


Abstract
Opioids mediate their analgesic effects by activating μ-opioid receptors (MOR) not only within the central nervous system but also on peripheral sensory neurons. The peripheral analgesic effects of opioids are best described under inflammatory conditions (e.g., arthritis). The present study investigated the effects of inflammation on MOR binding and G-protein coupling of full versus partial MOR agonists in dorsal root ganglia (DRG) of primary afferent neurons. Our results show that Freund's complete adjuvant (FCA) unilateral hindpaw inflammation induces a significant up-regulation of MOR binding sites (25 to 47 fmol/mg of protein) on DRG membranes without affecting the affinity of either full or partial MOR agonists. In our immunohistochemical studies, the number of MOR-immunoreactive neurons consistently increased. This increase was mostly caused by small-diameter nociceptive DRG neurons. The full agonist DAMGO induced MOR G-protein coupling in DRG of animals without FCA inflammation (EC50 = 56 nM; relative Emax = 100%). FCA inflammation resulted in significant increases in DAMGO-induced MOR G-protein coupling (EC50 = 29 nM; relative Emax = 145%). The partial agonist buprenorphine hydrochloride (BUP) showed no detectable G-protein coupling in DRG of animals without FCA inflammation; however, partial agonist activity of BUP-induced MOR G-protein coupling was detectable in animals with FCA inflammation (EC50 = 1.6 nM; relative Emax = 82%). In behavioral studies, administration of BUP produced significant antinociception only in inflamed but not in noninflamed paws. These findings show that inflammation causes changes in MOR binding and G-protein coupling in primary afferent neurons. They further underscore the important differences in clinical studies testing peripherally active opioids in inflammatory painful conditions.
Opioid analgesia is not mediated exclusively within the central nervous system but also in the periphery. This has been shown in many animal models, including unilateral hindpaw inflammation induced by intraplantar injection of Freund's complete adjuvant (FCA) (Stein et al., 1988a). Moreover, controlled clinical trials have reported peripheral analgesic effects of opioids in both short-term postoperative and long-term arthritic pain (Stein et al., 2001). The peripheral analgesic effects of opioids are elicited by activation of opioid receptors on primary afferent neurons. This is best described under local inflammatory conditions (Stein et al., 1989). In addition, it has been shown in clinical studies that the effects of exogenous opioids in peripheral antinociception were enhanced in inflamed tissue (Stein et al., 2001). It was suggested that an increase in antinociception during inflammation might be related to an increase in the number of μ-opioid receptors (MOR) in dorsal root ganglia (DRG) (Ji et al., 1995). However, it remains unclear whether inflammation alters intracellular signaling (e.g., G-protein coupling and ligand binding of MOR on peripheral sensory neurons). Therefore, this study compares animals with and without inflammation to examine whether FCA inflammation 1) alters MOR binding in DRG neurons, 2) leads to immunohistochemical differences in the distribution and density of MOR on DRG neurons, 3) causes differences in the potency and efficacy of agonists coupling to MOR of DRG neurons, or 4) reveals differences in behavioral studies for the partial MOR agonist BUP.
Materials and Methods
Guanosine-5′-O-(3-[35S]thio)-triphosphate ([35S]GTPγS) (1250 Ci/mmol) was purchased from PerkinElmer Life Sciences (Boston, MA). [3H][d-Ala2,N-Me-Phe4, Gly5-ol]enkephalin (DAMGO; 56 Ci/mmol) was purchased from Amersham Biosciences (Little Chalfont, Buck-inghamshire, UK). DAMGO, buprenorphine hydrochloride, naloxone, and d-Phe-Cys-Tyr-d-Trp-Orn-Thr-Pen-Thr-NH2 (CTOP) were purchased from Sigma RBI (Taufkirchen, Bayern, Germany). Scintillation fluid was obtained from PerkinElmer Wallac (Turku, Finland). Antibodies for immunohistochemistry were obtained from Vector Laboratories (Burlingame, CA). Synthetic peptide for MOR was obtained from Gramsch Laboratories (Schwabhausen, Bayern, Germany). Dibutylpthalate polystyrene xylene was provided by Merck (Darmstadt, Hessen, Germany). Tissue Tek compound (OCT) was provided by Miles (Elkhart, IN). Anesthesia was performed with halothane from Willy Rüsch GmbH (Böblingen, Baden Würtemberg, Germany). FCA was obtained from Calbiochem (San Diego, CA).
Subjects. Experiments were performed in male Wistar rats (180–200 g) individually housed in cages lined with sawdust. Standard laboratory rodent chow and water were available ad libitum. Room temperature and relative humidity were maintained at 22 ± 0.5°C and 60%, respectively. A 12-h:12-h light/dark cycle was used. All testing was conducted in the light phase, employing separate groups of animals. The guidelines on ethical standards for investigations of experimental pain in animals were followed (Zimmermann, 1983).
Induction of Inflammation. Unilateral hindpaw inflammation was induced by injection of 0.15 ml of FCA into the right hindpaw under brief halothane anesthesia. A detailed description of the time course and magnitude of the inflammatory reaction is given elsewhere (Stein et al., 1988b). The inflammation remained confined to the inoculated paw and all experiments were performed 96 h (4 days) after FCA inoculation.
