG channels expressed in Xenopus laevis oocytes. We find that this drug alters the rate of onset and voltage dependence of steady-state inactivation with no significant effects on the voltage dependence for activation or recovery from inactivation of hERG channels. The effect of the drug on mutant hERG channels revealed that it is also a weak blocker. Thus, NS1643 is a partial agonist.

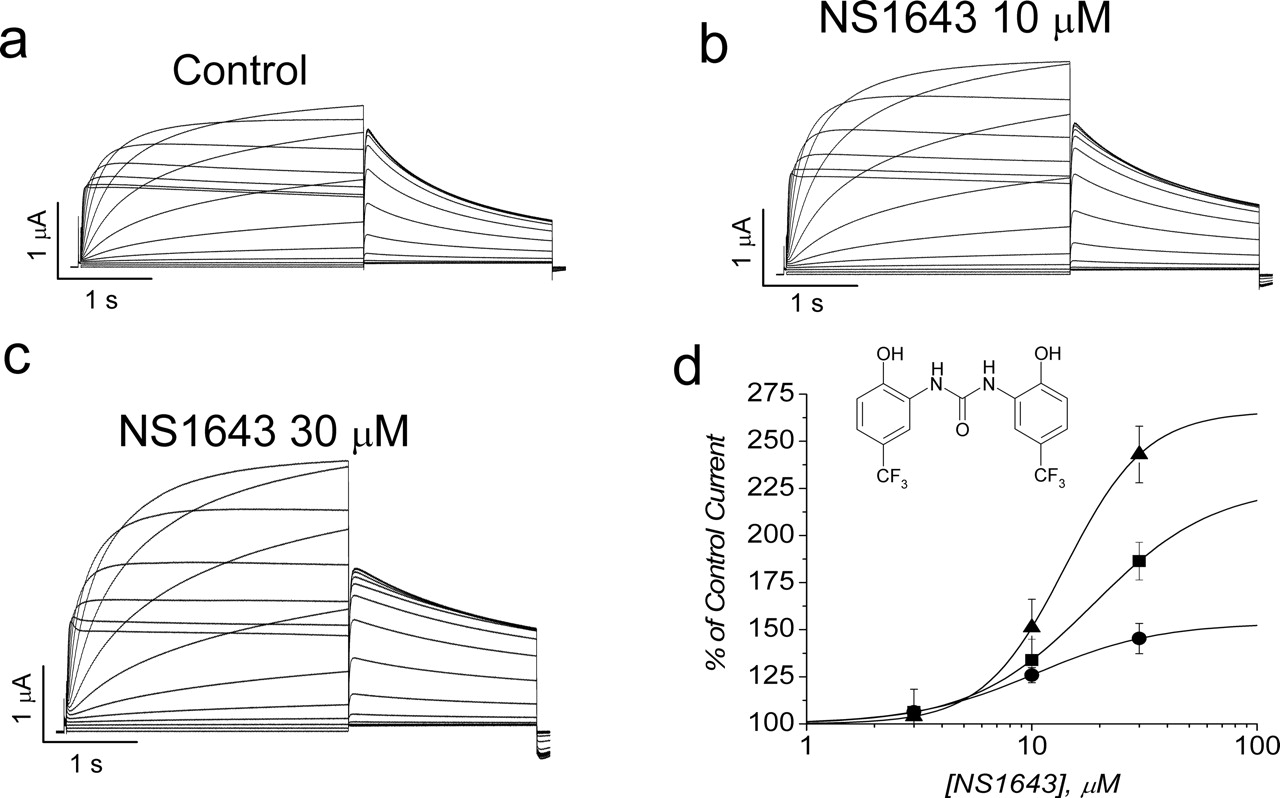
NS1643 increases hERG current in Xenopus oocytes. a to c, representative example of currents recorded in a single oocyte in control (a) and after achieving a steady-state level of increase with 10 μM (b) and 30 μM (c) NS1643. d, concentration-response relationship for NS1643. The percentage of increase in hERG current measured at the end of 2-s pulses to variable potentials was plotted as a function of [NS1643] and fitted with a Hill equation. The EC50 values determined from the fits were 10.4 ± 1.2 μM at –10 mV (•), 18.6 ± 3.2 μM at 0 mV (▪), and 14.0 ± 0.8 μM at +10 mV (▴) (n = 6). Inset shows chemical structure of NS1643.
