


Abstract
Metabotropic glutamate (mGlu) receptors are a family of G-protein-coupled receptors that play central roles as modulators of both glutamatergic and other major neurotransmitter systems in CNS. Using molecular modeling, site-directed mutagenesis, [3H]LY354740 binding, [35S]GTPγS binding, and activation of GIRK current, we have been able to identify residues crucial for the binding of LY354740 and glutamate to rat mGlu2 receptors. Several of the crucial residues located in the binding site (Arg-57, Tyr-144, Tyr-216, Asp-295) have not been identified previously. We propose that the γ-carboxyl group of LY354740 forms H-bonds to Arg-57, whereas the α-carboxyl group forms an H-bond with the hydroxyl group of Ser-145. The α-amino group of LY354740 forms H-bonds to Asp-295 and to the side-chain hydroxyl group of Thr-168. In addition, Tyr-144 may establish a hydrophobic (C-H/π)–interaction with the bicyclo-hexane ring of LY354740. Furthermore, the mutation of residues Ser-148 and Arg-183, which are too remote for a direct interaction, affected the ligand affinity dramatically. These results suggest that Ser-148 and Arg-183 may be important for the 3D structure and/or are involved in closure of the domain. Finally, Asp-146, which is also remote from the binding site, was shown to be involved in the differential binding affinity of [3H]LY354740 for mGlu2 versus mGlu3 receptors. All the mGlu receptors except mGlu2 are activated by Ca2+ and have serine instead of aspartic acid at this position, which suggests a critical role of this aspartic acid residue in the binding properties of this unique receptor.
The majority of excitatory synapses in the central nervous system employ glutamate as their neurotransmitter. After its release, glutamate binds to and activates two distinct classes of receptors, ionotropic (NMDA, AMPA, and kainate) (Monaghan et al., 1989; Nakanishi, 1992) and metabotropic (mGlu) (Pin et al., 1999, Pellicciari et al., 2000). The mGlu receptor family comprises eight receptors that are divided into three classes on the basis of their sequence similarities, signal transduction, and agonist rank order of potency. Group I receptors (mGlu1 and -5) are coupled to phosphoinositide hydrolysis; group II receptors (mGlu2 and -3) are negatively coupled to cAMP production and are not stimulated byl-(+)-2-amino-4-phosphono butyric acid (l-AP4); and group III receptors (mGlu4, -6, -7, and -8) are also negatively coupled to cAMP production but are activated by l-AP4 (Conn and Pin, 1997, Pin et al., 1999, De Blasi et al., 2001). Because of their critical role as modulators of synaptic transmission, ion channel activity, and synaptic plasticity (Nakanishi, 1994, Anwyl, 1999,Holscher et al., 1999), mGlu receptors are implicated in the pathology of major neurological disorders such as Alzheimer's and Parkinson's disease as well as depression, schizophrenia, anxiety, and pain (Nicoletti et al., 1996; Bruno et al., 1998; Bordi and Ugolini, 1999).
The mGlu receptors belong to a new family of G-protein coupled receptors (GPCRs) designated family 3 (Bockaert and Pin, 1999). The other members of this family include the GABABreceptor, the Ca2+-sensing receptor and putative pheromone receptors (Bockaert and Pin, 1999). They have an unusually large extracellular amino-terminal domain (ATD) (∼ 500 to 600 aminos acids) with no sequence homology to other families of GPCRs. Although the sequence homology among family 3 members is low (∼ 20% amino acid identity), they are structurally related. O'Hara et al. (1993)observed the similarity between a region of the mGlu1 ATD and a family of bacterial periplasmic amino acid-binding proteins, in particular the leucine-, isoleucine- and valine binding protein (LIVBP) (Sack et al., 1989). Based on the crystal structure of LIVBP, they proposed a bilobal structure for the agonist-binding pocket of mGlu receptors in which the glutamate is bound in a “venus flytrap” mechanism. Furthermore, mutagenesis studies verified that two residues, Ser-165 and Thr-188 of mGlu1, were indeed crucial for binding to glutamate and quisqualate as predicted by this model (O'Hara et al., 1993). Using molecular modeling, site-directed mutagenesis, and chimeric receptors, many other researchers have subsequently reported on the structural similarity of the ligand-binding site of both NMDA receptors (Kuryatov et al., 1994) and iGlu receptors (Stern-Bach et al., 1994) to the two lobes of the bacterial lysine-arginine-ornithine binding protein. The recent X-ray structure of the rat iGlu2 receptor extracellular domains (Armstrong et al., 1998) also strongly supports the mGlu/periplasmic amino acid-binding protein-like model (O'Hara et al., 1993) for the glutamate-binding site. Furthermore, site-directed-mutagenesis of the GABAB R1 (Galvez et al., 1999) has identified critical residues in the ligand-binding pocket, the localization of which supports the “venus flytrap” model. Biochemical studies of the purified extracellular ligand-binding region of mGlu1 (Okamoto et al., 1998), mGlu4 (Han and Hampson, 1999), and, recently, GABAB R1 (Malitschek et al., 1999) have indicated that these truncated soluble receptors retain ligand-binding properties similar to wild-type receptors. However, compared with wild-type receptors, the mGlu4 ATD displayed higher affinities for agonists and lower affinities for antagonists (Han and Hampson, 1999). Therefore, the amino acids involved in ligand binding must be localized in the ATD of these receptors. The long-awaited crystal structure of the ATD of the mGlu1 receptor complexed with glutamate has recently been reported (Kunishima et al., 2000) depicting a “clamshell”-like structure similar to that found in X-ray structure of iGlu2 receptor (Armstrong et al., 1998) and LIVBP (Sack et al., 1989).
In the present study, we used tritiated (+)-2-aminobicyclo-[3.1.0]-hexane-2,6-dicarboxylate (LY354740), which has a nanomolar potency at mGlu2 and -3 and no effect at mGlu1, -5, -8, -4 and -7 receptors up to 300 μM (Schoepp et al., 1997; Schaffhauser et al., 1998, Malherbe et al., 1999, Schweitzer et al., 2000). Its selectivity, combined with site-directed mutagenesis and molecular modeling, provided a valuable tool to study amino acids involved in agonist binding to mGlu2 and -3 receptors. Measurements of function [GTPγ35S binding and activation of G-protein-coupled inwardly rectifying potassium channel (GIRK) current] were performed to evaluate the consequences of mutations on agonist efficacy and selectivity. In addition, the inhibitory effect of two compounds derived from LY354740 were assessed on [3H]LY354740 binding.
