


Abstract
The purposes of the present studies were to examine the androgen receptor (AR) binding ability and in vitro functional activity of multiple series of nonsteroidal compounds derived from known antiandrogen pharmacophores and to investigate the structure-activity relationships (SARs) of these nonsteroidal compounds. The AR binding properties of sixty-five nonsteroidal compounds were assessed by a radioligand competitive binding assay with the use of cytosolic AR prepared from rat prostates. The AR agonist and antagonist activities of high-affinity ligands were determined by the ability of the ligand to regulate AR-mediated transcriptional activation in cultured CV-1 cells, using a cotransfection assay. Nonsteroidal compounds with diverse structural features demonstrated a wide range of binding affinity for the AR. Ten compounds, mainly from the bicalutamide-related series, showed a binding affinity superior to the structural pharmacophore from which they were derived. Several SARs regarding nonsteroidal AR binding were revealed from the binding data, including stereoisomeric conformation, steric effect, and electronic effect. The functional activity of high-affinity ligands ranged from antagonist to full agonist for the AR. Several structural features were found to be determinative of agonist and antagonist activities. The nonsteroidal AR agonists identified from the present studies provided a pool of candidates for further development of selective androgen receptor modulators (SARMs) for androgen therapy. Also, these studies uncovered or confirmed numerous important SARs governing AR binding and functional properties by nonsteroidal molecules, which would be valuable in the future structural optimization of SARMs.
The role of androgens in the development and maintenance of male sexual phenotype is mediated by the androgen receptor (AR), a member of the steroid/thyroid hormone receptor superfamily (Tsai and O'Malley, 1994;Zhou et al., 1994). Upon androgen binding, AR undergoes a conformational change, binds to specific DNA sequences called androgen response elements, and modulates the transcription of target genes (Zhou et al., 1994). For decades, AR has been a target of drug development, aiming at the therapy of diseases caused by altered androgen levels/responsiveness or the improvement of physical performance and regulation of male fertility.
Chemicals that modulate the transcriptional activity of AR can be divided into two structural (steroidal and nonsteroidal) and two functional (androgenic and antiandrogenic) classes. Steroidal androgens, mainly testosterone and its derivatives, have been used clinically as replacement therapy for androgen-deficiency (Wu, 1992;Bagatell and Bremner, 1996). Antiandrogens are used to counteract the undesirable actions of excessive androgens (e.g., to treat acne, hirsutism, male-pattern baldness, and androgen-dependent prostatic hyperplasia and carcinoma) (Neumann, 1982; McLeod, 1993). Nonsteroidal antiandrogens, such as flutamide (Eulexin), nilutamide (Anandron), and bicalutamide (Casodex), are often referred to as “pure antiandrogens” because they bind exclusively to the AR and, therefore, are devoid of antigonadotropic, antiestrogenic, and progestational effects. These agents are advantageous over steroidal antiandrogens (e.g., megestrol acetate, cyproterone acetate) in terms of specificity, selectivity and pharmacokinetic properties (Neri et al., 1979; Cockshott et al., 1990; Teutsch et al., 1994).
In recent years, there has been growing interest in the development of nonsteroidal modulators for steroid hormone receptors as therapeutic agents. In addition to the above-discussed nonsteroidal antiandrogens, selective estrogen receptor modulators (SERMs) and nonsteroidal modulators for progesterone receptor have been successfully developed (Hamann et al., 1998; Mitlak and Cohen, 1999; Weryha et al., 1999; Zhi et al., 1998, 2000). These nonsteroidal ligands are known for their better receptor selectivity than steroidal ligands, and are more flexible in structural modification for optimal physicochemical, pharmacokinetic, and pharmacologic properties. More importantly, with these nonsteroidal ligands, it is possible to achieve tissue-selective actions and thus to generate agents with diverse activity profiles meeting specific therapeutic needs. SERMs, for example, are well known to demonstrate tissue-selectivity (Mitlak and Cohen, 1999; Weryha et al., 1999). SERMs, such as tamoxifen and raloxifene, are nonsteroidal estrogen receptor (ER) ligands that manifest distinctive agonist and antagonist actions in various target tissues. Both tamoxifen and raloxifene are ER antagonists in breast but agonists in bone. However, unlike tamoxifen, raloxifene has no ER agonist activity in the uterus.
Whereas nonsteroidal antiandrogens have been used clinically for many years, nonsteroidal androgens were only recently conceptualized. In the search for AR affinity ligands, our laboratories discovered a group of nonsteroidal androgens that are electrophilic derivatives of bicalutamide and hydroxyflutamide (Dalton et al., 1998). Also, several analogs of quinoline-based AR antagonists were reported by other research groups to have AR agonist activity (Edwards et al., 1998,1999; Hamann et al., 1999; Higuchi et al., 1999; Zhi et al., 1999). These studies marked the emergence of a novel category of pharmacological agents with potential applications in androgen therapy. The discovery of nonsteroidal androgens not only provides an opportunity to identify agents with superior therapeutic index and pharmacokinetic profiles to steroidal androgens but also implicates the possibility to obtain tissue-selective AR modulators (SARMs), the counterpart of SERMs.
Although nonsteroidal androgens bearing different pharmacophores have been reported, the general structural elements of nonsteroidal ligands that lead to optimal agonist activity remain poorly defined. Systematic studies to explore the structure-activity relationships (SARs) of nonsteroidal ligands for AR binding and agonist activity are of crucial importance for the optimization of chemical structures for maximal functional activity and for the ultimate development of SARMs. Toward a better understanding of the SARs, our laboratories designed and synthesized several series of nonsteroidal compounds that incorporated a variety of structural features known or unknown to influence the ligand-AR interaction and evaluated their biological properties. We present herein the results of our AR binding and in vitro functional activity studies with these synthetic molecules and the SARs revealed by these compounds.
Materials and Methods
Organic Synthesis
Compounds were prepared in our laboratories and the purities were greater than 99% (He et al., 2002). The structures of synthesized compounds were confirmed using elemental analyses and spectroscopic data (1H and 13C NMR, mass spectroscopy, and infrared spectroscopy).
Chemicals and Animals
[17α-methyl-3H]Mibolerone ([3H]MIB, 84 Ci/mmol) and unlabeled MIB were purchased from PerkinElmer Life Sciences (Boston, MA). Triamicinolone acetonide, phenylmethylsulfonyl fluoride (PMSF), Tris base, sodium molybdate, dihydrotestosterone (DHT),o-nitrophenyl-β-d-galactopyranoside, and ATP were purchased from Sigma Chemical Co. (St. Louis, MO). Hydroxyapatite (HAP) was purchased from Bio-Rad Laboratories (Hercules, CA). EcoLite (+) scintillation cocktail was purchased from ICN Research Products Division (Costa Mesa, CA). Ethyl alcohol (USP grade) was purchased from AAPER Alcohol and Chemical Company (Shelbyville, KY). Minimal essential medium (MEM), Dulbecco's modified Eagle's medium (DMEM), penicillin-streptomycin, trypsin-EDTA, and LipofectAMINE reagent were purchased from Invitrogen (Carlsbad, CA). Fetal bovine serum (FBS) was obtained from Atlanta Biologicals, Inc. (Norcross, GA). Adult male Sprague-Dawley rats, weighing approximately 250 g, were purchased from Harlan Biosciences (Indianapolis, IN).
Buffers
Homogenization buffer contained 10 mM Tris, 1.5 mM disodium EDTA, 0.25 M sucrose, 10 mM sodium molybdate, and 1 mM PMSF and was adjusted to pH 7.4. PMSF was prepared as a stock solution of 200 mM in ethanol and added to other components immediately before use. HAP wash buffer contained 50 mM Tris and 1 mM KH2PO4 and was adjusted to pH 7.4.
β-Galactosidase assay buffer was 200 mM sodium phosphate buffer, pH 7.3, containing 2 mM MgCl2, 100 mM β-mercaptoethanol, and 1.33 mg/mlo-nitrophenyl-β-d-galactopyranoside. Luciferase assay buffer consisted of 25 mM glycylglycine, 15 mM MgCl2, 5 mM ATP, and 0.5 mg/ml bovine serum albumin, and was adjusted to pH 7.8 with 1 M sodium hydroxide.