Materials and Methods
Molecular Biology. Site-directed mutation of HERG subcloned into the pSP64T oocyte expression vector was performed using the megaprimer method (Sarkar and Sommer, 1990). Restriction mapping and DNA sequencing of the polymerase chain reaction-amplified segment were used to confirm the presence of the desired mutation. cRNA for injection into oocytes was prepared with SP6 Capscribe (Roche Diagnostics, Indianapolis, IN) after linearization with EcoRI. Estimates of cRNA quality and quantity were determined by gel electrophoresis and UV spectroscopy.
Isolation, Injection, and Voltage Clamp of Oocytes. Stage IV and V X. laevis oocytes were isolated and injected with cRNA encoding wild-type (WT) or mutant hERG channels. Oocytes were injected with 5 to 20 ng of HERG cRNA and then cultured in Barth's solution supplemented with 50 μg/ml gentamicin and 1 mM pyruvate at 18°C for 1 to 3 days before use in voltage-clamp experiments. Barth's solution contained 88 mM NaCl, 1 mM KCl, 0.4 mM CaCl2, 0.33 mM Ca(NO3)2, 1 mM MgSO4, 2.4 mM NaHCO3, and 10 mM HEPES, pH 7.4. For voltage-clamp experiments, oocytes were bathed in a modified ND96 solution containing 96 mM NaCl, 4 mM KCl, 1 mM MgCl2, 1 mM CaCl2, and 5 mM HEPES, pH 7.6. Currents were recorded at room temperature (23–25°C) with standard two microelectrode voltage-clamp techniques (Stuhmer, 1992). The holding potential was –80 mV, and the interpulse interval for all voltage-clamp protocols was 10 s or slower. To obtain current-voltage (I-V) relationships and activation curves, 2-s voltage steps were applied in 10-mV increments to potentials that varied from –90 to +50 mV. After each step depolarization, the oocyte was repolarized to –70 mV to record deactivating tail currents.
Data Analysis. pCLAMP 8 (Molecular Devices, Sunnyvale, CA), and Origin 7 (OriginLab Corp, Northampton, MA) software were used for data acquisition and analysis on a Dell Optiplex GX150 personal computer. The voltage dependence of hERG channel activation was determined by analysis of peak tail currents recorded at –70 mV. The plot of normalized tail current amplitude versus test potential was fit to a Boltzmann equation to obtain the half-point (V1/2) and slope factor (k) for channel activation: In = 1/(1 + exp[(V1/2–Vt)/k]). Other voltage pulse protocols are described under Results and in figure legends. Data are expressed as mean ± S.E.M. (n is number of oocytes).
Drugs. NS1643 was synthesized by the Department of Chemistry at NeuroSearch (Ballerup, Denmark). Its structure is shown in Fig. 1d. The drug was prepared as a 100 mM stock solution in dimethyl sulfoxide and stored at –20°C.
Results
Increase in hERG Current by NS1643 Is Concentration- and Voltage-Dependent. Oocytes were voltage clamped to a holding potential of –80 mV, and hERG channel current was elicited with 2-s depolarizations to potentials ranging from –90 to +50 mV (Fig. 1a). Each test pulse was followed by a 2-s pulse to –70 mV to measure current deactivation and to determine the voltage dependence of activation. The effects of NS1643 on a single oocyte at concentrations of 10 and 30 μM are illustrated in Fig. 1, b and c. The currents were increased in a concentration-dependent manner by the drug, and this effect was most notable at the more positive test potentials. The percentage of increase in tail currents measured at –70 mV after a pulse to +10 mV was 8.8 ± 0.05% with 10 μM and 19.5 ± 0.2% with 30 μM drug (n = 6). The percentage of increase in hERG current was measured at the end of a 2-s pulse and plotted as a function of [NS1643] in the bath solution (Fig. 1d). The EC50 for NS1643, determined by fitting these data to a Hill equation, was similar for all three voltages examined (Fig. 1d).

Voltage-dependent increase of hERG current by NS1643. a, I-V relationship for normalized WT hERG current determined before (control; n = 13) and after treatment of oocytes with three concentrations of NS1643 (n = 6). Current values were normalized to the peak of the control current at –10 mV (3.86 ± 0.53 μA). b, voltage dependence of hERG activation is not shifted by NS1643. The half-point (V1/2) and slope factors (k) determined from fitting data to a Boltzmann function were as follows: WT (V1/2 = –31.2 ± 0.2 mV; k = 7.6 ± 0.2 mV), 10 μM (V1/2 = –29.3 ± 0.2 mV; k = 7.8 ± 0.2 mV), and 30 μM (V1/2 = –30.2 ± 0.3 mV; k = 8.9 ± 0.3 mV).