Membrane Preparations. Rats were killed by halothane anesthesia after 96-h treatment with saline or FCA and lumbar (L3–L5) DRGs were removed. In animals treated with FCA, DRG on the inflamed and contralateral sites were removed separately. The tissue was placed immediately on ice in cold assay buffer (50 mM Tris-HCl, 1 mM EGTA, 5 mM MgCl2, pH 7.4). Membrane preparations were made by pooling DRG tissue from 10 rats. Tissue was homogenized with a Polytron homogenizer (Kinematica AG, Littau, Switzerland) and centrifuged at 48,000g at 4°C for 20 min. The pellet was resuspended in assay buffer followed by a 10-min incubation at 37°C to remove endogenous ligands. The homogenate was centrifuged again at 48,000 g and resuspended in assay buffer. Membranes were aliquoted and stored at -80°C.
Preparation of Sciatic Nerves. Rats were anesthetized with halothane 48 h after FCA or saline injections. The right sciatic nerve was surgically exposed, dissected away from the surrounding tissue, and ligated with nonabsorbable silk at the midfemoral position (5 mm below the sciatic notch) in animals with FCA inflammation or saline treatment. The incision was then closed with wound clips. After 96 h of FCA inflammation, rats were killed, and the proximal part of sciatic nerve was removed and membranes prepared as described above.
Opioid Receptor Binding. Membranes were diluted in assay buffer. Saturation analysis of [3H] DAMGO binding was performed by incubating 50 μg of membrane protein with 0.02 to 2 nM [3H]DAMGO in the presence and absence of 10 μM unlabeled naloxone (NLX) to determine nonspecific binding. Affinity (inhibition constants, Ki) of DAMGO and BUP at DRG membranes of animals with and without FCA inflammation were determined in [3H]DAMGO competition binding experiments. In animals with FCA inflammation, the contralateral side of DRG was removed and MOR binding sites were detected by incubating 50 μg of membrane protein with 2 nM [3H]DAMGO in the presence and absence of 10 μM unlabeled NLX. The accumulation of MOR binding sites in the sciatic nerve was detected by incubating 75 μg of membrane protein with 2 nM [3H]DAMGO in the presence and absence of 10 μM unlabeled NLX. Membranes were incubated for1hat 30°C in assay buffer. The reactions were terminated by rapid filtration under vacuum through Whatman GF/B glass fiber filters, followed by four washes with cold buffer (50 mM Tris-HCl, pH 7.4). Bound radioactivity was determined by liquid scintillation spectrophotometry after overnight extraction of the filters in 3 ml of scintillation fluid.
Immunohistochemistry. Four days after FCA treatment, six rats were deeply anesthetized with halothane and transcardially perfused with 60 ml of warm saline, followed by 300 ml of 4% (w/v) paraformaldehyde with 0.2% (v/v) picric acid in 0.16 M phosphate buffer solution, pH 6.9. The ipsilateral and contralateral L5 DRG were removed, postfixed in the same fixatives for 90 min, and then placed in 15% (w/v) sucrose solution at 4°C overnight. The tissue was embedded in Tissue Tek compound (OCT; Miles), frozen and cut in 14-μm sections. The sections were incubated overnight with anti-MOR (1:1000) (kindly provided by Drs. Stefan Schulz and Volker Höllt, Department of Pharmacology and Toxicology, Otto-von-Guericke University, Magdeburg, Germany). The sections were incubated for 90 min with the appropriate biotinylated secondary antibody and with avidin-biotin-conjugated peroxidase. Finally, the sections were washed and stained with 3′,3′-diaminobenzidine tetrahydrochloride containing 0.01% H2O2 in 0.05 M Tris-buffered saline, pH 7.6, for 3 to 5 min. After the enzyme reaction, the sections were washed in tap water, mounted onto gelatin-coated slides, dehydrated in alcohol, cleared in xylene, and mounted in dibutylpthalate polystyrene xylene. To demonstrate specificity of staining, the following controls were included: 1) preabsorption of diluted antibody against MOR with a synthetic peptide for MOR (Gramsch Laboratories, Schwabhausen, Germany) for 24 h at 4°C and 2) omission of either the primary antisera, the secondary antibodies, or the avidin-biotin complex. These control experiments did not show MOR staining.
The method of quantification for DRG staining has been described previously (Ji et al., 1995). Briefly, we stained every fourth section of DRG that was serially cut at 14 μm. The total number of MOR-containing neurons was counted by an observer blinded to the experimental protocol. This number was divided by the total number of neurons in each DRG section, and the percentage of MOR immunoreactive neurons was calculated. Percentages from four sections of each DRG were averaged. Five rats per group (inflamed and noninflamed) were used for analysis. The cell body diameter was measured with the nucleus in the focal plane and was estimated from the average length and width determined with a calibrated micrometer. A total number of 30 immunoreactive neurons with nucleus were measured in each animal.