Experimental Procedures
Materials.
(+)-α-Methyl-4-carboxyphenylglycine, 2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine, (+)- and (−)-LY354740, and 2-amino-bicyclo-[3.1.0]hex-3-ene-2,6-dicarboxylate were synthesized at Hoffmann-La Roche Ltd by Drs. G. Adam and J. Wichmann. [3H]LY354740 (specific activity, 35 Ci/mmol) was synthesized by Dr. P. Huguenin at the Roche chemical and isotope laboratories following a synthetic route devised by Dr. H. Stadler. Guanosine-5′-O-(3-thiotriphosphate) (GTPγS) was obtained from Sigma. l-Glutamate was from RBI/Sigma (Natick, MA). GTPγ35S (specific activity, 1000 Ci/mmol) was obtained from Amersham Pharmacia Biotech (Piscataway, NJ).
Construction of Point-Mutants.
cDNAs encoding the rat mGlu2 and mGlu3 receptors in pBlueScript II were generously given to us by Prof. S. Nakanishi (Kyoto, Japan). All point-mutants were constructed using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer's instructions and using pBlueScript II-mGlu2 (or -mGlu3) as a DNA template. Complementary 36-mer oligonucleotide primers (sense and antisense) containing the site of mutation in the middle were synthesized by Amersham Pharmacia Biotech. The following polymerase chain reaction conditions were used for repeated extensions of the plasmid template: 95°C for 1 min and 20 cycles of 95°C for 30 s, 55°C for 1 min, and 68°C for 18 min using 50 ng of plasmid DNA, 100 ng of each primer, and 2.5 units Pfu Turbo DNA polymerase (Stratagene). The entire coding regions of all positive point-mutants were sequenced from both strands using an automated cycle sequencer (Applied Biosystems, Foster City, CA). The cDNAs with positive point-mutants were then subcloned into the mammalian expression vector pcDNA3 (Invitrogen, Carlsbad, CA) downstream from the cytomegalovirus promoter.
Cell Culture, Transfection, and Membrane Preparation.
HEK-293 cells were maintained in minimum essential medium supplemented with 100 μg/ml penicillin, 100 μg/ml streptomycin, 10 mM HEPES, and 10% fetal calf serum. Three days before transfection, the cells were seeded at high density (200,000 cells/ml). The transfection of various receptor point-mutants was performed using LipofectAMINE Plus reagent (Invitrogen) according to the manufacturer's instruction. Six hours after transfection, the DNA-transfection mixture was removed and the cells were maintained in minimum essential medium with reduced l-glutamine (0.4 mM final), 10% dialyzed fetal calf serum and the mGlu antagonist, (+)-α-methyl-4-carboxyphenylglycine (0.32 mM final). After 48 h, the cells were harvested and washed three times with cold PBS and frozen at −80°C. The pellet was suspended in cold 20 mM HEPES-NaOH buffer containing 10 mM EDTA, pH 7.4, and homogenized with a Polytron homogenizer (Kinematica AG, Basel, Switzerland) for 10 s at 10,000 rpm. After centrifugation at 48,000g for 30 min at 4°C, the pellet was washed once with 20 mM HEPES-NaOH buffer containing 0.1 mM EDTA, pH 7.4, respun and resuspended in a smaller volume of a cold 50 mM Tris-HCl, 2 mM MgCl2binding buffer at pH 7.4. The membrane suspension was frozen at −80°C before use. Protein content was measured using the Pierce method (Socochim, Lausanne, Switzerland) using bovine serum albumin as the standard.
[3H]LY354740 Binding.
After thawing, the membranes were diluted in the binding buffer to a final assay concentration of 25 μg of protein/ml. Saturation isotherms were determined by addition of various [3H]LY354740 concentrations (1–1000 nM) to these membranes for 1 h at room temperature. To analyze the effect of Ca2+ on the mGlu2 wild-type receptor and D146S mutant, the saturation isotherms were performed in the absence or presence of 2 mM CaCl2. At the end of the incubation, membranes were filtered onto Whatman GF/C glass fiber filters (Whatman, Maidstone, UK) and washed five times with cold binding buffer. Nonspecific binding was measured in the presence of 10 μM 2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine. The radioactivity was measured by liquid scintillation counting after transfer of the filter into plastic vials containing 10 ml of Ultima-gold (Packard, Meriden, CT) in a Tri-Carb 2500 TR counter (Packard). Saturation experiments were analyzed with the iterative nonlinear curve fitting software Origin (Microcal Software Inc., Northampton, MA) using the rectangular hyperbolic equation derived from the equation of a bimolecular reaction and the law of mass action,B = (B max × [F])/(K D + [F]), where B is the amount of ligand bound at equilibrium,B max is the maximum number of binding sites, [F] is the concentration of free ligand, andK D is the ligand dissociation constant. The experiments were performed at least three times in triplicate and the mean ± S.D. of the individual K Dvalues were calculated and are reported in the Table1. For inhibition experiments, membranes containing the wild-type receptor or the Y144S mutant of the mGlu2 receptor were incubated with 10 or 100 nM [3H]LY354740 (for wild-type and Y144S, respectively) and various concentration of the inhibitory compound. IC50 and Hill coefficient values were derived from the inhibition curve and Ki values were calculated according to the equation K i = IC50/(1+[L]/KD) where [L] is the concentration of [3H]LY354740 andK D is its dissociation constant at the receptor, derived from the saturation isotherm.
Dissociation constants (K D) for [3H] LY354740 binding to the HEK-293 cell membranes transfected with the mGlu2 and -3 wild-type and point-mutated receptors. Values are mean ± S.D. of the K Dvalues, calculated from at least three individual experiments, performed in triplicate.
GTPγ35S Binding.