Effect of NS1643 on amplitude and kinetics of hERG currents. a and b, tail currents measured using the voltage protocol illustrated above the current traces. Note that current scaling is not the same for the two sets of tracings. c and d, time constants (τdeact) for fast and slow components of current deactivation in the absence (n = 12) and presence of 10 and 30 μM NS1643 (n = 7 each).
The I-V relationships for hERG current measured at the end of 2-s test pulses before and after treatment of oocytes with NS1643 are plotted in Fig. 2a. At 3 μM, a very small increase in current amplitude was observed. Progressively larger increases in hERG current were induced by 10 and 30 μM (Fig. 2a). Regardless of the concentration, NS1643 had only a small effect on currents activated by pulses applied to potentials between –70 and –30 mV, whereas an obvious enhancement of currents was observed for test potentials greater than –10 mV. For example, NS1643 increased hERG current by 17% at 10 μM and 22% at 30 μM when measured at –30 mV. In contrast, at +20 mV NS1643 increased outward current by 69% at 10 μM and 166% at 30 μM. At 100 μM, the agonist activity of NS1643 was attenuated at higher test potentials. At this high concentration, the increase in current was 68 ± 7% at –30 mV, 47 ± 10% at 0 mV, and 38 ± 15% at +20 mV (n = 5; not shown), suggesting that the drug is a partial agonist. Thus, the agonist activity of NS1643 on WT hERG channels was voltage- and concentration-dependent. In theory, voltage-dependent partial agonist activity might be explained by a voltage-independent agonist effect plus a drug-induced shift in the voltage dependence of channel gating. However, NS1643 at 10 and 30 μM had no effect on the voltage dependence of hERG activation determined from plots of tail current amplitudes (normalized to the peak current under each condition) versus voltage (Fig. 2b). These findings suggest that the block observed at high concentrations was voltage-dependent and that the partial agonist activity results from two opposing effects (agonist and block) on the channel.
HERG tail currents were measured at potentials between –140 and +40 mV after channel activation induced by a 1-s pulse to +40 mV (Fig. 3a, inset). Currents were inward and rapidly deactivating at potentials negative to –90 mV, reversed near –90 mV, and outward and deactivated more slowly at potentials positive to –90 mV (Fig. 3, a and b). The time course of current deactivation was determined at potentials varying from –120 to –60 mV. Deactivation was best described by a two-exponential function, and only 30 μM NS1643 caused a significant slowing in the time constants for both fast and slow deactivation (Fig. 3, c and d). Thus, NS1643 increased the amplitude of hERG without causing a shift in the voltage dependence of channel activation and slowed deactivation was observed at 30 μM.

Effects of NS1643 on rectification and inactivation of hERG. a, NS1643 slows the rate of hERG current inactivation. Voltage-clamp protocol used to measure hERG inactivation during third voltage step is shown above current traces. The second pulse to –120 mV was 12 ms in duration. Traces show inactivation of hERG current at 0 mV for a single oocyte. The initial 2.2 ms of the current traces were blanked to remove capacitance transient. The time constants for inactivation (τinact) in this example were 14.6 ms (control) and 18.9 ms (drug). b, NS1643 slowed the rate of hERG inactivation at all potentials examined (n = 6). c, fully activated I-V relationship for hERG. Currents were normalized to the peak of the outward control current (average, 1.51 ± 0.1 μA; n = 12). Amplitudes of currents in the presence of drugs were normalized to their matched control; n = 4 for 3 μM and n = 7 for 10 and 30 μM drug. d, voltage dependence of rectification, determined as described under Results. V1/2 was –47 ± 8 mV in control, –26 ± 4 mV at 10 μM, and –12 ± 2 mV at 30 μM NS1643. The slope factors were unchanged (control, 23.3 ± 0.9 mV; 10 μM, 22.1 ± 1.5 mV; and 30 μM, 22.7 ± 1.6 mV).