Measurement of Agonist Efficacy and Potency at MOR in DRG Membranes. Membranes were thawed, homogenized, and centrifuged at 48,000g for 10 min. Membranes were resuspended in [35S]GTPγS assay buffer (50 mM Tris-HCl, pH7.4, 5 mM MgCl2, 0.2 mM EGTA, 100 mM NaCl, and 1 mM DTT). The buffer composition was similar to that used by Newman-Tancredi et al. (2000). Concentration-effect curves were generated by incubating the appropriate concentration of membranes (50 μg) in assay buffer with 0.1% bovine serum albumin, various concentrations of BUP or DAMGO (10-12–10-4 M), with 50 μM GDP and 0.05 nM [35S]GTPγS in a total volume of 800 μl. Basal binding was assessed in the absence of agonist, and nonspecific binding was measured in the presence of 10 μM unlabeled GTPγS. The reaction was incubated for 2 h at 30°C.
[35S]GTPγS Saturation Binding at MOR of DRG Membranes. Saturation analysis of DAMGO and BUP-stimulated [35S]GTPγS binding to DRG membranes was performed. In the presence (DAMGO or BUP 10 μM) or absence (H2O) of agonists, membranes were incubated with 0.05 to 2 nM [35S]GTPγS in assay buffer for 2 h at 30°C. Unstimulated [35S]GTPγS binding was subtracted from agonist-stimulated binding at each measurement point. The incubations for all experiments were terminated by filtration under vacuum through Whatman GF/B glass fiber filters, followed by four washes with cold buffer (50 mM Tris-HCl, pH 7.4). Bound radioactivity was determined by liquid scintillation spectrophotometry after extraction overnight in scintillation fluid.
Measurement of Paw Pressure Threshold. Four days after FCA inoculation, nociceptive thresholds were assessed before (baseline) and after drug administration using the paw pressure algesiometer (modified Randall-Selitto test; Ugo Basile, Comerio, Italy). The pressure required to elicit paw withdrawal, the paw pressure threshold (PPT) (cutoff at 250 g), was determined by averaging three consecutive trials separated by 10 s (Stein et al., 1988b). The sequence of left and right paws was alternated between animals to avoid bias. Drugs were administered intraplantarly (100 μl), and antagonists were given concomitantly with agonist in a total volume of 200 μl. Control animals received saline in the same volume. The experimenter was blind to the treatment.
Data Analysis. All ligand binding and [35S]GTPγS binding data are reported as mean ± S.E. values of at least three experiments, each of which was performed in duplicate. [3H]DAMGO ligand binding experiments and [35S]GTPγS saturation binding experiments were fitted to a one-site binding hyperbola using Prism (GraphPad, San Diego) to determine Kd and Bmax values. For saturation analysis of stimulated [35S]GTPγS binding, basal [35S]GTPγS binding was subtracted from agonist (10 μM DAMGO or 10 μM BUP)-stimulated [35S]GTPγS binding. Stimulated [35S]GTPγS binding is defined as agonist-stimulated minus basal [35S]GTPγS binding. Efficacy (Emax) is defined as the maximum percentage stimulation by an agonist, as determined by nonlinear regression analysis of concentration-effect curves. Relative Emax values are expressed as a percentage of maximal stimulation with DAMGO in animals without inflammation. Nonspecific binding was subtracted from all [35S]GTPγS binding data. Statistical differences between animals with and without FCA inflammation were determined by the nonpaired Student's t test and Mann-Whitney rank sum tests. Amplification factors were defined by DAMGO-activated G-protein Bmax/MOR Bmax. Behavioral data are represented as mean ± S.E.M. Dose-response curves were assessed by analysis of variance followed by a post hoc Dunnett test. Time course data were analyzed by two-way repeated-measure analysis of variance (treatment × time) followed by a post hoc Dunnett test. Differences were considered significant at p < 0.05. All tests were performed using Sigma Stat 2.03 (SPSS Science, Chicago, IL) statistical software
Results
Binding Affinities and Opioid Receptor Numbers in DRG Membranes. Saturation binding of [3H]DAMGO displaced by naloxone showed similar binding affinities (Kd) in DRG membranes of animals without (Kd, 0.2 ± 0.03 nM) and with (Kd, 0.3 ± 0.04 nM) FCA inflammation. The number of opioid receptors (Bmax) increased significantly in animals with (Bmax,47 ± 2.1 fmol/mg of protein) compared to animals without (Bmax, 25 ± 1.1 fmol/mg of protein) FCA inflammation (t test, p < 0.05) (Fig. 1A). This increase in MOR binding sites was restricted to the inflamed side and was not detectable on the contralateral side of inflammation (data not shown). Competitive inhibition experiments of [3H]DAMGO by unlabeled DAMGO and BUP were performed to determine the inhibition constant Ki. There was no significant difference detectable between DRG of animals with and without FCA inflammation (DRG of animals without inflammation: Ki DAMGO, 5 ± 0.4 nM, Ki BUP, 0.18 ± 0.01 nM; DRG of animals with inflammation: Ki DAMGO 8 ± 1.4 nM, Ki BUP 0.17 ± 0.02 nM) (Mann-Whitney rank sum test PDAMGO/BUP > 0.05) (Fig. 1B).

A, saturation binding was performed with [3H]DAMGO to DRG membranes of animals with and without inflammation. Nonspecific binding was determined with 10 μM naloxone. Data shown are means of duplicates from at least three independent experiments. B, displacement of [3H]DAMGO binding to DRG membranes of animals with and without inflammation. Nonradioactive DAMGO and BUP was tested as a displacer at 10-11 to 10-4 M concentrations. One representative curve of four independent experiments is shown.