Preparation of Rat Prostate Cytosolic AR
Cytosolic AR was prepared from ventral prostates of castrated male Sprague-Dawley rats as described previously (Mukherjee et al., 1999). Briefly, rats were surgically castrated via a scrotal incision before the removal of prostates. One day after castration, rats were anesthetized with a mixture of ketamine/xylazine (87:13; v/v) at 1 ml/kg body weight. Ventral prostates were excised, weighed, and immersed immediately in ice-cold homogenization buffer. The prostates were minced with scissors and homogenized (Model Pro 200 homogenizer; Pro Scientific, Monroe, CT) in homogenization buffer (1:2, w/v). The homogenate was then centrifuged at 114,000g, 0°C for 1 h in an ultracentrifuge (Model L8-M; Beckman Instruments Inc., Palo Alto, CA). The supernatant (cytosol), containing AR proteins, was collected and stored at −80°C until use.
AR Competitive Binding Assay
The AR binding affinity of synthesized nonsteroidal compounds was determined using a radioligand competitive binding assay. An aliquot of AR cytosol preparation (50 μl) was incubated with a saturating concentration (1 nM) of [3H]MIB and 1 μM of triamcinolone acetonide at 4°C for 18 h in the absence or presence of increasing concentrations of the compound of interest (10 different concentrations ranging from 10−1nM to 104 nM). MIB is a synthetic high-affinity ligand for the AR. Triamcinolone acetonide (1 μM) was included in the incubate to block the interaction of [3H]MIB with glucocorticoid and progesterone receptors. Nonspecific binding of [3H]MIB was determined separately by adding an excess of unlabeled MIB (1000 nM) to the incubate. After incubation, the protein-bound radioactivity was separated from free radioactivity by HAP precipitation. HAP was prepared as a slurry in 50 mM Tris, pH 7.2 (1:15, w/v) after being washed twice with HAP wash buffer (1:15, w/v). An aliquot (500 μl) of HAP slurry was added to the incubate and gently agitated for 15 min at 4°C. The mixture was centrifuged at 2000g, 4°C for 1 min, and the HAP pellet was obtained and washed three times with 1 ml of 50 mM Tris, pH 7.2. The bound radioactivity was then extracted from HAP by incubating the HAP pellet with 1 ml of ethanol at room temperature for 1 h. After centrifugation at 2500g for 1 min, 0.8 ml of the ethanolic supernatant was added to 5 ml of scintillation cocktail. The radioactivity was counted in a Beckman LS6800 liquid scintillation counter (Beckman Coulter, Fullerton, CA).
Cell Culture
Monkey kidney fibroblast-like CV-1 cells were obtained from American Type Culture Collection (Manassas, VA). The cells were grown at 37°C in a humidified atmosphere with 5% carbon dioxide, and maintained in minimal essential medium supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin.
Cotransfection and Enzyme Assays
The in vitro functional activities of nonsteroidal ligands, as assessed by the ability of each ligand to induce or repress AR-mediated transcriptional activation of the luciferase reporter gene, were examined in transiently transfected CV-1 cells. One day before transfection, CV-1 cells were seeded into DMEM supplemented with 10% fetal bovine serum at a density of 2 × 105cells/well in 12-well tissue culture plates. For the following steps, DMEM without phenol red was used. Transient transfection of plated cells was carried out in serum-free medium using LipofectAMINE according to the manufacturer's instructions. Cells in each well were transfected with 50 ng of a human AR expression construct (pCMVhAR; generously provided by Dr. Donald J. Tindall, Mayo Clinic and Mayo Foundation, Rochester, MN), 1 μg of an androgen-dependent luciferase reporter construct (pMMTV-Luc; generously provided by Dr. Ronald Evans at The Salk Institute, San Diego, CA), and 1 μg of a β-galactosidase expression construct (pSV-β-galactosidase; Promega Corporation, Madison, WI) for constitutive expression of β-galactosidase using 6 μl of LipofectAMINE. After 10 h of transfection, cells were washed once with DMEM, and then recovered in fresh DMEM supplemented with 0.2% FBS for 10 to 12 h before the start of treatments.
To determine the AR agonist activity, the transfected cells were treated with increasing concentrations of the ligand of interest. To determine the AR antagonist activity, the cells were treated simultaneously with increasing concentrations of the ligand of interest and 1 nM DHT. Controls, where cells were treated with 1 nM DHT alone or vehicle alone, were included in each experiment. To measure any AR-independent effect, a parallel experiment was also performed in which cells cotransfected with pMMTV-Luc and pSV-β-galactosidase only were treated with 500 nM of the ligand of interest. All drugs were initially dissolved in 100% ethanol, and then serially diluted to the desired concentration using DMEM containing 0.2% FBS. The volume of ethanol in the final solutions was ≤0.05%. The final concentrations of the compound of interest were 1, 10, 100, and 500 nM. Drug treatments continued for 48 h, during which all drug-containing solutions were replaced at a 24-h interval to minimize the effect of possible chemical degradation.
After treatment, the cells were washed twice with phosphate-buffered saline and lysed with 200 μl/well of 1× Reporter Lysis Buffer (Promega Corporation) at room temperature for 30 min. Cell lysates were placed in 1.5 ml of polypropylene centrifuge tubes and centrifuged at 12,000g and 4°C for 2 min. For β-galactosidase assays, an aliquot (50 μl) of supernatant from each cell extract was transferred to a 96-well plate and incubated with 50 μl of β-galactosidase assay buffer at 37°C for 3 h. An aliquot (150 μl) of 1 M Na2CO3 was then added to each incubate to stop the enzyme reaction, and absorbance at 414 nm was measured using a 96-well plate reader (Titertek Multiskan MCC/340; Labsystems Inc., Franklin, MA). For luciferase assays, an aliquot (100 μl) of supernatant from each cell extract was added with an equal volume of luciferase assay buffer. Luminescence was then measured in an automated luminometer (AutoLumat LB953; PerkinElmer Wallac Inc., Gaithersburg, MD) immediately after injecting 100 μl of 1 mM beetle luciferin to each sample.
Data Analyses
AR Binding.
The specific binding of [3H]MIB at each concentration of the compound of interest (B) was obtained after subtracting the nonspecific binding of [3H]MIB, and expressed as the percentage of the specific binding in the absence of the compound of interest (B0). The concentration of compound that reduced the specific binding of [3H]MIB (B0) by 50% (IC50) was determined by computer-fitting the data to the following equation using WinNonlin (Pharsight Corporation, Mountain View, CA): B= B 0 × [1 −C/(IC50 + C)], whereC was the concentration of the compound of interest.
The equilibrium binding constant (K i) of the compound of interest was calculated byK i =K d × IC50/(K d +L), where K d was the equilibrium dissociation constant of [3H]MIB (0.19 ± 0.01 nM; determined in preliminary experiments as described previously) (Mukherjee et al., 1996), and L was the concentration of [3H]MIB used in the experiment (1 nM). Statistical analyses were performed using single factor analysis of variance, with p values less than 0.05 being considered as statistically significant differences.
Transcriptional Activation.
Transcriptional activation in each well was calculated as the ratio of luciferase activity to β-galactosidase activity to normalize the variance in cell number and transfection efficiency. Transcriptional activation induced by each ligand of interest, in the absence or presence of 1 nM DHT, was expressed as a percentage of that induced by 1 nM DHT. The efficacy of agonist activity for a ligand of interest was the maximal percentage of transcriptional activation induced by that ligand in the absence of DHT. All experiments were performed in triplicate or greater, and data were expressed as the mean ± S.D. in representative experiments.
Results
Androgen Receptor Binding of Nonsteroidal Compounds.