NS1643 Slows the Rate of Inactivation and Reduces the Extent of hERG Channel Rectification. The increase in WT hERG current by NS1643 was greatest at positive potentials, suggesting that the drug might decrease the extent of channel inactivation. The onset rate of hERG current inactivation was determined using a three-pulse protocol described previously (Smith et al., 1996; Spector et al., 1996). In brief, current was first activated and inactivated by a 1-s pulse to +40 mV. During the second pulse to –120 mV for 12 ms, channels were allowed to recover from inactivation. A third pulse was applied to a potential that was varied from –40 to +50 mV to observe the re-onset of current inactivation (Fig. 4a, inset). The currents during the third pulse were fitted to a single exponential function to estimate the time constants for inactivation (Fig. 4b). NS1643 at 10 and 30 μM slowed the rate of hERG inactivation throughout the voltage range examined. The V1/2 for the voltage dependence of recovery from inactivation was –74.1 ± 3.8 mV under control conditions and –73.1 ± 2.5 mV in the presence of 10 μM drug (n = 3). The slope factor for this relationship was also not altered by the drug (19.6 ± 1.3 mV for control versus 18.9 ± 1.3 mV after drug). A higher concentration of NS1643 (30 μM) was reported to cause a +11 -mV shift in V1/2 (Hansen et al., 2006). Thus, NS1643 slowed the onset of inactivation but did not alter the voltage dependence for recovery from inactivation sufficiently to explain its agonist activity.
Slow activation coupled with rapid inactivation of channels results in rectification of the I-V relationship for hERG (Smith et al., 1996; Spector et al., 1996). The fully activated I-V for hERG (Fig. 4c) was determined by measuring the peak amplitude of tail currents over a wide range of membrane potentials after a 1-s pulse to +40 mV. NS1643 at 10 and 30 μM increased tail current amplitudes at test voltages positive to the reversal potential, reducing the rectification of hERG current. This effect was quantified by measuring the rectification factor, defined as the deviation (reduction) of current amplitudes relative to that expected from extrapolation of the linear portion of the I-V relationship (between –140 and –110 mV) to more positive potentials. NS1643 shifted the half-point of the rectification-voltage relationship by +21 mV at 10 μM and +35 mV at 30 μM (n = 5; Fig. 4d). Thus, the agonist activity of NS1643 was associated with a decrease in the rate of onset and extent of hERG channel inactivation.