This assay was essentially performed as described previously (Cartmell et al., 1998). Briefly, after thawing, the membranes were washed once and resuspended in cold 20 mM HEPES-NaOH buffer containing 10 mM MgCl2and 100 mM NaCl, pH 7.4. Wheat germ agglutinin scintillation proximity assay beads (RPNQ0001; Amersham) were suspended in the same buffer (40 mg of beads/ml). Membranes and beads were mixed (beads: 13 mg/ml; membranes: 200 μg of protein/ml) and incubated with 2 μM GDP at room temperature for 1 h, with mild agitation. GTPγ35S binding was performed in 96-well microplates (picoplate; Packard) in a total volume of 180 μl with 15 μg of membrane protein and 0.3 nM GTPγ35S. Nonspecific binding was measured in the presence of 10 μM cold GTPγS. In order to study the effect of glutamate and LY354740, GTPγ35S binding was stimulated with various concentrations of glutamate (1 to 300 μM) or of LY354740 (1 to 300 nM). Plates were sealed and agitated at room temperature for 2 h. The beads were then allowed to settle and the plate counted in a Top-Count (Packard) using quench correction. The stimulation curves were fitted with a four parameter logistic equation giving EC50 values and Hill coefficients. The experiments were performed at least three times in triplicate and the mean ± S.D. of the EC50 values was calculated. Unfortunately, we were unable to set up conditions to measure reliably the stimulatory effect of agonists using membranes from mGlu3 transfected cells.
Electrophysiology.
A Chinese hamster ovary (CHO) cell line stably expressing human GIRK1-GIRK2 dimer was cotransfected with a 1:1 (w/w) mixture of mGlu2:enhanced green fluorescent protein plasmids using LipofectAMINE 2000 (Invitrogen). GIRK channel currents were recorded 24 to 96 h after cell transfection using the whole-cell configuration of the patch-clamp technique. Pipettes were pulled from borosilicate glass with resistances from 2 to 3 MΩ, when filled with a solution containing 130 mM KCl, 1 mM MgCl2, 10 mM HEPES, 5 mM K4BAPTA, 3 mM Na2ATP, 0.3 mM Na2GTP, 5 mMd-Glucose, adjusted to pH 7.2 with KOH, and osmolarity adjusted to 310 mOsM with sucrose. The cells were superfused with a solution containing 149 mM NaCl, 3.25 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 10 mM HEPES, 11 mM d-Glucose, adjusted to pH 7.4 with NaOH, and osmolarity adjusted to 340 mOsM with sucrose. For GIRK channel current recordings, external Na+ was replaced by an equal amount of K+ to reach 30 mM K+. Whole-cell currents were amplified with an Axopatch 200A amplifier (Axon Instruments, Foster City, CA), filtered at 1 KHz and acquired at 500Hz with a Digidata 1200A-acquisition board (Axon Instruments) for subsequent storage on a Dell personal computer. Cells were held at −70 mV and the recordings were made under conditions in which K+ currents would be inward ([K+]i = 150 mM, [K+]o = 30 mM).
Stock solutions of glutamate (Fluka, Buchs, Switzerland) and LY354740 were prepared in H2O and diluted in the external solution to their final concentration before use. All other chemicals were from Sigma. The drugs were applied locally to the cell by fast perfusion from a double-barreled pipette assembly. The rate of solution exchange was around 20 ms. The concentration-response curves were obtained by applying 20-s pulses of varying concentrations of the agonists to the cells every 90 s. The maximum current amplitudes from individual cells were first fitted separately using the Hill equation [I = I max / (1 + (EC50 / [Agonist])nH), where I is agonist-evoked current, I max is the maximum of the fit, EC50 is the agonist concentration evoking the half-maximum current, and n H is the Hill coefficient. The individual concentration-response curves were then normalized to I max and the mean ± S.E.M. values calculated from the normalized data for each concentration were plotted and fitted with the logistic equation.
Western Blot.
For Western blot analysis, 1 μg of membrane proteins were resuspended in Laemmli buffer containing 20 mM dithiothreitol and heated at 50°C for 5 min. Proteins were separated by 7% SDS-polyacrylamide gel and electroblotted onto a polyvinylidene difluoride membrane. After blocking, the blot was incubated with a mGlu2/3 commercially available antibody (AB 1553; Chemicon, Temecula, CA), in Tris-buffered saline/Tween-20 supplemented with 1% low-fat dry milk, for 1 h at room temperature. A horseradish peroxidase-conjugated sheep anti-mouse antibody (Amersham Pharmacia Biotech) was used as a second antibody at a dilution of 1:400 in Tris-buffered saline/Tween-20 supplemented with 1% low fat dry milk, and was incubated with the filter for 30 min at room temperature. The signal was revealed using the Lumi-Light Western Blotting Kit (Amersham Pharmacia Biotech).
Results
Generation of Point Mutations and Their Expression in HEK-293 Cells.
A model of the extracellular part of rat mGlu2 was built by homology to a bacterial periplasmic binding protein (Escherichia coli leucine/isoleucine/valine binding protein, LIVBP, PDB code2liv, http://www.rcsb.org/pdb) (Sack et al., 1989). Distant sequence similarity between both proteins has been detected previously (O'Hara et al., 1993). We have confirmed this relationship by using PSI-Blast (Altschul et al., 1997). Our sequence alignment between LIVBP and mGlu2 is essentially the same as that of O'Hara et al. (1993). Figure1 shows the alignment of rat mGlu receptors and their limited sequence homology (<20%) to bacterial Leu/Ile/Val-binding protein. To determine the amino acid residues that control glutamate and LY354740 affinity, efficacy and selectivity, 23 point mutations, 21 in mGlu2 and 2 in mGlu3, were introduced by site-directed mutagenesis. Because the Ser-165 and Thr-188 of mGlu1 receptor have been shown previously to be crucial for the binding to glutamate, our mutated residues were chosen from two regions proximal to these residues (Fig. 1). The model also predicted that the residues Arg-57 and Asp-295 might be located in the binding pocket. For mutants that did not bind [3H]LY354740, cell membrane preparations were subjected to immunoblotting using an anti-mGlu2/3 antibody. For these mutants and for the wild-type receptor, an immunoreactive band around 100 kDa was detected that was absent in the mock-transfected cell membranes, demonstrating the expression of the mutant receptors in the transfected cells (Fig.2). Additionally, strong bands around 200 kDa were detected that are most likely to be receptor dimers.