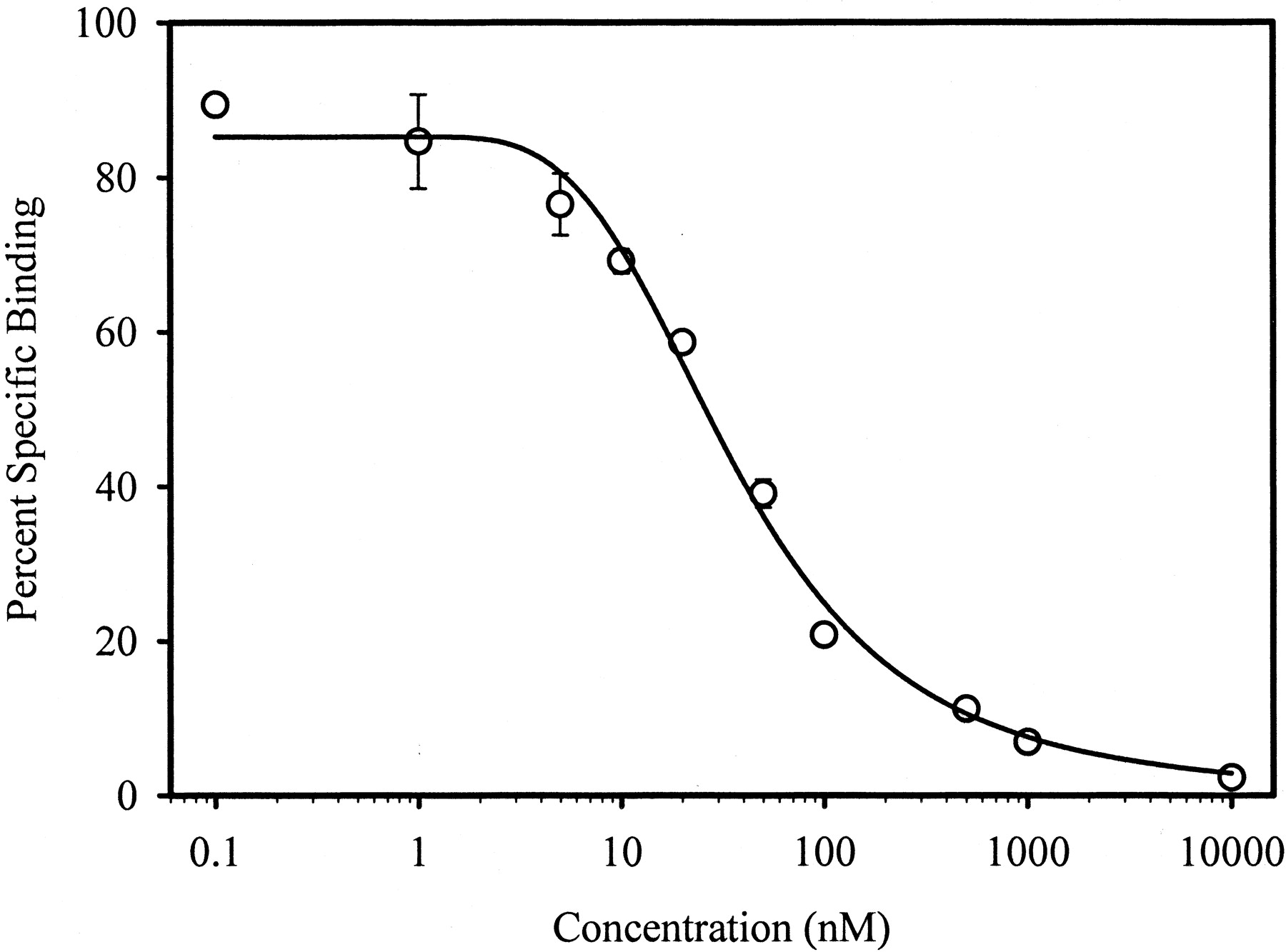
We designed and synthesized several series of nonsteroidal compounds, based on the SARs for nonsteroidal AR binding obtained from the literature and previous studies in our laboratories (Glen et al., 1986;Tucker et al., 1988; Morris et al., 1991; Mukherjee et al., 1996, 1999;Dalton et al., 1998; Kirkovsky et al., 2000). Bicalutamide, hydroxyflutamide, or nilutamide pharmacophores (Fig.1) were investigated. The AR binding affinities of these synthetic molecules, reported asK i values, were determined by a radioligand competitive binding assay using rat prostates as an AR source. A representative binding displacement curve (withR-5) is shown in Fig. 2. The lower the K i value, the greater the receptor binding affinity.

Structures of nonsteroidal antiandrogen pharmacophores.
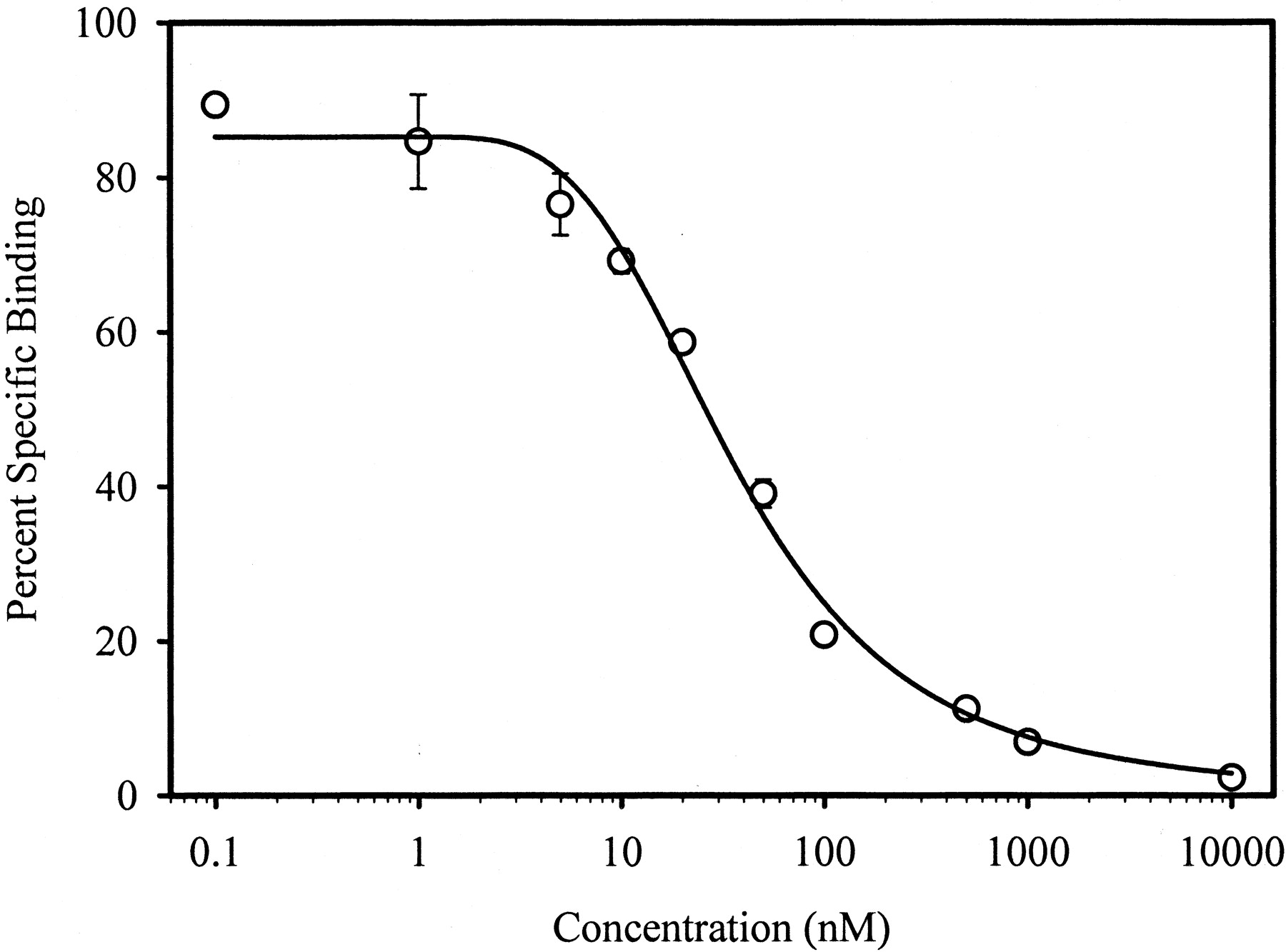
A representative AR binding displacement curve withR-5. Rat prostate cytosolic AR was incubated with 1 nM of [3H]MIB, 1 μM of triamcinolone acetonide at 4°C for 18 h, in the presence of increasing concentrations of testing ligand or in the absence of testing ligand, as described underMaterials and Methods. Nonspecific binding was determined separately by including 1000 nM of unlabeled MIB in the incubate. Each data point (○) represents the mean ± S.D. in a representative experiment of triplicate measurements. The smooth line is the fitted curve by the inhibitory effect sigmoidE max model.