NS1643 Does Not Affect Noninactivating hERG Channels. HERG channels with impaired inactivation were used to confirm the importance of altered inactivation as the mechanism of action for NS1643. Inactivation of hERG can be completely removed by the double mutation G628C/S631C (Smith et al., 1996). Because G628C/S631C hERG channels do not inactivate, current is increased progressively when the membrane is depolarized to potentials positive to 0 mV (Fig. 5a). In contrast to WT hERG, NS1643 had no effect on current magnitude of G628C/S631C channels (Fig. 5b). The normalized I-V relationships for this mutant channel before and after treatment of oocytes with 10 or 100 μM drug are plotted in Fig. 5c. NS1643 was without effect on currents at all potentials tested between –90 and +50 mV.
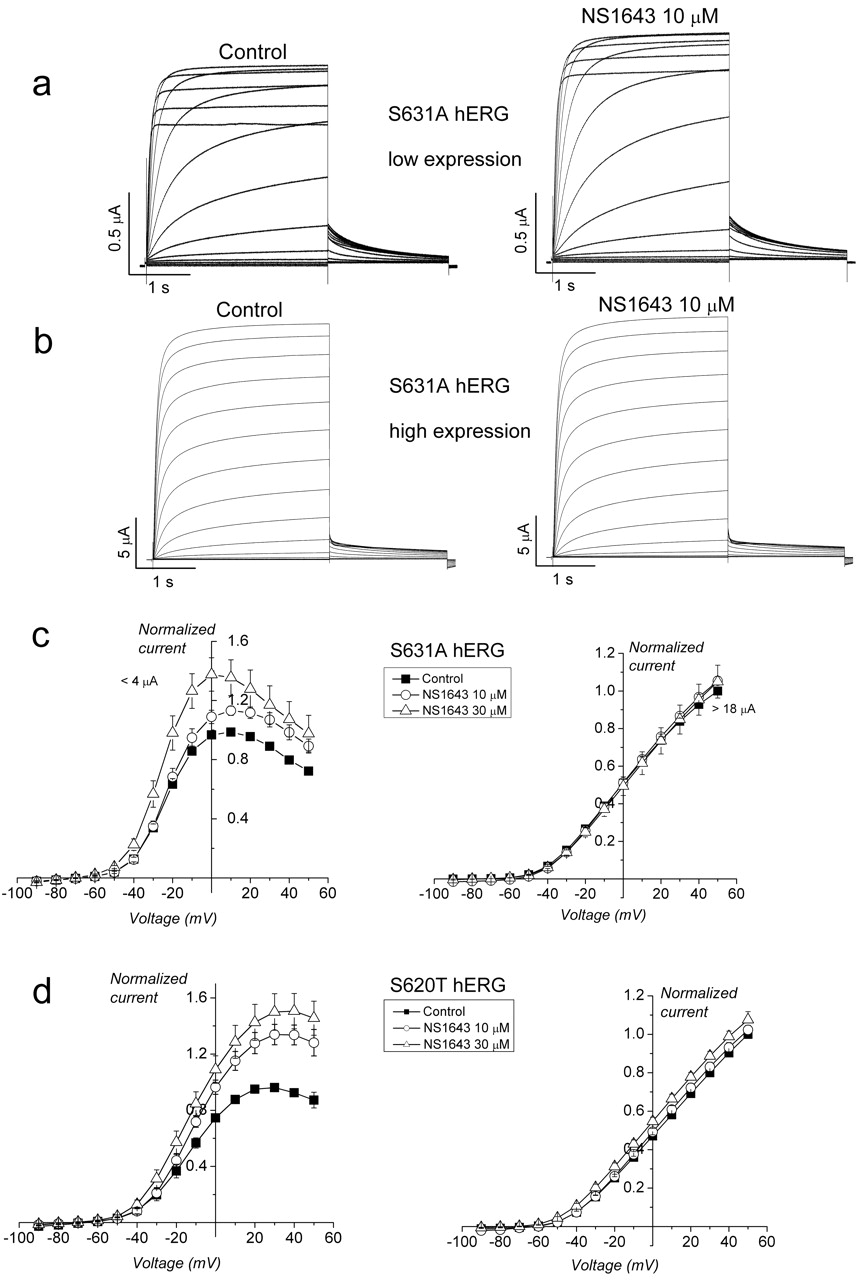
Activation of large outward K+ currents can cause extracellular K+ accumulation within invaginations of the plasma membrane and between the plasma and vitelline membranes in oocytes. High external [K+] reduces inactivation of hERG channels (Smith et al., 1996). HERG inactivation is also hindered, but not eliminated, by point mutation of Ser residues located in the pore helix (S620T) or in the linker between the selectivity filter and the S6 domain (S631A). When either of these mutant channels was highly overexpressed in oocytes, the resulting extracellular [K+] accumulation that is predicted to occur during prolonged depolarizations nearly eliminated fast inactivation. As a result, NS1643 enhanced small S631A channel currents (<4 μA at +10 mV, the peak of the I-V relationship) that exhibited inactivation (Fig. 6a). However, inactivation was not observed when currents were large (>18 μA at +50 mV, the peak of the I-V relationship) (Fig. 6b). The averaged I-V relationships are summarized in Fig. 6c where currents were normalized to the peak values measured under control conditions in each cell. Although NS1643 enhanced small S631A hERG currents, the drug had no effect on large currents. A similar relationship between current magnitude, resulting inactivation, and sensitivity to NS1643 was observed for S620T hERG (Fig. 6d). Thus, enhancement of hERG current by NS1643 is only observed when channels are capable of inactivation.

NS1643 did not increase current magnitude of a mutant (G628C/S631C) hERG channel that does not inactivate. a and b, currents recorded before and after treatment of a single oocyte with 10 μM NS1643. c, I-V relationships for G628C/S631C hERG channel current. Currents in the presence of 10 μM(n = 5); 100 μM(n = 3) drug was normalized to the peak value measured in control (3.2 ± 0.9 μA; n = 5).