MOR in Sciatic Nerve Membrane Preparations. In the sciatic nerve of unligated rats, almost no specific binding was detectable (data not shown). In the absence of inflammation, an accumulation of binding sites was shown proximal to the ligature (7 ± 1.3 fmol/mg of protein). However, after 96 h of FCA inflammation, a significantly higher accumulation of MOR-specific binding sites was detected in proximal parts of the ligature (17 ± 2.5 fmol/mg of protein, t test, p < 0.05).
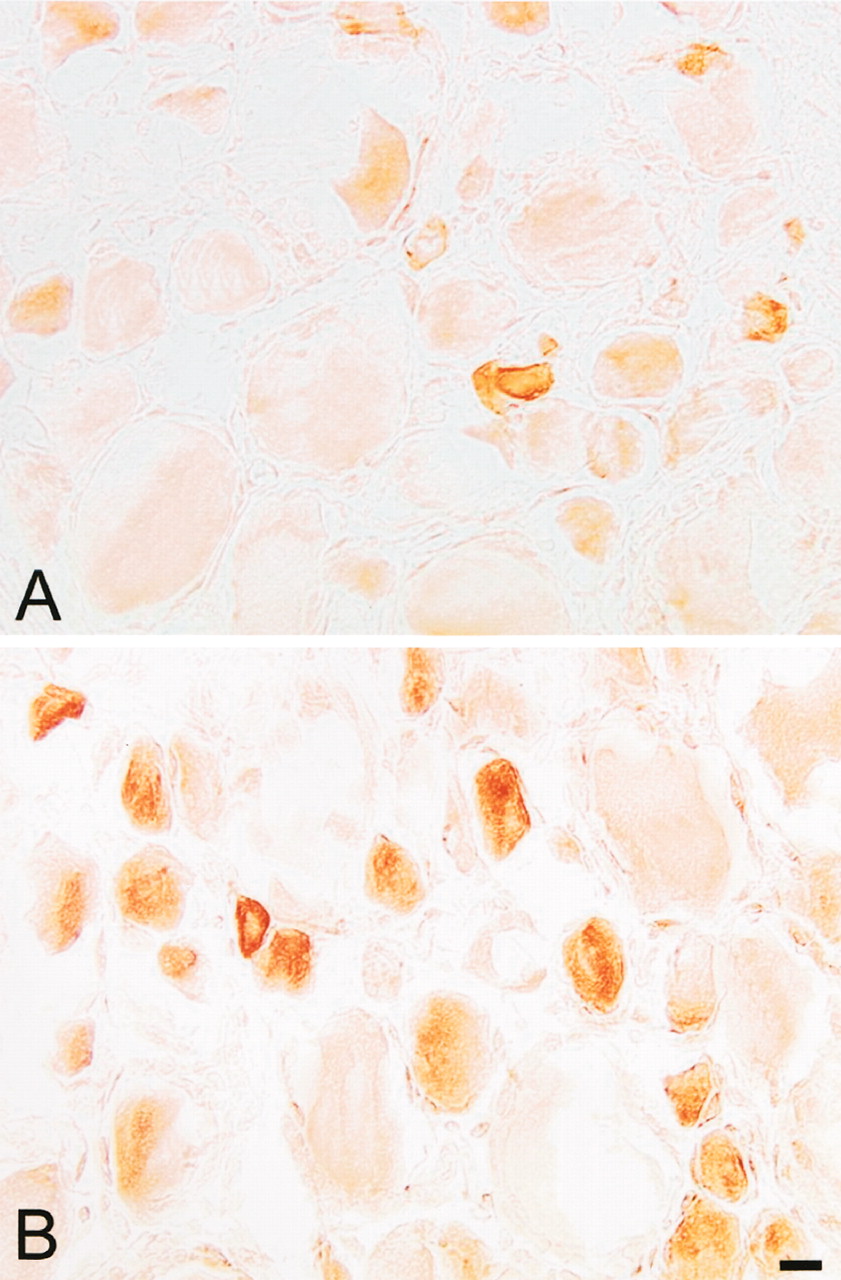
Immunohistochemistry. In nontreated rats, some DRG neurons contained MOR-immunoreactivity (MOR-IR) (Fig. 2A). These neurons were mainly of small diameter. The mean cell body diameter of MOR-positive neurons was 37.4 + 1.1 μm; the majority lay between small- and medium-diameter neurons (23–51 μm). Of all neurons, 17.2 ± 0.9% were MORIR. Four days after FCA, there was a noticeable increase in the number of MOR-positive DRG neurons on the inflamed side (Fig. 2B) and this increase in MOR was not detectable on the contralateral side of inflammation (data not shown). Of all DRG neurons, 25 ± 1.3% were MOR-positive, which represents a 45.3% relative increase (p < 0.01, Mann-Whitney rank sum test). There was no significant difference in the mean diameter of MOR-positive neurons between animals with and without FCA inflammation (p > 0.05), suggesting that any increase in MOR synthesis was not caused by a change in cell size.

Bright-field micrographs showing MOR positive neurons in L5 DRGs of rats without FCA inflammation (A) and in DRGs of rats with FCA inflammation (B). MOR-IR is mainly seen in small DRG neurons. Scale bar, 20 μm.