Multiple alignment of the extracellular domain of rat mGlu receptors with E. coli Leu/Ile/Val-binding protein (2livBP) using the Pileup program. The numbers on the right refer to the amino acids of rat mGlu1–8 including the signal peptide and mature LIVBP. Asterisks indicate identical residues among mGlus and LIVBP. Colons indicate identical residue between mGlu2 and LIVBP. The residues which have been site mutated in mGlu2 and -3 are underlined. The mGlu2 crucial residues involved in the binding to glutamate and LY354740 are indicated (↑). The important residues in the proximity of hinge region that might control domain closure and/or stability of 3D-structure are identified (♦). The residues found in the mGlu1 crystal structure (Kunishima et al., 2000) to interact directly and/or via a water molecule with glutamate are highlighted.
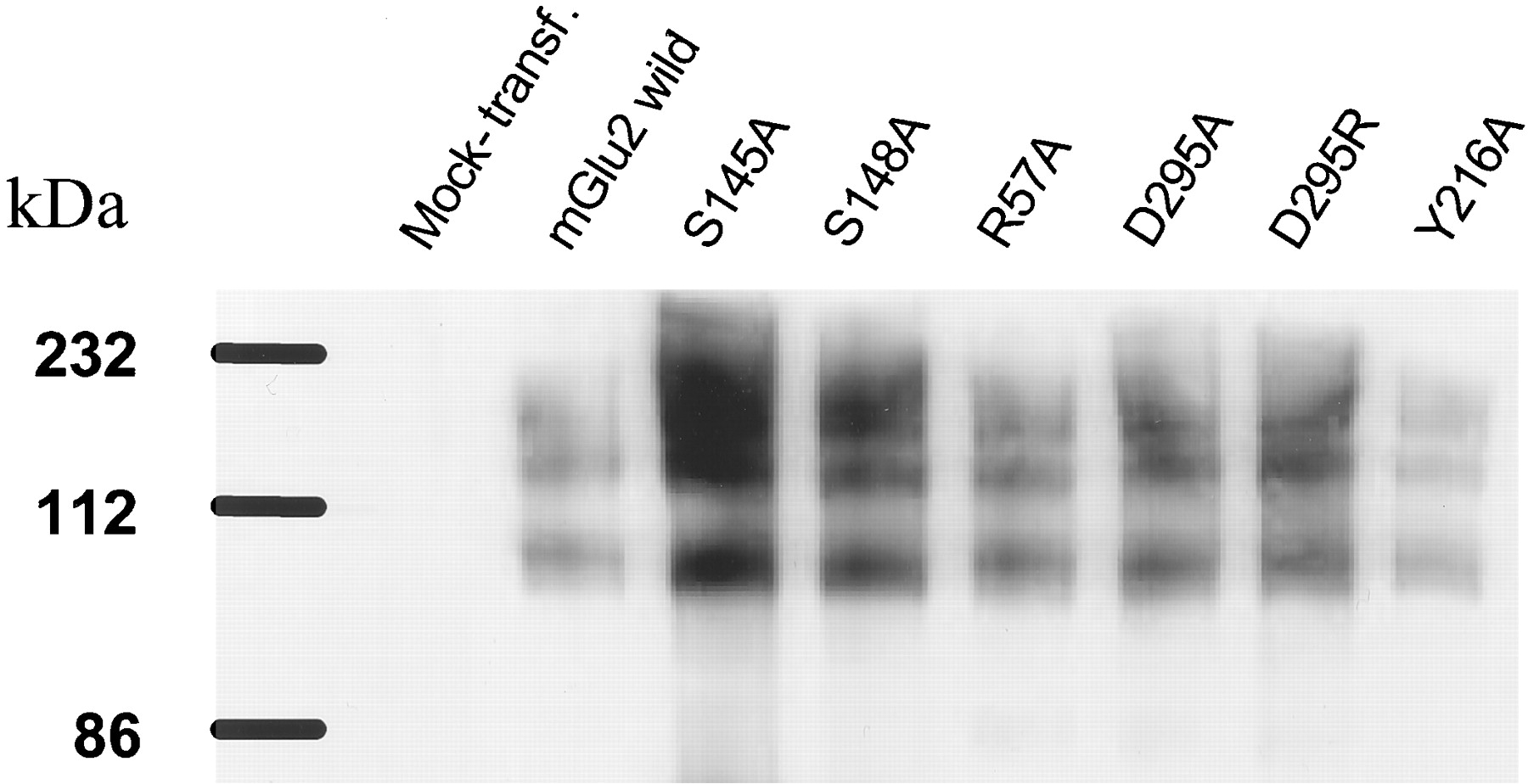
Immunoblot analysis of HEK-293 cells transfected with the mGlu2 wild-type receptors and different point-mutants. Membrane protein (1 μg) was loaded on each lane. The C-terminal mGlu2/3 antibody from Chemicon was used. The pcDNA3 was used for mock-transfected control.
Effect of Point Mutations on the [3H]LY354740 Binding.