The binding results of a series of bicalutamide derivatives with modifications in the aromatic B-ring or the X-linkage are shown in Table 1. Previous studies from our laboratories found that bicalutamide displayed enantioselective AR binding; the binding affinity of the R-isomer was 30-fold higher than that of the S-isomer (Mukherjee et al., 1996). Thus, all compounds in this series were prepared as pure optical isomers (R or S). Consistent with our previous finding, all R-isomers showed at least 4-fold higher AR binding affinity (lower K i values) than their corresponding S-isomers. Because theS-isomers are essentially of no use for our purposes, the following discussion of SARs for binding is focused entirely onR-isomers. In this series, R-5, R-12, and R-13 exhibited higher AR binding affinity than the lead compound (R)-bicalutamide (R-1; p< 0.05). AR binding affinity in this series of compounds was influenced by the X-linkage group and B-ring substituents. When thepara-substitution in the aromatic B-ring was an amino group, the sulfone (X = SO2, R-4) showed greater affinity than the sulfide (X = S, R-2), whereas the sulfoxide (X = SO, R-3) had no binding affinity. However, when the nitrogen molecule in the para-amino group was further substituted, sulfides demonstrated higher binding than sulfones (compare R-5 with R-7, andR-12 with R-13). In compounds with the same X-linkage, the B-ring substituent clearly played an important role in AR binding. The overall effect seemed to be determined by a delicate balance of factors, including nature, size, and position of the substituent. Introduction of a chloroacetamido group at thepara- position (R-12 and R-13) resulted in the highest binding affinity in each of the sulfide and sulfone sets of compounds (compare R-12 with R-5,R-8, R-10, and R-9; R-13 with R-1, R-4, R-6, andR-16), indicating a beneficial role of an electrophilic group at this position in AR binding. However, the beneficial effect of an electrophilic group might also be offset by the negative effect of its bulky size on receptor binding. For instance, R-10, which bears a bromoacetamido group at the para-position, showed lower binding affinity than R-5 (with apara-acetamido substitution) and R-8 (with apara-propionamido substitution), probably because of the bulkiness of the bromoacetamido moiety. However, it is also important to note that the bromoacetamido group is less electrophilic than the chloroacetamido group. Excluding those compounds that bear strong electrophilic substituents and R-4 (with an amino group), there was a general observation that an increase in the size of the substituent above a yet undefined limit decreased the binding affinity. This effect can be illustrated by comparison of the binding affinities of R-5, R-6, R-8, and R-9 in the sulfides and R-7 and R-16 in the sulfones. In addition, as shown by comparisons of R-10 withR-11 and R-13 with R-14,para-substitution of the B-ring was superior tometa-substitution in terms of AR binding. Tucker et al. (1988) proposed that the tertiary hydroxy group was involved in direct interaction with the AR. This hypothesis was corroborated by our observation that introduction of an acetyl group to that hydroxy moiety in R-17 abolished the receptor binding of R-9.
AR binding affinity of bicalutamide B-ring derivatives
We previously reported that a nitro group, instead of a cyano group, in the para-position of the anilide ring (corresponding to the aromatic A-ring in bicalutamide derivatives) promoted AR binding and agonist activity of some hydroxyflutamide analogs (Dalton et al., 1998). We tested whether the same SAR could be extended to our bicalutamide derivatives. Thus, the second series of bicalutamide derivatives, with a para-nitro group in the A-ring and various substituents in the para-position of the B-ring or a modified X-linkage group, was evaluated for their AR binding affinity (Table 2). Confirming the previous results, these compounds showed enantioselective binding. AllR-isomers had receptor affinity at least 9-fold higher than their corresponding S-isomers. As far as theR-isomers were concerned, the introduction of a nitro group in the A-ring generally improved, or at least maintained, the binding affinity compared with analogs bearing the cyano moiety at this position (compare R-18 with R-2, R-19 with R-5, R-20 with R-7, andR-24 with R-13). A single exception to this trend, where the nitro substitution slightly decreased the receptor binding (compare R-23 with R-12), was noted. Among the present series, a total of five R-isomers (R-19, R-21, R-22, R-23, and R-24) were superior to R-bicalutamide in terms of AR binding (p < 0.05). Sulfides in most cases showed at least a 2-fold higher binding affinity than corresponding sulfones (compare R-19 with R-20, R-21 with R-22, and R-23 with R-24). However, this relationship was reversed when the B-ring substituent was a NHSO2CH3 group, where the binding affinity of R-26 (sulfone) was 3-fold higher than that of R-25 (sulfide). These results further indicated that the B-ring substituents largely determined the effect of the X-linkage on AR binding. A trifluoroacetamido, chloroacetamido, or acetamido substituent at the para-position of B-ring led to superior AR binding, regardless of the X-linkage.
AR binding affinity of bicalutamide A- and B-ring derivatives
Morris et al. (1991) showed that hydroxyflutamide analogs require strong hydrogen bond donor ability of the tertiary hydroxy group for efficient AR binding. To examine the effect of an increased hydrogen bond donor ability of the tertiary hydroxy group on the binding affinity of our bicalutamide analogs, we synthesized and evaluated a group of compounds bearing a trifluoromethyl group connected to the chiral carbon (Table 3). These compounds were prepared as racemic mixtures of R- andS-isomers. Among this series, compounds 29, 30, and 31 demonstrated very high binding affinities for the AR, each similar to the binding affinity of the corresponding R-isomer in the previous series, in which a methyl group was connected to the chiral carbon (i.e., R-19, R-23, and R-24, respectively). Considering that the S-isomers generally have poor receptor binding, the binding affinities of pureR-isomers in these racemic compounds would thus be expected to be even higher. Although a definitive conclusion can be made only by directly comparing binding affinities of parallel pure isomers, it seemed that the replacement of methyl group with trifluoromethyl group improved AR binding affinity. Moreover, the introduction of an electron-withdrawing nitro group into the B-ring (compounds 27 and 28) was apparently not beneficial for AR binding in this series.
AR binding affinity of racemic bicalutamide derivatives
Our laboratories reported previously that hydroxyflutamide derivatives 32 and 33 (Table 4) were high- affinity AR ligands, and that ligand 33 was a potent AR agonist (Dalton et al., 1998). We thus further evaluated the effects of various combinations of electron-withdrawing substitutions in the aniline ring on the binding affinity (compounds 34 to 39). As shown in Table 4, different substitution patterns led to compounds with a wide range of binding affinities. However, none of the newly synthesized compounds showed higher binding than ligands 32 and 33. Nitro substitution in the 2-position of the aniline ring resulted in decreased AR binding (compare 34 with 35). The 3,5-dinitro substitution (compound 37) was superior to the 2,4-dinitro substitution (compound 35) in terms of AR binding. The binding affinity was improved with the presence of electron-withdrawing groups in the para- andmeta-positions of the aniline ring (compare 33 with 34 and 32-33 with 38-39). We also determined the binding affinities of a group of compounds (compounds 40–44) that differed from hydroxyflutamide in the aromatic ring substituents. None of these compounds bound to the AR, suggesting that bisubstitution in this ring was essential for high AR binding affinity.
AR binding affinity of hydroxyflutamide derivatives
Recently, a series of nonsteroidal compounds based on tricyclic quinolinones (Fig. 3A) were shown to produce AR agonist activity (Edwards et al., 1998; Hamann et al., 1999;Higuchi et al., 1999; Zhi et al., 1999). Because the quinolinone skeleton is structurally different from the previously discussed pharmacophores, we were interested to determine whether hybrids of these two sets of lead structures bound to the AR and elicited agonist activity. Meanwhile, Ekena et al. (1998) showed that a proton-accepting group in the ligand is important for AR binding. We hypothesized, therefore, that replacement of the NH in the aromatic ring system (carbostyril ring, Fig. 3B) of quinolinones with an oxygen molecule, a proton acceptor in hydrogen bonding, might improve the receptor binding. The change from NH to oxygen would convert the carbostyril ring to a coumarin ring (Fig. 3C). This rationale led to our design and synthesis of compounds bearing the coumarin ring and segments from bicalutamide, hydroxyflutamide, or nilutamide analogs. To our surprise, the introduction of a coumarin ring into the bicalutamide, hydroxyflutamide, or nilutamide derivatives was detrimental to AR binding affinity. As shown in Table 5, in the series of bicalutamide analogs whose aromatic A-ring was replaced with a coumarin ring, the highest binding affinity was observed for compound 47 (K i = 80 ± 16 nM). The replacement of aromatic A-ring with coumarin resulted in decreased binding affinities (compare compounds 47, 51, and 52 withR-1, R-2, and R-5, respectively), and the relative magnitudes of reduction were related to thepara-substituent in the B-ring. Particularly, the largest drop in binding was observed when there was a para-acetamido group in the B-ring (compare 52 with R-5). Data in Table6 showed that compounds combining a coumarin ring with segments from hydroxyflutamide or nilutamide derivatives also exhibited poor AR binding. Most of the compounds in the latter series (series B) had no affinity for the AR.