Potencies and Efficacies of DAMGO and BUP for [35S]GTPγS Binding in DRG. It has been shown that agonist efficacy for stimulation of [35S]GTPγS binding is dependent on the concentration of GDP (Traynor and Nahorski, 1995; Selley et al., 1997; Newman-Tancredi et al., 1999). Among various concentrations (5–200 μM) of GDP tested, 50 μM GDP achieved the maximal percentage stimulation by DAMGO in DRG membranes and was used in all subsequent studies. EC50 and Emax values are shown in Table 1 and Fig. 3. Relative Emax values were expressed as a percentage of maximal DAMGO stimulation in DRG membranes of animals without FCA inflammation. After 96 h of FCA inflammation, DAMGO induced a significant increase in efficacy (Emax) (Mann-Whitney rank sum test, p < 0.05) and a nonsignificant, leftward shift in potency in DRG membranes (Table 1, Fig. 3A). In contrast, the partial agonist BUP did not induce any detectable G protein activation in DRG membranes of animals without FCA inflammation. However, after 96 h of FCA inflammation, BUP showed effective G-protein coupling (Table 1, Fig. 3B).
[35S]GTPγS intrinsic efficacies (Emax), potencies (EC50), and relative Emax of DAMGO and BUP in DRG membranes of animals without (Control) and with (FCA) inflammation
Data are mean values ± S.E.M. of at least three independent experiments as described in Materials and Methods. Emax values are percentage stimulation over basal and relative Emax values are expressed as a percent of maximal stimulation with DAMGO in animals without inflammation.

Stimulation of [35S]GTPγS binding to DRG membranes of animals without (Control) and with (FCA) inflammation. Concentration-response curves were determined for DAMGO (A) and BUP (B), as described under Materials and Methods. [35S]GTPγS binding is expressed as percentage of maximal stimulation with DAMGO (100%) obtained in noninflamed animals. Nonspecific binding was determined using 10 μM unlabeled GTPγS and was subtracted from each data set. Basal [35S]GTPγS binding in the absence of added drugs was 4000 to 6000 cpm in both groups. Each value represents the mean ± S.E.M. of at least three independent experiments performed in duplicate.
[35S]GTPγS Saturation Binding Experiments. [35S]GTPγS saturation binding exhibited high affinities for G-proteins at MOR (Table 2) after DAMGO (10 μM) stimulation, but no significant differences were detectable between Kd Gprotein in DRG membranes of animals with and without FCA inflammation (p > 0.05) (Table 2). In animals with FCA inflammation, a significant increase in DAMGO-stimulated Bmax Gprotein was detected (Fig. 4, Table 2) (p < 0.05). After stimulation with the partial agonist BUP, Bmax Gprotein was only measurable in animals with FCA inflammation (Table 2 and Fig. 4). Bmax determination of G-proteins in animals with FCA inflammation revealed that after BUP stimulation, only 34% (145 fmol/mg) of G-proteins were activated compared with 100% (425 fmol/mg) of G-proteins activated by the full agonist DAMGO (Table 2, Fig. 4). The amplification factors (amount of G-protein bound/number of opioid receptors expressed on the surface) was calculated according to Selley et al. (1998). No significant difference in amplification factors was detectable between animals with (amplification factor 9) and without (amplification factor 11) FCA inflammation.
Characterization of G-protein binding to MOR
Affinity (Kd Gprotein) and number (Bmax Gprotein) of G-proteins for net agonist-stimulated [35S]GTPγS binding in DRG membranes of animals without (Control) and with (FCA) inflammation after DAMGO and BUP stimulation. Membranes were incubated with varying concentrations of [35S]GTPγS as described under Materials and Methods. Data are mean Bmax and Kd values ± S.E.M., obtained from at least three independent experiments.

Saturation analysis of DAMGO and BUP-stimulated [35S]GTPγS binding to DRG membranes of animals with and without inflammation. Saturation binding of [35S]GTPγS was performed in the absence and presence of 10 μM DAMGO or BUP. Unstimulated and stimulated [35S]GTPγS binding were subtracted.
Behavioral Studies. Intraplantar injection of BUP in a dose of up to 5 μg in normal rats did not show any significant changes in PPT (p > 0.05) (Fig. 5B). Injection of higher doses of BUP, such as 10 μg, increased PPT not only in the injected paw, but also in the contralateral paw of rats with and without FCA inflammation, indicating a systemic (central) site of action (data not shown). In contrast, administration of BUP into inflamed paws resulted in significant elevations of PPT (p < 0.05) (Fig. 5A). No PPT changes were observed in the contralateral noninflamed paws (p > 0.05) indicating that the site of action is restricted to the inflamed paw (data not shown). PPT elevations in inflamed paws increased dose dependently. BUP in a dose of 1 μg showed a peak effect at 5 min, whereas BUP in a dose of 3 and 5 μg showed maximum effect at 30 min. This antinociception was very long-lasting (up to 2 h), and by 4 h, the PPT returned to baseline values (Fig. 5A). The peripheral antinociceptive effect produced by BUP in inflamed paws (5 μg at 30 min) was dose-dependently antagonized by intraplantar coadministration of naloxone (30 μg, p < 0.05) (Fig. 6A) and CTOP (120 μg, p < 0.05) (Fig. 6B).