Activation of hERG by NS1643 Is Enhanced by Disruption of the Binding Site for hERG-Blocking Drugs. Many drugs that block hERG channels interact with specific residues (i.e., Y652 and F656) that are located on the S6 transmembrane domain in positions that face toward the central cavity of the channel (Lees-Miller et al., 2000; Mitcheson et al., 2000; Sanchez-Chapula et al., 2002, 2003; Fernandez et al., 2004). Mutation of these residues, especially F656, reduces the potency of blockers. Therefore, we determined whether Y652A or F656V mutations might also reduce the effectiveness of NS1643 to enhance hERG current. The effect of NS1643 at 10 and 30 μM on Y652A hERG (Fig. 7a) was nearly identical to the effect of the drug on WT hERG (compare with Fig. 2a). Similar to WT hERG, the increase in Y652A hERG current was most evident at potentials greater than –20 mV, and 30 μM increased outward current by an average of 64 ± 2% at 0 mV. Thus, mutation of Y652 did not alter the agonist activity of NS1643. By contrast, NS1643 enhanced F656V hERG channels at all potentials, and the peak increase obtained with 30 μM at 0 mV was much larger (208 ± 75%; Fig. 7b). Similar results were obtained with F656T and F656M hERG channels (Fig. 7, c and d). Mutation of F656 to another aromatic residue (Tyr) resulted in agonist activity that was intermediate compared with WT and the other F656 mutant channels (Fig. 7e). Thus, mutation of residues known to be important for drug binding did not prevent the action of NS1643, and in fact, mutation of F656 to nonaromatic residues potentiated the agonist activity of the drug by more than 3-fold.
Discussion
Treatment of acute drug-induced and congenital LQTS is inadequate. HERG channel activators might be useful for the treatment of both forms of LQTS. It has been proposed that specific activation of hERG channels might prevent arrhythmia by suppressing action potential duration alternans (Hua and Gilmour, 2004; Hua et al., 2006). Three different compounds have recently been reported to possess such activity: RPR260243 (Kang et al., 2005), PD-118057 (Zhou et al., 2005), and NS1643 (Hansen et al., 2006). Understanding the molecular mechanism of action of these structurally diverse compounds could facilitate the discovery and development of additional compounds. The development of safe and effective hERG channel activators may provide a pharmacological treatment for LQTS and perhaps surpass the effectiveness of magnesium sulfate for this purpose (Tzivoni et al., 1984; Martinez, 1987).

Effect of NS1643 on mutant hERG channels with partially impaired inactivation gating. a, NS1643 enhances S631A hERG currents when expression is low. b, NS1643 does not enhance S631A hERG when currents are large. c, I-V relationships for S631A hERG when expressed at low (left, peak current <4 μA at +10 mV) or high (right, average peak current 18 μA at +50 mV) levels in oocytes. Left, S631A hERG peak control current, 2.07 ± 0.7 μA (n = 6); 10 μM (n = 6); and 30 μM (n = 3). Right, hERG peak control current, 21.3 ± 1.07 μA (n = 3 for all). d, I-V relationships for S620T hERG when expressed at low (left) or high (right) levels in oocytes. Left, peak control current, 2.2 ± 0.5 μA (n = 7); 10 μM (n = 6); and 30 μM (n = 5). Right, peak control current, 14.4 ± 1.0 μA (n = 6); 10 μM (n = 5); and 30 μM (n = 4).
Activation of hERG channels and shortening of action potential duration can be achieved by different mechanisms. RPR260243 was reported to cause a slight slowing of activation and approximately a 20% increase in hERG current magnitude. The main mechanism of action potential shortening by this drug is the pronounced delay in channel closure (deactivation); it is without any significant effects on the voltage dependence of activation or inactivation (Kang et al., 2005). PD-118057 (Zhou et al., 2005) and NS1643 (Hansen et al., 2006) also have no effect on the voltage dependence of activation. However, in contrast to RPR260243, NS1643 activates hERG primarily by altering the voltage dependence of channel inactivation with little (present study) or no effect (Hansen et al., 2006) on the rate of deactivation. Similar to NS1643, the hERG agonist activity of PD-118057 was voltage-dependent, and the drug did not significantly alter the voltage dependence of activation or recovery from inactivation of channels (Zhou et al., 2005). The agonist effects of PD-118057 were attributed to an unspecified alteration of single channel activity; however, the decrease in rectification suggests that this compound may have a mechanism similar to that described here for NS1643.
NS1643 reduced the rectification of hERG, an effect that was best revealed by measurement of the fully activated I-V relationship before and after treatment of oocytes with the drug (Fig. 4a). NS1643 also slowed the onset rate of hERG current inactivation. Rectification is caused by fast channel inactivation. Thus, the relatively minor effect (+10-mV shift) by the drug on the voltage dependence of recovery from inactivation was unexpected (Hansen et al., 2006). However, we have previously reported that mutations of hERG can also differentially affect the onset and recovery from inactivation of hERG. A mutation in the pore of hERG (S631A) shifts the peak of the isochronal (1-s) I-V relationship by approximately +30 mV, a measure of steady-state inactivation. However, S631A shifts the voltage dependence for recovery from inactivation, determined with a triple-pulse protocol, by +102 mV (Zou et al., 1998). Thus, it is possible to differentially affect the onset and recovery from inactivation by drugs or by mutation of the channel.