Chemical structures of the tricyclic quinolinone skeleton (A), carbostyril ring (B), and coumarin ring (C).
AR binding affinity of bicalutamide derivatives bearing coumarin ring
AR binding affinity of hydroxyflutamide and niluramide derivatives bearing coumarin ring
In Vitro Functional Activity of Nonsteroidal AR Ligands.
Previous experiments in our laboratories determined that the potency of DHT (i.e., the lowest DHT concentration that produced the maximal transcriptional activation of the AR) was 1 nM (Dalton et al., 1998). Therefore, in the present study, the transcriptional activation induced by this concentration of DHT was set as 100%, and used as the reference for quantifying the agonist and antagonist activity of other testing ligands.
According to their pharmacological activity, receptor ligands can be classified into full agonists, partial agonists, and antagonists. For convenience of comparison, we adopted the following arbitrary standards to define our AR ligands: 1) full AR agonists refer to those ligands that induced a level of transcriptional activation not significantly lower than that of 1 nM DHT in the agonist assay; 2) partial AR agonists refer to those ligands that induced at least 10% of transcriptional activation of 1 nM DHT but significantly lower than that of 1 nM DHT in the agonist assay; 3) AR antagonists refer to those compounds that induced less than 10% of transcriptional activation of 1 nM DHT in the agonist assay but significantly suppressed the DHT-induced transcriptional activation in the antagonist assay.
Figure 4 shows the AR agonist (A) and antagonist (B) activities of a series of (R)-bicalutamide derivatives with modifications in the B-ring or the X-linkage.R-2, with a para-amino group in the B-ring, demonstrated the lowest AR binding affinity (K i = 91 ± 16 nM) in this series of compounds in previous experiments. This compound was not able to stimulate AR-mediated transcriptional activation with concentrations up to 500 nM in the agonist assay. However, R-2 significantly inhibited DHT-induced transcriptional activation at higher concentrations. Therefore, R-2 was classified as an AR antagonist. Bearing an N-alkylamido substituent in thepara-amine of B-ring, all other ligands in this series induced the expression of luciferase in a concentration-dependent fashion in the agonist assay. The AR must mediate the activity of these ligands, because 500 nM of each ligand had no effect on luciferase expression when there was no AR expression plasmid transfected in the cells. These results clearly demonstrated that R-5,R-7, R-8, and R-13 function as AR agonists in the mammalian cell context. R-5 (acetothiolutamide), the ligand with the highest AR binding affinity (K i = 4.9 ± 0.2 nM) in this series, exhibited the most potent and efficacious agonist activity. Nevertheless, R-7, which demonstrated higher AR binding affinity than R-8, was inferior to R-8 in agonist activity, indicating that higher AR binding affinity was not necessarily a predictor of greater AR agonist activity. Interestingly, a change of the linkage group from sulfur in R-5 to sulfone in R-7 switched a full agonist to a partial agonist. In accordance with previously identified SARs for AR binding, an increase in the size of R1 resulted in decreased agonist activity (compareR-5 with R-8). However, this SAR was not true when a strong electrophilic function was introduced at thepara-position of B-ring (compare R-7 withR-13). It is also important to note that higher concentrations of R-5 and R-13 resulted in a lesser degree of AR-mediated transcriptional activation when coincubated with 1 nM DHT during our studies for antagonist activity (Fig. 4B). This phenomenon was previously reported for androgen agonists (i.e., testosterone, R1881, and mibolerone) during in vitro transcriptional activation assays (Kemppainen et al., 1999) and may be related to squelching of intracellular factors, such as coactivators and some transcriptional factors, in the cellular system under excessive ligand stimulation. Similar results were observed withR-19 and R-23 (Fig.5B).
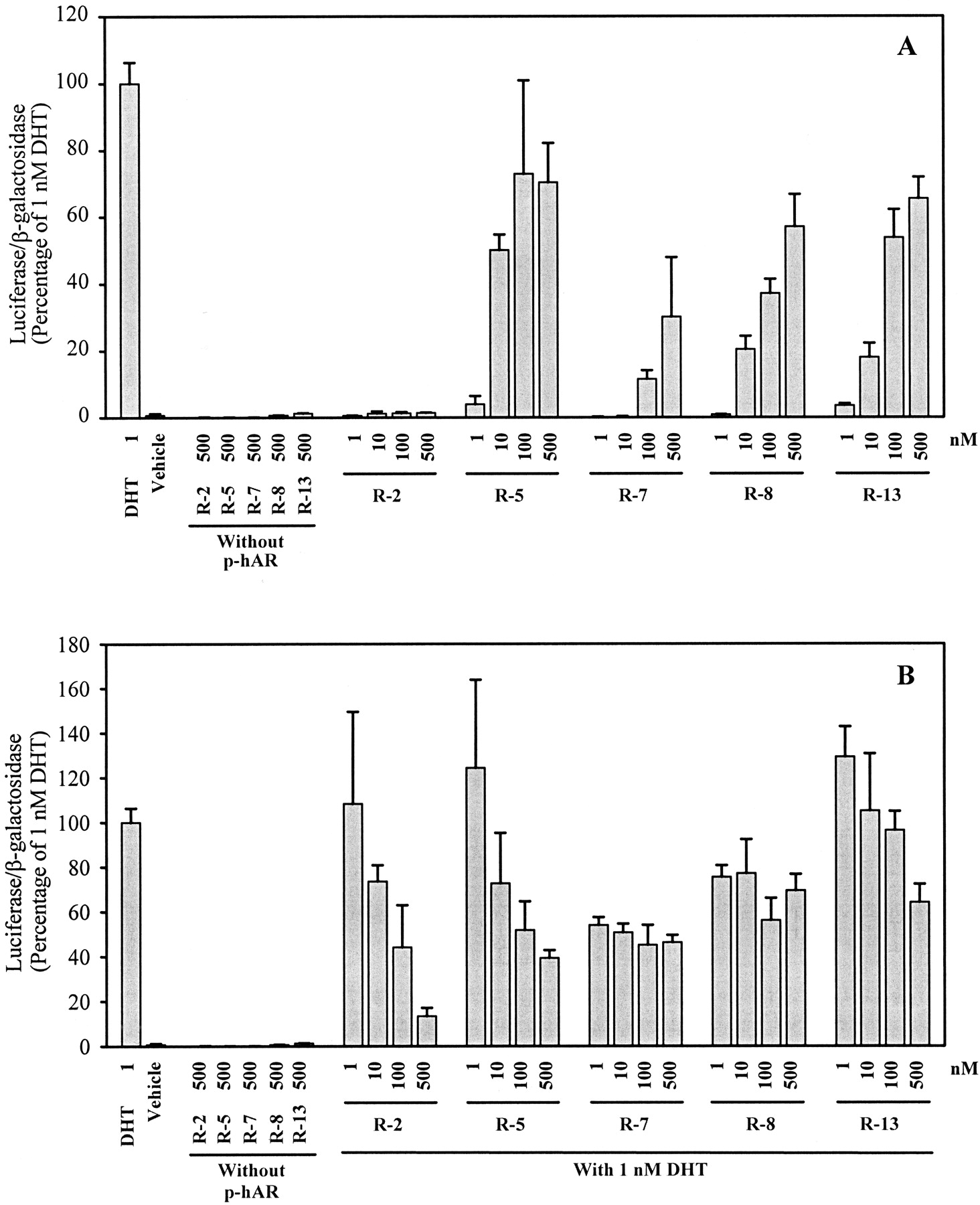
AR-mediated transcriptional activation ofR-2, R-5, R-7,R-8, and R-13. A, AR agonist activity. B, AR antagonist activity. CV-1 cells plated at 2 × 105cells/well in 12-well tissue culture plates were transfected with a human AR expression construct, an androgen-responsive luciferase reporter construct and a constitutively expressed β-galactosidase construct using LipofectAMINE, as described under Materials and Methods. Transfected cells were incubated with 1 nM DHT alone, vehicle, or increasing concentrations of ligand alone or together with 1 nM DHT for 48 h. Luciferase activity in each well was standardized according to β-galactosidase activity, and then expressed as the percentage of that produced by 1 nM DHT. Values represent mean ± S.D. (n = 3).