The [3H]LY354740 (Fig.3A) saturation isotherms for the mGlu2 and -3 wild-type and selected mutated receptors are shown in Fig.4 and the dissociation constants and maximum number of binding site (B max) in Table 1. For the mGlu2 receptor, six mutations (Y144F, A166S, S167A, S169A, S164A, and R271A) did not significantly affect the ligand affinity compared with the wild-type mGlu2 receptor (Table 1). However, the mutations S145A, R57A, R57K, R57Y, Y216A, Y216F, D295A, and D295R completely abolished [3H]LY354740 binding. The mutation S148A led to a dramatic reduction in [3H]LY354740 affinity, although the effect was less marked than observed with the eight previous mutations. Two other mutations, T168A and R183A, led to 21- and 11-fold decreases in affinity, respectively (Fig. 4A, Table 1). The conversion of the tyrosine 144 to an alanine (Y144A) or a serine (Y144S) led to a statistically significant 10-fold decrease of [3H]LY354740 affinity (P < 0.05, Student's t test; Fig. 4A, Table 1). The conversion of this tyrosine to a glycine (Y144G) also led to a statistically significant 6-fold reduction in affinity (P < 0.05, Student's t test). However, its conversion to a phenylalanine (Y144F) had no effect on [3H]LY354740 affinity (Table 1). Replacing the aspartic acid in position 146 with a serine (D146S) led to a significant decrease of affinity (75 ± 13 versus 20 ± 2 nM, for the mutant and the wild-type mGlu2 receptor, respectively,p < 0.01 Student's t test, Fig. 4A) and the affinities of this mGlu2 mutant and the wild-type mGlu3 receptor were not statistically different. Interestingly, the addition of 2 mM CaCl2 had no effect on the wild type mGlu2 saturation isotherm, but had a tendency to increase the affinity of [3H]LY354740 for the D146S mutant (K D values of 20 ± 2 and 18 ± 6 for the wild-type and 75 ± 18 and 42 ± 5 nM for the mutant receptors in the absence or presence of 2 mM CaCl2, respectively). The conversion of the serine residue at position 152 of the mGlu3 receptor to an aspartic acid (S152D), which corresponds to the reciprocal mutation performed in the mGlu2 receptor (D146S), led to a significant increase of [3H]LY354740 affinity for the mutant (K D values of 82 ± 7 and 27 ± 2.5 nM for the wild-type and the mutant receptors, respectively,P < 0.01, Student's t test, Fig. 4B). The affinities of the S152D mGlu3 mutant and of the wild-type mGlu2 receptors were not significantly different. Moreover, the conversion of this serine 152 to a histidine residue (S152H) did not change the affinity of the mutant compared with the wild-type mGlu3 receptor (K D value of 76 ± 4 nM; Fig. 4B).

Chemical structures. A, [3H]LY354740. B, (−)-2-aminobicyclo-[3.1.0]hexane-2,6-dicarboxylate. C, 2-amino-bicyclo-[3.1.0]hex-3-ene-2,6-dicarboxylate.
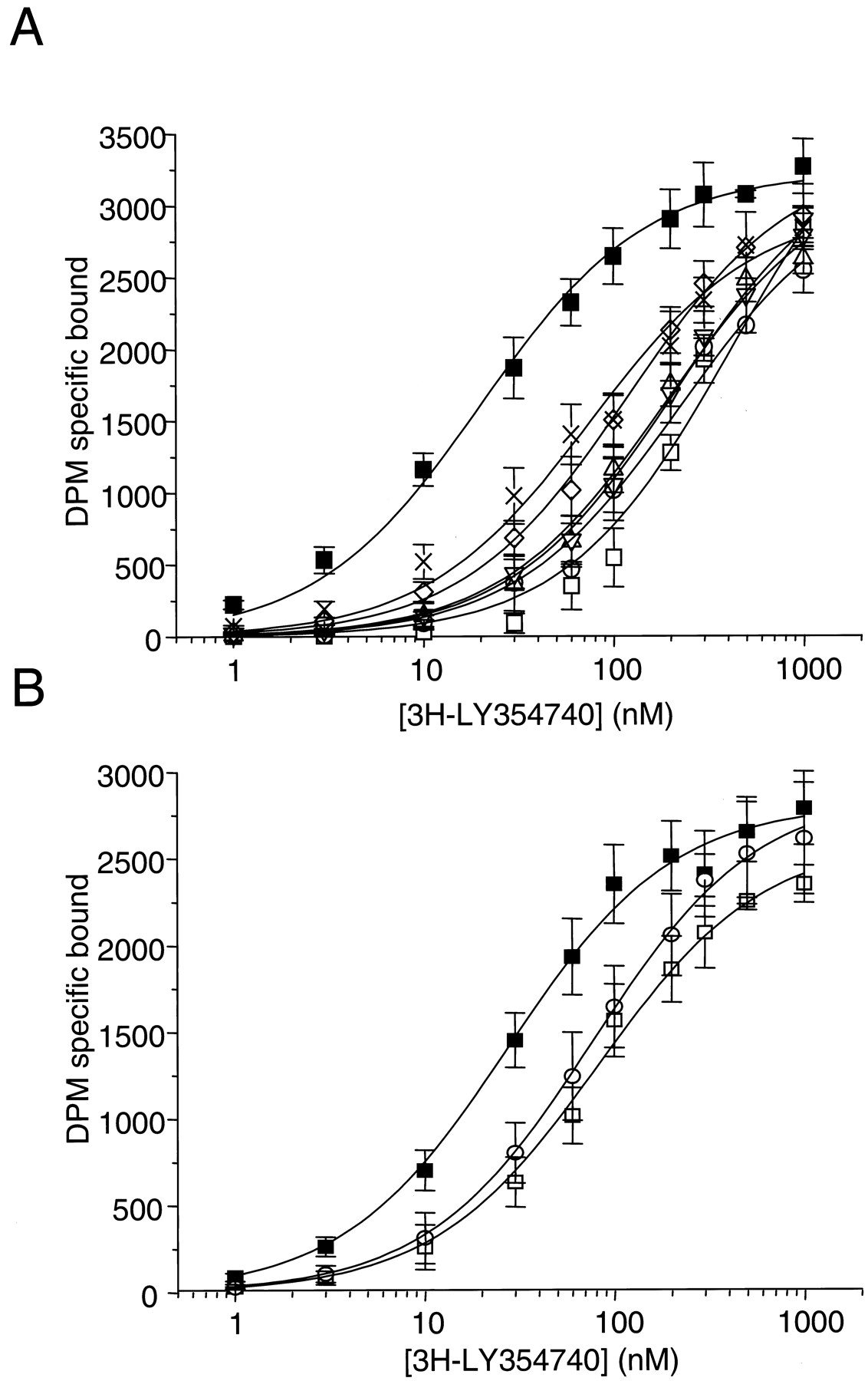
Saturation isotherms of [3H]LY354740 binding to the wild-type (▪) and various point-mutated mGlu2 receptors: T168A (■), R183A (○), Y144A (▵), Y144S (▿); Y144G (⋄), D146S (×) (A) and the wild-type (■) and point-mutated mGlu3 receptors: S152D (▪) and S152G (○) (B). Data are mean ± S.D. (bars) of at least three separate experiments, performed in triplicate.