Time course and dose-response of the antinociceptive effects of BUP in injected paw of (A) rats with inflamed hindpaws and (B) normal rats. *, significantly different from saline treated group (related to 1, 3, and 5 μg BUP).
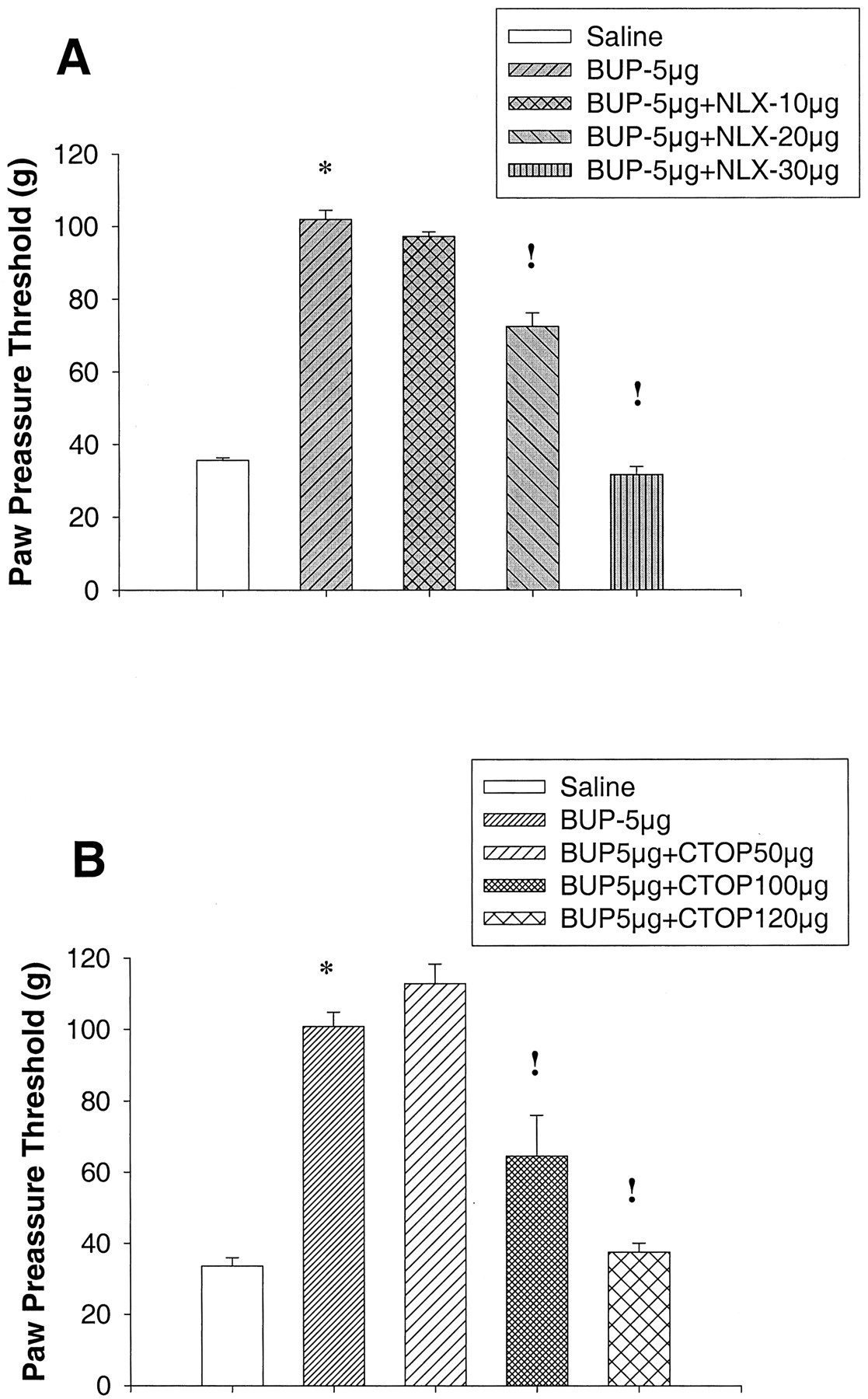
Blocking effect of Naloxone (A) and CTOP (B) on the antinociceptive action of BUP. *, significantly different from saline treated group;!, significantly different from BUP (5 μg)-treated group.
Discussion
Our results show that inflammation enhances the efficacy of G-protein coupling of full and partial MOR agonists by an increased binding and G-protein coupling at MOR of primary afferent neurons. First, FCA inflammation induces an upregulation of MOR binding sites on DRG membranes without affecting binding affinities. Consistently, the number of MOR per DRG as well as the number of MOR-ir neurons that are predominantly of small diameter increases after FCA inflammation. Second, the full agonist DAMGO but not the partial agonist BUP induces MOR G-protein coupling in DRG of animals without FCA inflammation. FCA inflammation results in significant increases in DAMGO-induced MOR G-protein coupling and in partial agonist activity of BUP-induced MOR G-protein coupling. Third, BUP injection into inflamed, but not normal hindpaws elicits potent and long-lasting antinociceptive effects.