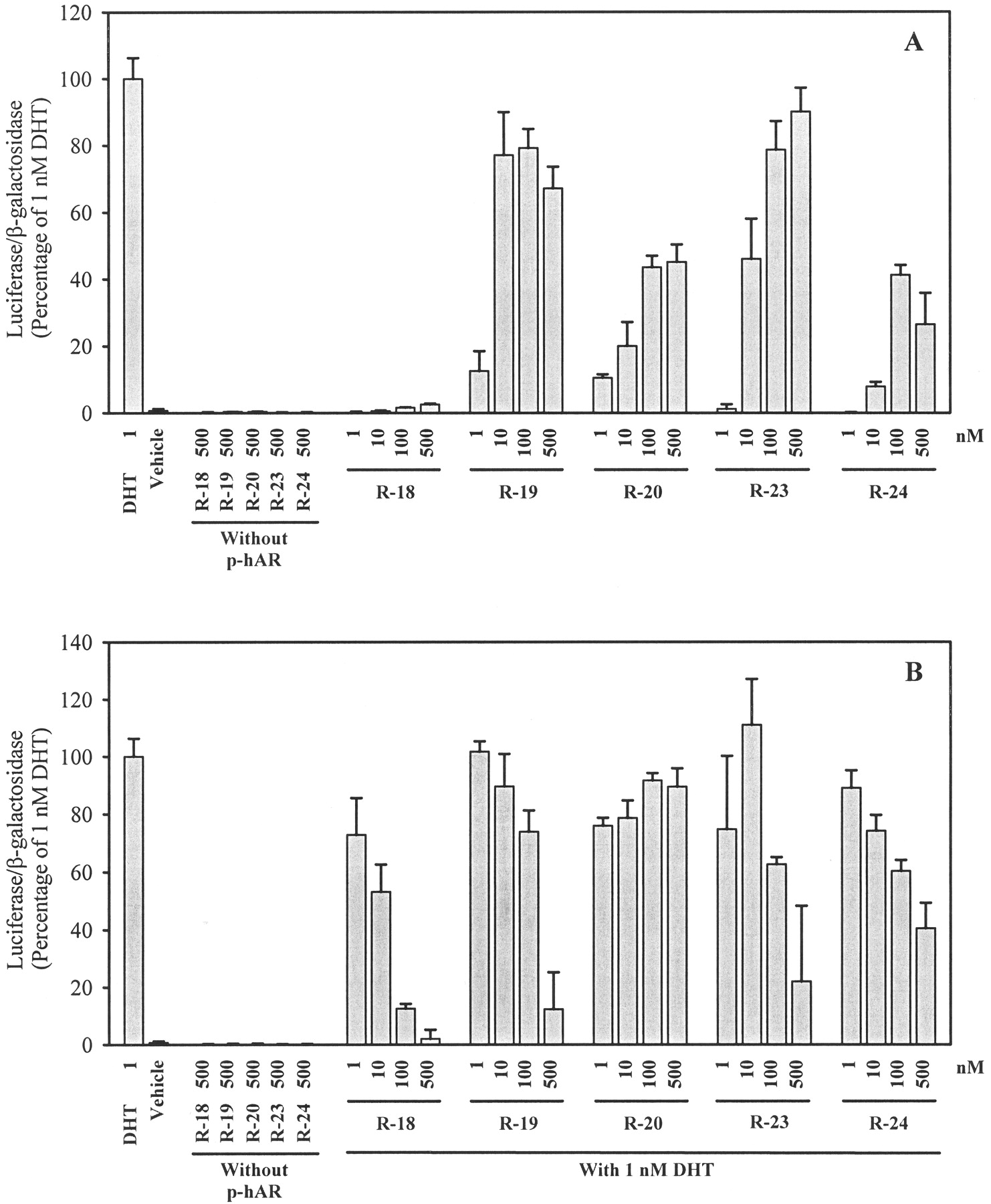
AR-mediated transcriptional activation ofR-18, R-19, R-20,R-23, and R-24. A, AR agonist activity. B, AR antagonist activity. CV-1 cells plated at 2 × 105 cells/well in 12-well tissue culture plates were transfected with a human AR expression construct, an androgen-responsive luciferase reporter construct, and a constitutively expressed β-galactosidase construct using LipofectAMINE, as described under Materials and Methods. Transfected cells were incubated with 1 nM DHT alone, vehicle, or increasing concentrations of ligand alone or together with 1 nM DHT for 48 h. Luciferase activity in each well was standardized according to β-galactosidase activity, and then expressed as the percentage of that produced by 1 nM DHT. Values represent mean ± S.D. (n = 3).
Dalton et al. (1998) previously showed that hydroxyflutamide analogs with a para-nitro group in the anilide ring possess greater AR agonist activity than those with a para-cyano substituent. In AR binding studies, we also observed that the change from a para-cyano to a para-nitro in the A-ring of bicalutamide derivatives in most cases improved AR binding affinity. Here, we further examined the effect of this structural change on functional activities of bicalutamide derivatives. Shown in Fig. 5 are the agonist (A) and antagonist (B) activities of a series of bicalutamide derivatives with a para-nitro group in the A-ring. Similar to the observation with ligands shown in Fig. 4,R-18, with a para-amino group in the B-ring, functioned as an AR antagonist, whereas N-alkylamido substituents at this position conferred agonist activity to other ligands in this series. Among the four agonists, the change of a sulfide to a sulfone in the X-linkage converted full AR agonists to partial agonists, as shown by a comparison of R-19 withR-20 and R-23 with R-24. Unlike those ligands with a para-cyano in the A-ring, the presence of an electrophilic group in the para-position of the B-ring slightly decreased the agonist activity in this series of ligands (compare R-19 with R-23, and R-20 withR-24). An interseries comparison of ligands in Fig. 5 with ligands in Fig. 4 indicated that the replacement of thepara-cyano with a para-nitro in the A-ring increased AR agonist activity whenever the binding was improved (compare R-19 with R-5, and R-20 withR-7). However, the agonist activity decreased when there was no notable enhancement in AR binding (compare R-24 withR-13).
Surprisingly, although the change from sulfide to sulfone led to decreased agonist activity in the previous series of ligands, it switched a full agonist (R-21) to an antagonist (R-22) when the para-substituent in the B-ring was a trifluoroacetamido group (Fig. 6). This change in functional activity was accompanied by a more than 2-fold decrease in binding affinity. R-22 differs fromR-20, a partial agonist, only in thepara-substituent in the B-ring. Despite a 1.5-fold higher binding affinity than R-20, R-22 functioned as an antagonist, again demonstrating that higher AR binding affinity does not necessarily predict greater agonist activity. The functional activities of another pair of sulfide and sulfone ligands,R-25 and R-26, are shown in Fig.7. R-25 showed a minimal level of agonist activity (15% at 500 nM), but antagonist activity at higher concentrations. R-26 showed no agonist activity at concentrations up to 500 nM and a weak antagonist activity. The change of the carbonyl link of the B-ring substituent in R-21 andR-22 to a sulfonyl link in R-25 andR-26 not only impaired the AR binding but also reduced the activities (compare agonist activity of R-21 withR-25 and antagonist activity of R-22 withR-26).

AR-mediated transcriptional activation ofR-21 and R-22. A, AR agonist activity. B, AR antagonist activity. CV-1 cells plated at 2 × 105cells/well in 12-well tissue culture plates were transfected with a human AR expression construct, an androgen-responsive luciferase reporter construct and a constitutively expressed β-galactosidase construct using LipofectAMINE, as described under Materials and Methods. Transfected cells were incubated with 1 nM DHT alone, vehicle, or increasing concentrations of ligand alone or together with 1 nM DHT for 48 h. Luciferase activity in each well was standardized according to β-galactosidase activity and then expressed as the percentage of that produced by 1 nM DHT. Values represent mean ± S.D. (n = 3).