Inhibition of [3H]LY354740 Binding by LY354740 Derivatives.
LY354740 was able to inhibit fully the binding of the labeled compound to the wild-type receptor and Y144S mutant mGlu2 receptor, with K i values of 0.013 ± 2 and 0.33 ± 0.03 μM, respectively. As might be predicted from the study of Monn et al. (1997), the corresponding (−)-enantiomer of LY354740 (Fig. 3B) was completely inactive. In addition, 2-amino-bicyclo-[3.1.0]hex-3-ene-2,6-dicarboxylate (Fig. 3C) inhibited the binding of [3H]LY354740 to the wild-type receptor with a K i value of 0.63 ± 0.06 μM, whereas it inhibited the binding of the labeled compound to the Y144S mutant with a K ivalue of 1.5 ± 0.05 μM.
Glutamate- and LY354740-Stimulated GTPγ35S Binding.
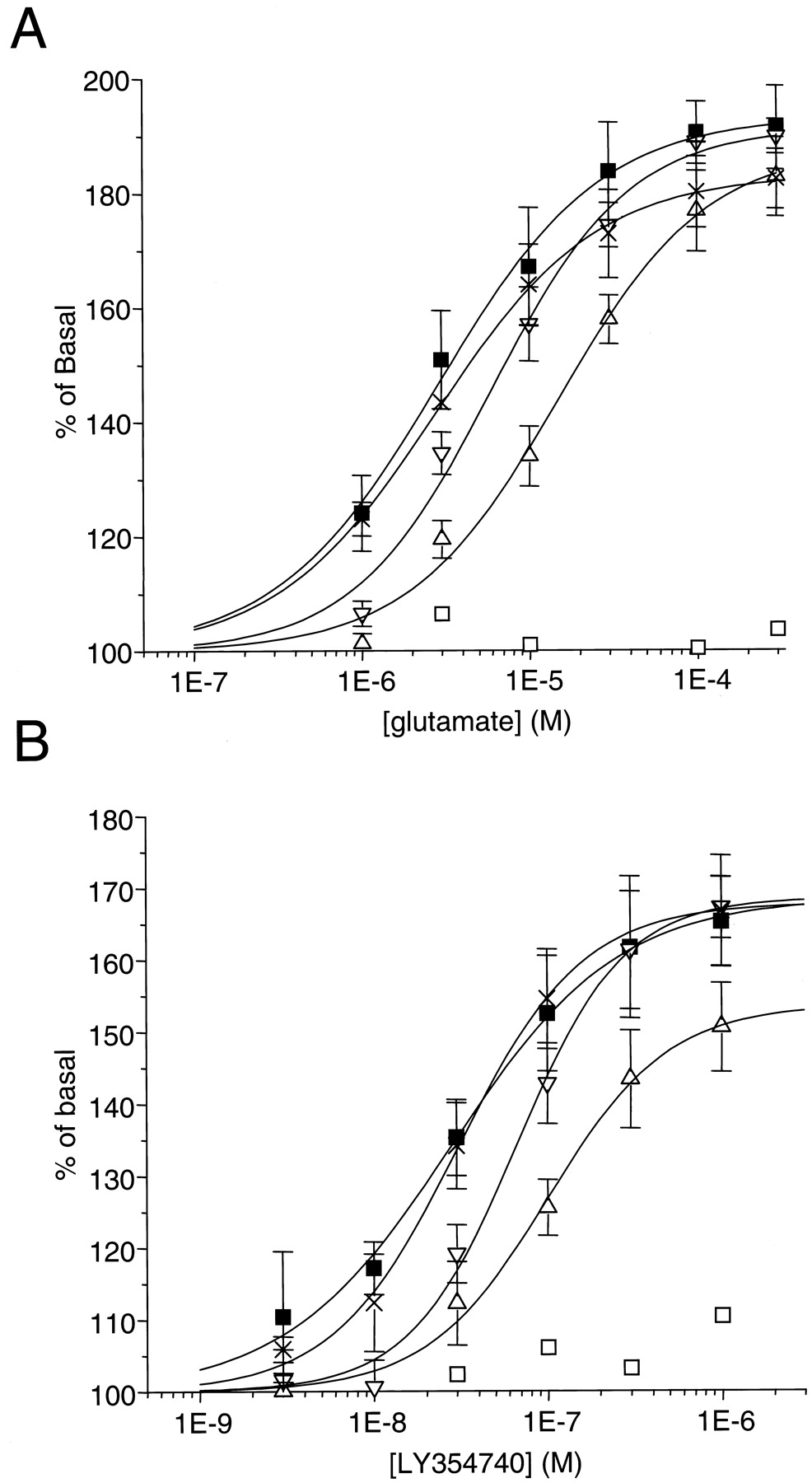
The functional effect of receptor activation was measured with the wild-type and selected mutant mGlu2 receptors using glutamate- and LY354740-stimulated GTPγ35S binding to the membranes of the transfected cells (Fig.5, Table2). In a test using membranes from cells expressing the wild-type mGlu2 receptor, glutamate and LY354740 induced a concentration-dependent increase in GTPγ35S binding that corresponded to nearly a doubling of the GTPγ35S binding in absence of agonist with glutamate and to a somewhat lower level of stimulation for LY354740. The EC50 values were 3 ± 1 μM and 28 ± 4 nM for glutamate and LY354740, respectively.