Peripheral opioid receptors are localized and expressed on primary sensory neurons. Primary sensory neurons offer the advantage of characterizing receptors in their native environment. In addition, the possibility to induce a locally applied inflammation allows study of the effects of MOR binding and signaling under pathological conditions. In animals with FCA inflammation, we found a significant increase in the number of MOR binding sites on DRG membranes, although the affinity of DAMGO to MOR remained unchanged. Displacement experiments of [3H]DAMGO with either nonlabeled DAMGO or the partial agonist BUP revealed similar inhibitory constants (Ki) in DRG membranes of animals with and without FCA inflammation. These results suggest that an inflammatory stimulus can increase the number of MOR on DRG membranes but does not change the affinity of MOR to opioids.
Axonal transport has been demonstrated for various neuroreceptors, including MOR in peripheral nerves (Laduron and Castel, 1990). We performed a set of experiments to show that an increase in MOR specific binding sites in the DRG is accompanied by an increase of the axonal transport of MOR to the periphery. Almost no MOR binding sites were detectable in the unligated sciatic nerve preparations. However, ligation of the sciatic nerve in the absence of inflammation resulted in an accumulation of MOR at the proximal part of the ligation, indicating an anterograde transport from the DRG to the noninflamed paw. In rats with inflamed paws, a significant increase in MOR-specific binding sites at the proximal part of the ligation was detected. Together with our binding data in DRG membranes, this indicates that inflammation can cause an increase in MOR-specific binding sites in both DRG and the sciatic nerve. This strongly suggests that an increase in MOR levels in the DRG would also be seen in the peripheral portions of the nerve after axonal transport to the periphery. This also confirms previous neuroanatomical evidence for the existence of MOR in the sciatic nerve and peripheral cutaneous nerve fibers (Hassan et al., 1993). It was shown recently that an increase in MOR number might be related to mediators of inflammation (e.g., IL-4, tumor necrosis factor) (Kraus et al., 2001).
Our immunohistochemical results confirmed an up-regulation of MOR after FCA inflammation. However, this increase was restricted to DRG on the inflamed side and was not detected in DRG on the contralateral side. An increase in Bmax (90%, as determined with [3H]DAMGO binding) and an increase in the number of MOR-positive DRG neurons (45%, as determined in our immunohistochemical studies) indicates that this up-regulation is caused by an increase in the number and density of MOR-positive neurons. Consistent with previous studies, under both normal and inflammatory conditions, MOR immunoreactive DRG neurons were mainly of small diameter, suggesting that the increase in MOR immunoreactivity is mainly restricted to nociceptive neurons (Ji et al., 1995; Mousa et al., 2001).
An important finding of the present study is that the efficacy of DAMGO-stimulated G-protein activation increased significantly in animals with FCA inflammation. This observation might explain why the application of exogenous opioids in peripheral antinociception is enhanced under inflammatory conditions (Stein et al., 2001). In addition, the mechanisms of μ-opioid agonist efficacy and inflammation were investigated using agonist-stimulated [35S]GTPγS binding. The advantages and disadvantages to this measurements have been described earlier (Newman-Tancredi et al., 1997a; Selley et al., 1997): Scatchard analysis of [35S]GTPγS binding measures the competition of a radiolabeled ligand ([35S]GTPγS) for a nonlabeled ligand (GDP) only under nonequilibrium conditions. At equilibrium, the [35S]GTPγS would displace as much of the GDP as possible, and no agonist-stimulated binding could be observed. This assay is therefore not quantitatively accurate in the sense that a given Bmax G protein represents the exact maximal number of G proteins; however, relative comparisons between inflamed and noninflamed tissue are possible. We found that the number of [35S]GTPγS binding sites that can be occupied after DAMGO stimulation increased 1.6-fold (from 266 to 425 fmol/mg of protein) in animals with FCA inflammation. The amplification factor (or number of G-proteins activated per MOR) decreased, suggesting that an increase in MOR during inflammation is not proportional with an increase in G-protein coupling. In addition, we found that DAMGO-occupied receptors on DRG membranes in animals with and without FCA inflammation revealed no differences in the DAMGO-induced guanine nucleotide affinities (measured as the Kd Gprotein value of [35S]GTPγS binding). Taken together, these results suggest that inflammation can cause an increase in receptor density per cell, which results in an increased number of activated G-proteins. Consistent with this notion, it was shown earlier that a drop in MOR in SH-SY5Y cells and in cannabinoid receptors in certain areas in the brain could cause an equivalent drop in the level of [35S]GTPγS binding (Sim et al., 1996; Remmers et al., 2000; Selley et al., 2001). These results appear in contrast to other studies in the 5-HT1A system, where an increase in receptor density did not change the activated G-protein number (Newman-Tancredi et al., 1997b). However, those studies were performed in highly expressing Chinese hamster ovary cells in which the number of G-proteins might be limited in comparison with the number of receptors. DRG membranes of noninflamed animals expressed 25 fmol/mg of protein MOR, which is clearly below Chinese hamster ovary cells expressing 1600 fmol/mg 5-HT1A receptors (Newman-Tancredi et al., 1997b). Therefore, an increase of MOR in animals with inflammation might explain the observed increase in G-protein activation. Although animals with FCA inflammation exhibited a 90% increase in expression of MOR, maximal stimulation of [35S]GTPγS binding by DAMGO was only 48% higher and therefore did not increase proportionally with receptor levels.