AR-mediated transcriptional activation ofR-25 and R-26. A, AR agonist activity. B, AR antagonist activity. CV-1 cells plated at 2 × 105cells/well in 12-well tissue culture plates were transfected with a human AR expression construct, an androgen-responsive luciferase reporter construct and a constitutively expressed β-galactosidase construct using LipofectAMINE, as described under Materials and Methods. Transfected cells were incubated with 1 nM DHT alone, vehicle, or increasing concentrations of ligand alone or together with 1 nM DHT for 48 h. Luciferase activity in each well was standardized according to β-galactosidase activity, and then expressed as the percentage of that produced by 1 nM DHT. Values represent mean ± S.D. (n = 3).
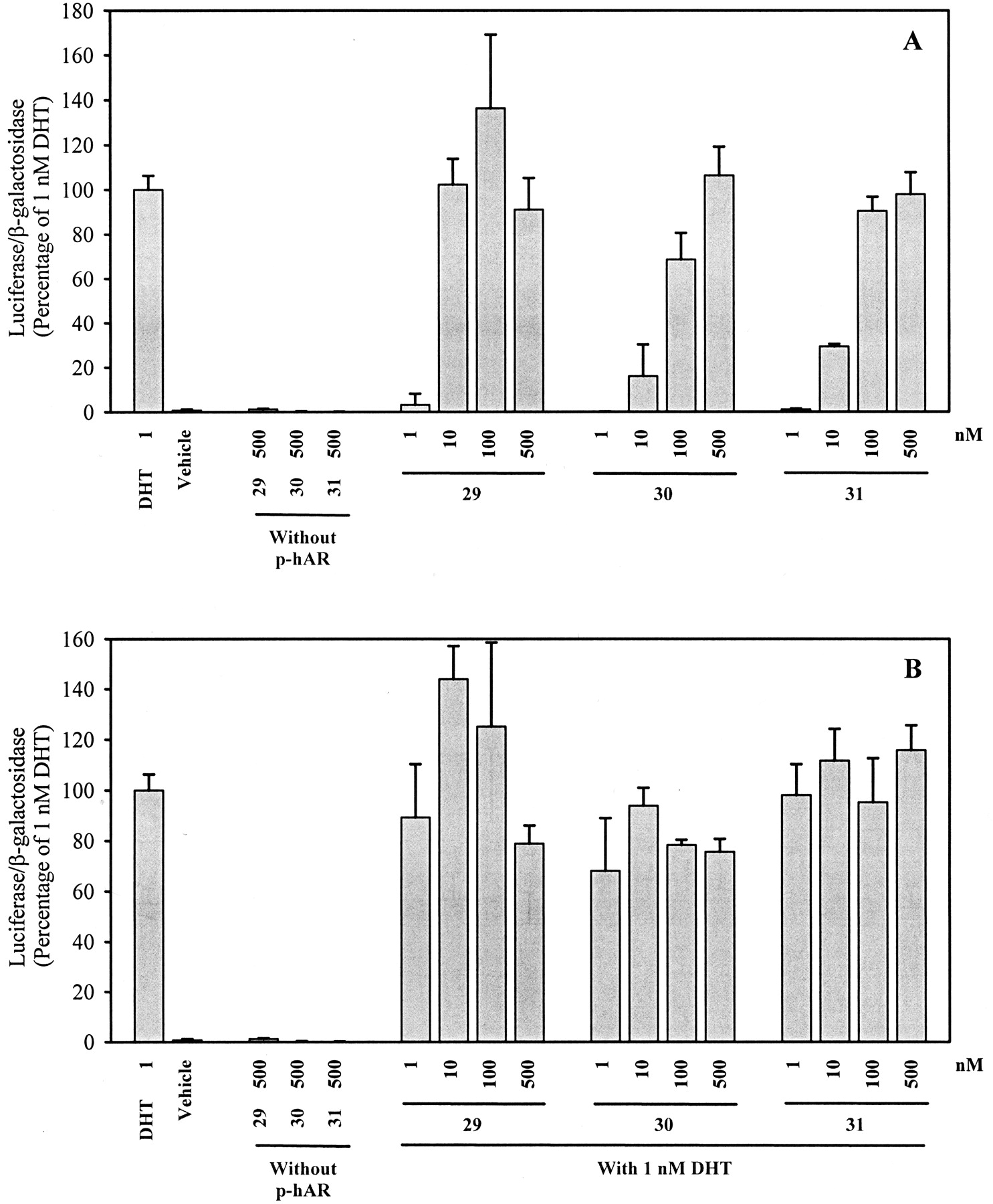
Ligands 29, 30, and 31 are racemic bicalutamide derivatives that bear a trifluoromethyl connected to the chiral carbon, and they exhibited very high binding affinity for the AR. As shown in Fig.8, all three ligands acted as full AR agonists in the agonist assay and none showed any antagonist activity in the antagonist assay. Ligand 29, which demonstrated higher AR binding affinity than the other two ligands, showed the greatest agonist activity within this series. At 10 nM, ligand 29 was equally efficient as 1 nM DHT in inducing the AR-mediated transcriptional activation.
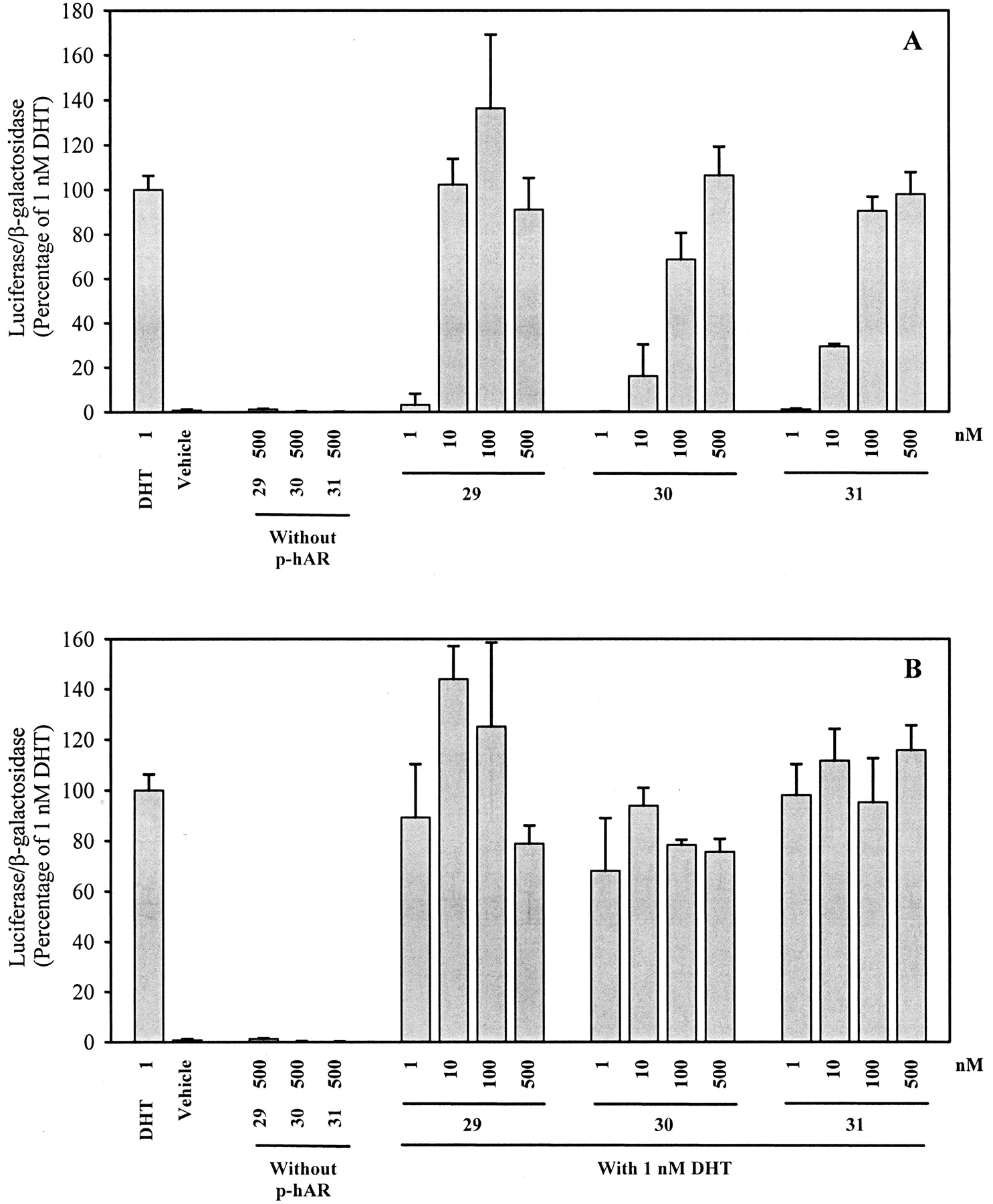
AR-mediated transcriptional activation of nonsteroidal ligands 29, 30, and 31. A, AR agonist activity. B, AR antagonist activity. CV-1 cells plated at 2 × 105cells/well in 12-well tissue culture plates were transfected with a human AR expression construct, an androgen-responsive luciferase reporter construct and a constitutively expressed β-galactosidase construct using LipofectAMINE, as described under Materials and Methods. Transfected cells were incubated with 1 nM DHT alone, vehicle, or increasing concentrations of ligand alone or together with 1 nM DHT for 48 h. Luciferase activity in each well was standardized according to β-galactosidase activity, and then expressed as the percentage of that produced by 1 nM DHT. Values represent mean ± S.D. (n = 3).