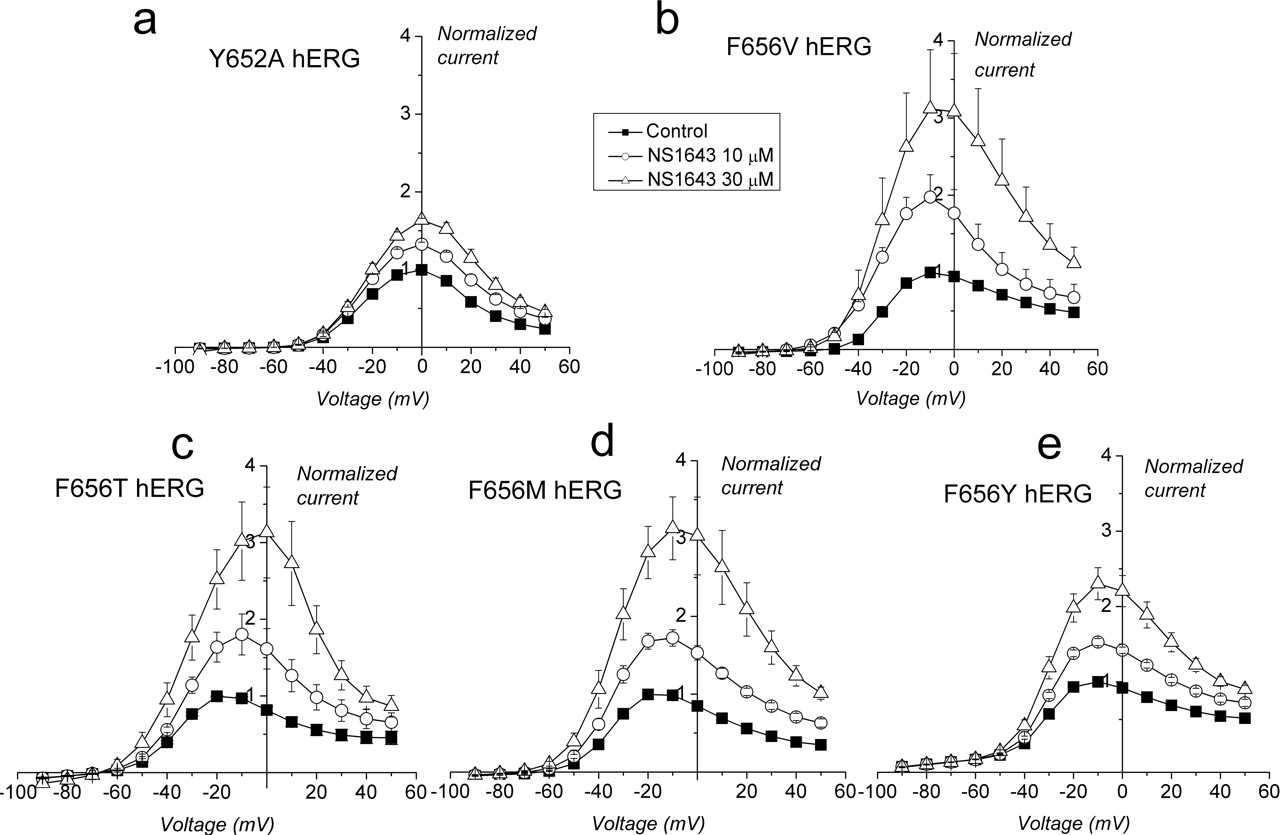
Effect of NS1643 on Y652A and F656 mutant hERG channels. a, I-V relationship for Y652A hERG for control (peak control current, 4.9 ± 0.5 μA; n = 5), 10 μM (n = 4), and 30 μM (n = 4). b, I-V relationship for F656V hERG (peak control current, 1.46 ± 0.2 μA; n = 9), 10 μM (n = 6), and 30 μM (n = 5). c, I-V relationship for F656T hERG (peak control current, 1.14 ± 0.28 μA; n = 5). d, I-V relationship for F656M hERG (peak control current, 0.65 ± 0.1 μA; n = 5). e, I-V relationship for F656Y hERG (peak current, 0.8 ± 0.04 μA; n = 5).