Suprisingly, BUP did not induce any detectable G-protein coupling in DRG membranes of animals without FCA inflammation. In contrast, BUP stimulated G-protein coupling in DRG membranes of animals with FCA inflammation. The extent of stimulation of [35S]GTPγS binding (EC50) in animals with FCA inflammation was lower for BUP than for DAMGO, as in many other in vitro (Huang et al., 2001; Zaki et al., 2000) and in vivo (Gopal et al., 2002; Traynor et al., 2002) systems. It has been suggested previously that a given biological effect requires the switching on (or off) of a certain number of effector molecules (Chavkin and Goldstein, 1984). Due to stoichiometric interactions between receptors and effectors, it might be that the partial agonist BUP could not activate a detectable amount of G-proteins in animals without FCA inflammation. However, because of the increase of MOR in DRG membranes of animals with FCA inflammation, the number of receptors appears sufficient to activate the pool of G-proteins.
Scatchard analysis of basal and agonist-stimulated [35S]GTPγS binding confirmed that BUP (low efficacy partial agonist) produced a lower affinity GTP-binding state in DRG of animals with inflammation (presumably in the guanine nucleotide binding site of μ receptor-coupled G protein α subunits) than DAMGO (higher efficacy agonist). Partial agonists do not fully shift the affinity of the G-proteins into a GTP-preferring state (Selley et al., 1998; Traynor et al., 2002), and this was clearly evident for BUP in DRG membranes of animals with FCA inflammation: Agonist-induced guanine nucleotide affinity for BUP (Kd Gprotein, 1.8 nM) compared to DAMGO (Kd Gprotein, 0.8 nM) was different and the catalytic activation of G proteins, as measured by the agonist-stimulated Bmax Gprotein of [35S]GTPγS binding, was lower for BUP (Bmax Gprotein, 145fmol/mg) than for DAMGO (Bmax Gprotein, 425fmol/mg).
It should be noted that an increase in maximal [35S]GTPγS binding as a function of receptor density does not necessarily result in a similar increase in the magnitude of a downstream response (Law et al., 1994; Prather et al., 1994). Therefore, we performed a set of behavioral experiments to test whether the results obtained with [35S]GTPγS binding and BUP showed functional consequences in antinociception.
We found that intraplantar injection of the partial agonist BUP in noninflamed paws did not change paw pressure thresholds compared with intraplantar saline injections. In contrast, local BUP injections in animals with inflamed paws produced PPT elevations (i.e., antinociception). These results indicate that BUP can act as an effective peripheral antinociceptive agent only in the presence of inflammation. The MOR-selective antagonists NLX and CTOP could block this antinociceptive effect of BUP, which clearly indicates that BUP mediates its antinociceptive activity through MOR. The contralateral paws showed no changes in PPT, suggesting that low doses of BUP induce only peripheral, not central, opioid analgesic effects. It was already shown earlier that opioid full agonists (e.g., fentanyl) produce dose-dependent elevations of PPT in animals with and without FCA inflammation; however, antinociception is smaller in noninflamed hindpaws compared with inflamed hindpaws (Antonijevic et al., 1995). There are many steps between MOR binding and antinociception (e.g., inhibition of cAMP, inhibition of calcium channel conductance) that can modulate the downstream responses. However, the observed antinociceptive action after BUP injection only in animals with FCA inflammation might be related to the lack of G-protein coupling observed in DRG membranes. This supports the hypothesis that in animals without FCA inflammation, there are not enough receptors present to develop opioid analgesia by the partial MOR agonist BUP.
In conclusion, inflammation is associated with an up-regulation of MOR, mainly in small-sized primary afferent neurons, and enhances the efficacy of full and partial MOR agonists in G-protein coupling. These changes might contribute to the occurrence of peripheral antinociceptive effects of the partial MOR agonist BUP, which are not present under normal conditions. These adaptive changes underscore the important differences in opioid receptor binding and signaling between normal and inflamed tissue. They strongly indicate that clinical studies testing peripherally active opioids are much more likely to yield positive results when they are performed in inflammatory painful conditions.
Footnotes
-
This work was supported by “Klinische Forschergruppe Grant from the Deutsche Forschungsgemeinschaft KFO 100/1”.
-
This work was previously presented at the International Narcotics Research Conference 2002; 2002 Jul 9–14; Asilomar, California. Abstract 30 (Available on the web at http://www.inrcworld.org/2002meeting/INRCFina.pdf).
-
ABBREVIATIONS: FCA, Freund's complete adjuvant; MOR, μ-opioid receptor; DRG, dorsal root ganglion; GTPγS, guanosine-5′-O-(γ-thio)triphosphate; BUP, buprenorphine hydrochloride; DAMGO, [d-Ala2,N-Me-Phe4, Gly5-ol]-enkephalin; CTOP, d-Phe-Cys-Tyr-d-Trp-Orn-Thr-Pen-Thr-NH2; NLX, naloxone; PPT, paw pressure threshold; IR, immunoreactivity.
- Received November 18, 2002.
- Accepted April 10, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}