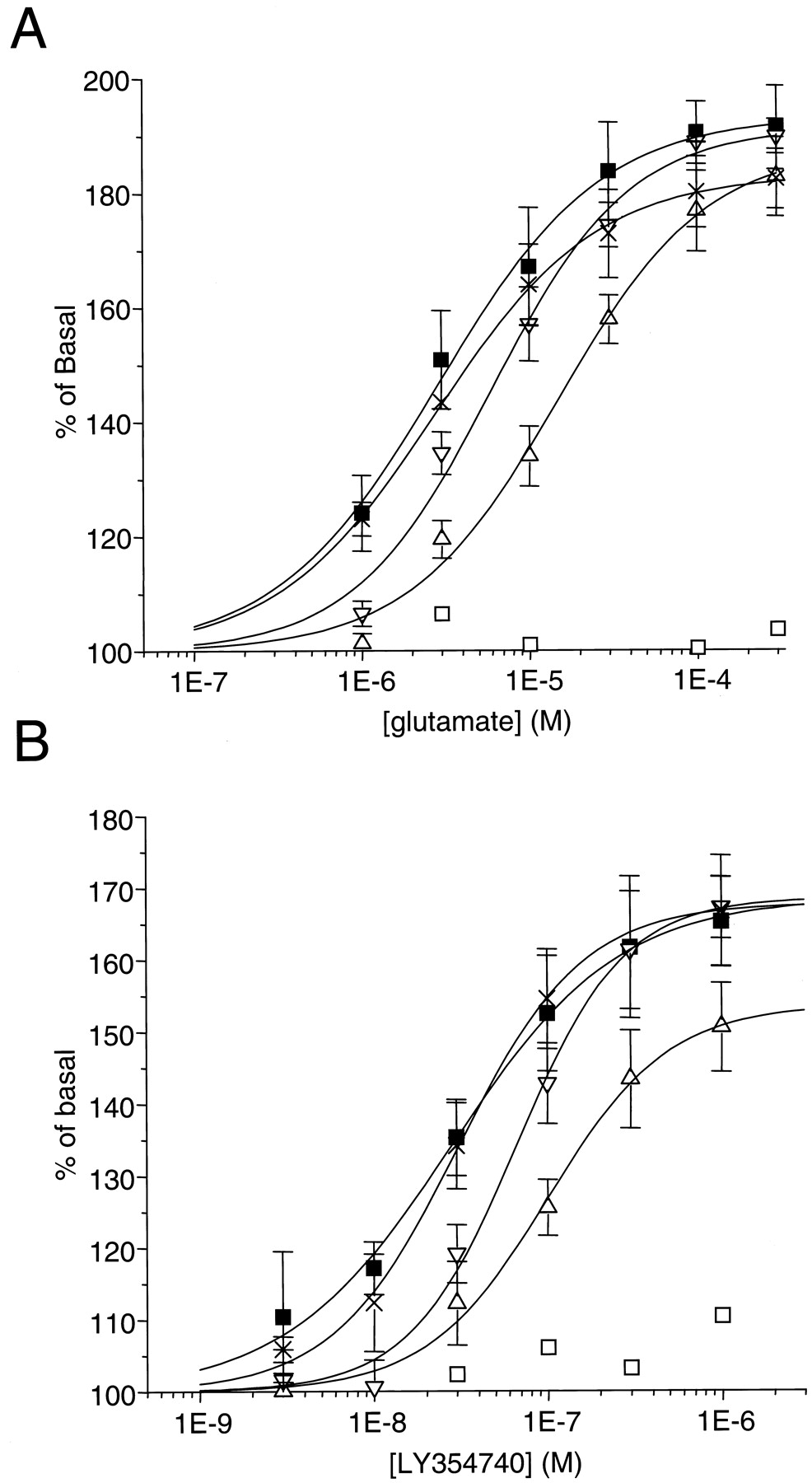
Concentration-dependent stimulation of GTPγ35S binding to mGlu2 wild-type (▪) and different point-mutants: T168A (■), Y144A (▵), Y144S (▿), D146S (×) by glutamate (A) and LY354740 (B). Results are expressed as percentage of basal and are mean ± S.D. of three individual experiments, performed in triplicate.
EC50 values for the glutamate- and LY354740 stimulated GTPγ35S binding on the HEK-293 cell membranes transfected with the mGlu2 wild-type and point-mutated receptors. Values are mean ± S.D. of the EC50 values, calculated from at least three individual experiments, performed in triplicate.
As shown in Table 2, the mutants A166S, S167A, and S169A, which did not exhibit significant changes in [3H]LY354740 affinity, did not display changes in affinity or efficacy in the functional assay with both agonists.
As expected, the S145A mutant, which did not bind [3H]LY354740, was not stimulated by the agonist. In agreement with the binding experiments, the mutant S148A was stimulated by LY354740 and glutamate with a much-reduced affinity; the T168A mutation, which led to a decrease of only 20-fold in [3H]LY354740 affinity, resulted in the total disappearance of functional responses with both agonists. The mutation of the tyrosine 144 (Y144S or Y144A) led to a decrease of both agonist potencies, although this difference was statistically significant only for the Y144A mutant. Finally, there was no effect of the mutation D146S on the EC50 values of glutamate and LY354740 in the functional assay despite a statistical significant effect on radioligand affinity.
Effect of Point Mutations on the GIRK Channel Current.