Hansen et al. (2006) reported that 10 μM NS1643 increased the magnitude of hERG tail currents by 45% when measured at –60 mV. The percentage of increase in tail currents in our study were only 8.8 ± 0.05% with 10 μM and 19.5 ± 0.2% with 30 μM drug. Hansen et al. (2006) measured hERG channel deactivation at –60 mV using a bath solution containing a [KCl] of 1 mM. In contrast, we measured tail currents at –70 mV and used a bath solution with 4 mM KCl. hERG inactivation is greater at –70 mV, and its voltage dependence is leftward shifted with low [K+]. Therefore, it is likely that the drug-induced positive shift in the voltage dependence of hERG inactivation resulted in a larger increase in current in the Hansen et al. (2006) experiments compared with our experiments. Moreover, as can be seen in Fig. 4c, the tail currents are larger when measured at –60 mV compared with –70 mV.
The agonist effect of NS1643 was greatly enhanced by mutation of F656 to Ala, Val, or Met and to a lesser extent by mutation to Tyr. In contrast, Y652A channels were affected by NS1643 in an almost identical manner as WT hERG channels. Most of the hERG channel blockers we have examined interact with both F656 and Y652 residues (Mitcheson et al., 2000; Sanchez-Chapula et al., 2002, 2004; Fernandez et al., 2004). However, quinidine was strongly influenced by mutation of F656 (i.e., 125-fold increase in IC50 for F656A) but not by mutation of Y652A hERG (3-fold increase in IC50 for Y652A) (Sanchez-Chapula et al., 2002). Thus, NS1643 most likely blocks hERG channels by low-affinity interactions with F656 but not Y652. This blocking activity is why we refer to NS1643 as a partial agonist. Furthermore, based on the intermediate effects of NS1643 on F656Y channels, the block of hERG by this drug may be favored by a π -π interaction involving F656 rather than a more generalized hydrophobic interaction described previously for potent blockers such as MK-499, terfenadine, and cisapride (Fernandez et al., 2004).
An important observation relative to specificity of RPR260243 and PD-118057 was that these drugs did not affect other cardiac currents, including ICaL, INa, IK1, or IKs. In addition, RPR260243 partially blocked, but did not affect the gating of ERG3, the channel most similar to hERG (ERG1). This differential effect of the drug on ERG1 and ERG3 should facilitate determination of the structural basis of the agonist effect. Future experiments will examine the binding sites for these drugs that are responsible for their distinct molecular mechanisms of hERG channel activation. More importantly, experiments in animals or isolated hearts are needed to determine whether the specific molecular mechanism of NS1643 provides unique advantages as an antiarrhythmic compared with other hERG agonists.
Acknowledgments
We thank Krista Kinard and Meng San Pun for technical assistance.
Footnotes
-
This work was supported by National Heart, Lung, and Blood Institute/National Institutes of Health Grant HL55236 (to M.C.S.) and Ministerio de Educacacion y Ciencia PR2005-0176 (to O.C.).
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.105.019943.
-
ABBREVIATIONS: LQTS, long QT syndrome; hERG, human ether-a-go-go-related gene; RL-3, L-364,373 (3-R)-1,3-dihydro-5-(2-fluorophenyl)-3-(1H-indol-3-ylmethyl)-1-methyl-2H-1,4-benzodiazepin-2-one; PD-118057, 2-{4-[2-(3,4-dichloro-phenyl)-ethyl]-phenylamino}-benzoic acid; RPR260243, (3R,4R)-4-[3-(6-methoxyquinolin-4-yl)-3-oxo-propyl]-1-[3-(2,3,5-trifluoro-phenyl)-prop-2-ynyl]-piperidine-3-carboxylic acid; NS1643, 1,3-bis-(2-hydroxy-5-trifluoromethyl-phenyl)-urea; WT, wild-type; I-V, current-voltage; MK-499, (+)-N-[1′-(6-cyano-1,2,3,4-tetrahydro-2(R)-naphthalenyl)-3,4-dihydro-4(R)-hydroxyspiro(2H-1-benzopyran-2,4′-piperidin)-6-yl]methanesulfonamide monohydrochloride.
- Received October 18, 2005.
- Accepted November 11, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}