Although the data are not shown, studies in our laboratories also examined the specificity of these nonsteroidal agonists over other steroidal hormone receptors. Results showed that these agonists had no effect on the other steroid receptor-mediated transcriptional activation and are therefore highly specific for the AR.
Discussion
By modifying the structures of known nonsteroidal AR ligands, we obtained several series of compounds that covered a wide range of binding affinity for the AR. These novel compounds were mainly based upon three structurally related antiandrogen pharmacophores: bicalutamide, hydroxyflutamide, and nilutamide. Earlier studies with these chemical scaffolds revealed several structural elements that are important for nonsteroidal AR binding and antiandrogen activity (Glen et al., 1986; Tucker et al., 1988; Morris et al., 1991). For compounds based on the bicalutamide and hydroxyflutamide pharmacophores, Glen et al. (1986) and Tucker et al. (1988) showed that an electron-deficient aromatic ring separated from the tertiary carbinol by an amide link was required for AR binding. Substitutions at the 3- and 4- positions of the anilide ring with electron-withdrawing groups, particularly with cyano or nitro as the 4-substituent and trifluoromethyl or chloro as the 3-substituent, improved AR binding affinity. Physicochemical studies also showed that a coplanar geometry between the amide bond and the tertiary hydroxyl group, along with electron-withdrawing groups in the anilide ring, enhanced the hydrogen bond donor ability of the tertiary hydroxyl group; hydrogen bonding is regarded as another crucial factor for receptor binding (Tucker et al., 1988; Morris et al., 1991). Results from present binding studies with novel nonsteroidal compounds were consistent with findings from these SAR studies. Besides, the present binding data revealed new AR binding SARs for the linkage group and the aromatic B-ring substitutions of the bicalutamide pharmacophore. A number of our novel compounds, mainly from the bicalutamide-related series, exhibited an AR binding affinity higher than their lead molecules. For the bicalutamide-related series, the AR binding was generally enhanced by: 1) R-isomer; 2) an electrophilic para-substituent in the aromatic B-ring; 3) a nitro group in the para-position of A-ring; and 4) a trifluoromethyl group linked to the chiral carbon. The AR binding of hydroxyflutamide-related series was improved in the presence of electrowithdrawing substituents at the para-andmeta-positions of the aniline ring. The introduction of a coumarin ring decreased the AR binding of bicalutamide, hydroxyflutamide, and nilutamide analogs.
The recently reported crystal structure of metribolone (R1881)-bound human AR ligand binding domain (LBD) showed that a total of 18 amino acid residues, scattering sparsely over two regions (amino acids 700 to 788, and 875 to 896) in the domain, interact directly with the ligand (Matias et al., 2000). These amino acids may form a ligand-binding pocket to accommodate the ligand. Most of these residues are hydrophobic in nature and interact mainly with the hydrophobic moieties in the ligand molecule, whereas a few residues are hydrophilic and may form hydrogen bonds with polar atoms in the ligand (Matias et al., 2000). Moreover, in vitro mutagenesis studies demonstrated that the last 12 carboxyl-terminal amino acid residues in the receptor are also required for ligand binding, because truncation of these residues abolishes the binding of ligands to the human AR (Jenster et al., 1991;Kuil et al., 1995). With the present nonsteroidal ligands, we found that the AR binding is influenced by structural properties such as stereoisomeric conformation, steric effect, and electronic effect in the molecule. These findings indicated indirectly that accessibility to the binding pocket, and interaction with amino acid residues in the pocket through hydrophobic interaction and hydrogen binding are also critical for nonsteroidal AR binding. It would be very interesting to determine whether these nonsteroidal ligands bind to the same binding pocket and amino acids in the LBD as those steroid ligands.
The AR agonist and antagonist activities of identified high-affinity AR ligands were determined in vitro using the cotransfection assay. These nonsteroidal ligands demonstrated a range of functional activity profiles, including antagonists, partial agonists, and full agonists. No general correlation was observed between the receptor binding affinity and functional activities with these ligands. In general, agonist activity was most often observed in bicalutamide derivatives with a sulfide linkage and an N-alkylamido substituent in the para-position of B-ring. The change from sulfide to sulfone in the linkage position resulted in attenuated agonist activity or even switched a full agonist to an antagonist.
The present functional activity data demonstrated that minor structural differences in these nonsteroidal molecules could greatly alter the nature of receptor-ligand interaction and lead to completely different pharmacological responses (i.e., agonist or antagonist activities). The divergent event triggering the differentiated activities of these structurally related molecules is most likely the ligand-induced conformational change in the receptor, an essential step in steroid hormone receptor signaling pathways. Compelling evidence shows that AR agonists and antagonists induce distinct conformational changes in the receptor, so that the receptor interacts differently with other transcriptional factors and/or coactivator/corepressors in the subsequent signaling pathway (Kallio et al., 1994; Kuil and Mulder, 1994, 1995; Kuil et al., 1995). It is possible, as was suggested for other steroid receptors (Brzozowski et al., 1997), that the AR may respond to agonists and antagonists by positioning helix H12, one of the 12 helices contained in the secondary structure of its LBD, to either seal (in the case of agonists) or open (in the case of antagonists) the ligand binding pocket. The full spectrum of functional activities displayed by our series of nonsteroidal ligands suggests the potential value of these compounds as tools in studying ligand-AR interactions at the molecular level.
Very importantly, the identification of numerous potent and efficacious full AR agonists by in vitro assay represents another major progress in our efforts toward defining the SAR necessary for AR agonist activity of bicalutamide-based nonsteroidal ligands. These SARs provided valuable guides for the eventual structural optimization for AR agonist activity and development of a new generation of tissue-selective nonsteroidal androgens, SARMs. With obvious advantages over steroid androgens, SARMs have great therapeutic implications in that they could not only be used as superior alternatives to steroidal androgens in androgen replacement therapy for male hypogonadism but also expand the scope of androgen therapy to treat wasting syndromes in critically ill patients, to prevent aging-related disorders, and to regulate male fertility.
In summary, the present studies examined the AR binding and functional activities of a group of novel nonsteroidal compounds with in vitro systems. From these studies, high-affinity AR ligands with diverse activity profiles were discovered, and SARs for AR binding and transcriptional activation were identified. Because in vitro cotransfection studies cannot account for the myriad of factors that govern in vivo activity, later studies in our laboratories have focused on characterization of the in vivo functional activity of selected full agonists. These studies and others detailing the preclinical pharmacology of SARMs are the topics of forthcoming reports from our laboratories.
Footnotes
- Received June 25, 2002.
- Accepted October 14, 2002.
-
These studies were supported by grants from the National Institute of Child Health and Human Development (R15-HD35329), National Institute of Diabetes and Digestive and Kidney Diseases (R01-DK59800), National Cancer Institute (R29-CA68096), the St. Francis of Assisi Foundation, and the Harriet S. Van Vleet Professorship in Pharmacy.
Abbreviations
- AR
- androgen receptor
- SAR
- structure-activity relationship
- SARM
- selective androgen receptor modulator
- SERM
- selective estrogen receptor modulator
- ER
- estrogen receptor
- MIB
- mibolerone
- PMSF
- phenylmethylsulfonyl fluoride
- DHT
- dihydrotestosterone
- FBS
- fetal bovine serum
- HAP
- hydroxyapatite
- LBD
- ligand binding domain
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}