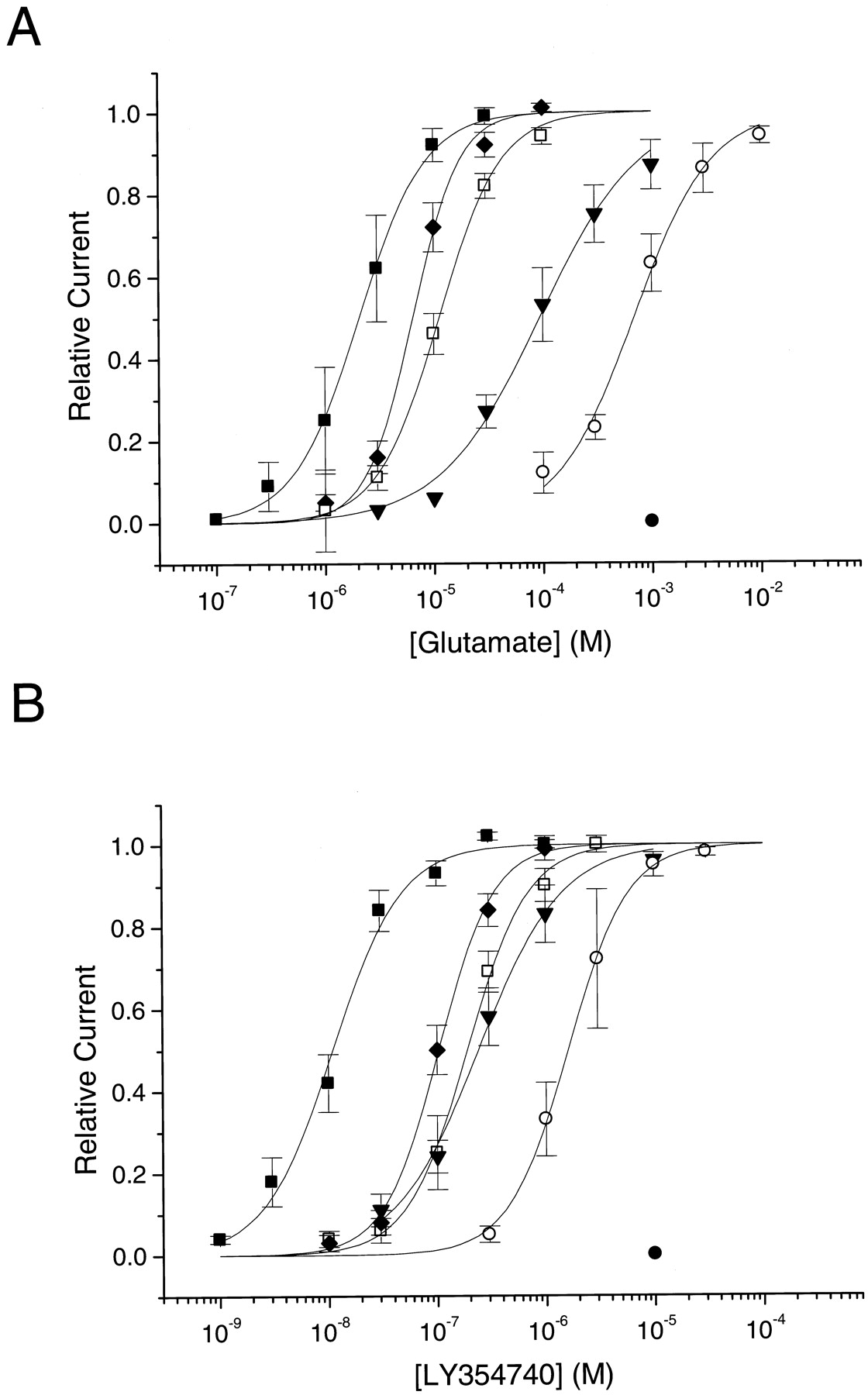
Members of the GPCR superfamily coupling through Go/Gi activate heterotetrameric G-protein-coupled GIRKs through a fast, membrane-delimited pathway involving the βγ subunits of heterotrimeric G-proteins. As shown previously for dopamine D2L- and D3- (Kuzhikandathil et al., 1998) and for mGlu receptors (Saugstad et al., 1996), GIRK activation by GPCRs can be used to assess pharmacological properties of GPCRs. Therefore, we transiently expressed the wild-type and selected critical point mutants of the mGlu2 receptor in a CHO line stably expressing concatenated GIRK1-GIRK2 channels. The concentration-response curves for GIRK current evoked by the glutamate and LY354740 are illustrated in Fig.6; their derivedI max, pEC50, and Hill coefficient (n H) values are shown in Table3. The concentration-response curves of glutamate and LY354740 obtained from cells expressing the mutated receptors were all shifted to the right (Fig. 6), reflecting a decrease in potency. The maximum currents were not significantly different for most mutations. Receptors carrying the D295A mutation were insensitive to glutamate and LY354740. The R183A mutation led to a loss in potency for glutamate and LY354740 and to lower Hill coefficients. Interestingly, mGlu2 receptors with the R183A mutation induced GIRK currents with a slower activation kinetic than that observed with wild-type mGlu2 receptors (data not shown). This suggests a regulatory role of Arg-183 in the agonist-induced conformational change of the mGlu2 receptor.

Glutamate (A) and LY354740 (B) concentration-response curves for GIRK current activation in CHO cells expressing different point-mutants of the mGlu2 receptor. The relative current amplitudes were measured and plotted for the cells expressing the mGlu2 wild-type (▪), Y144S (■), Y144A (♦), R183A (▿), R57A (○), D295A (●) receptors as described under Experimental Procedures. The points are mean values ± S.E.M. for three to six cells. The sigmoidal curves were generated with the mean EC50 and Hill coefficient values.
Imax, pEC50, and Hill coefficient (n H) values for the glutamate- and LY35474-evoked GIRK current in the CHO-GIRK1/2 stable line transfected with the mGlu2 wild-type and point-mutated receptors. The maximum current amplitudes from individual cells were fitted separately using the Hill equation (see Experimental Procedures). The values forI max, pEC50, and n Hrepresent the mean ± S.E.M. of three to six cells. The EC50 values of glutamate and LY354740 obtained from the mutated receptors are expressed as the fold difference from the EC50values obtained from the wild-type receptor.
Molecular Modeling of the ATD Domain of the mGlu2 Receptor.
As we described briefly above, we initially constructed a model of the mGlu2 ATD region by homology to the LIVBP (PDB code 2liv). Despite the limited homology (<20%), the model predicted many important residues. The recent publication of the X-ray structure of mGlu1 ATD region (Kunishima et al., 2000), with a 44% identity in the area of interest, offered an alternative template. Based on this crystal structure of mGlu1 (PDB code 1ewk), a 3D model of rat mGlu2 was built that starts at Lys-23 and ends at Pro-486. The segment Ala-108 to Pro-133 is not part of the model because the corresponding segment in mGlu1 is missing from the crystal structure. The alignment between mGlu1 and mGlu2 is obvious and none of the insertions/deletions seem to be near the binding site of the ligand. Where possible, the torsion angles of the amino acid side chains were kept similar to those in the crystal structure; otherwise, they were optimized in the Moloc force field (Gerber and Mueller, 1995). The model does not contain water molecules.
C-α representation of the homology model of mGlu2 is shown in Fig.7. LY354740 has been docked in the cleft between both domains of the ATC. Amino acids discussed in this work are highlighted. According to our model, they can be divided into two groups: (1) amino acids that are directly involved in the binding to glutamate and (2) amino acids that contribute to the overall stability of the three-dimensional structure of the domain and/or are involved in the closing mechanism.
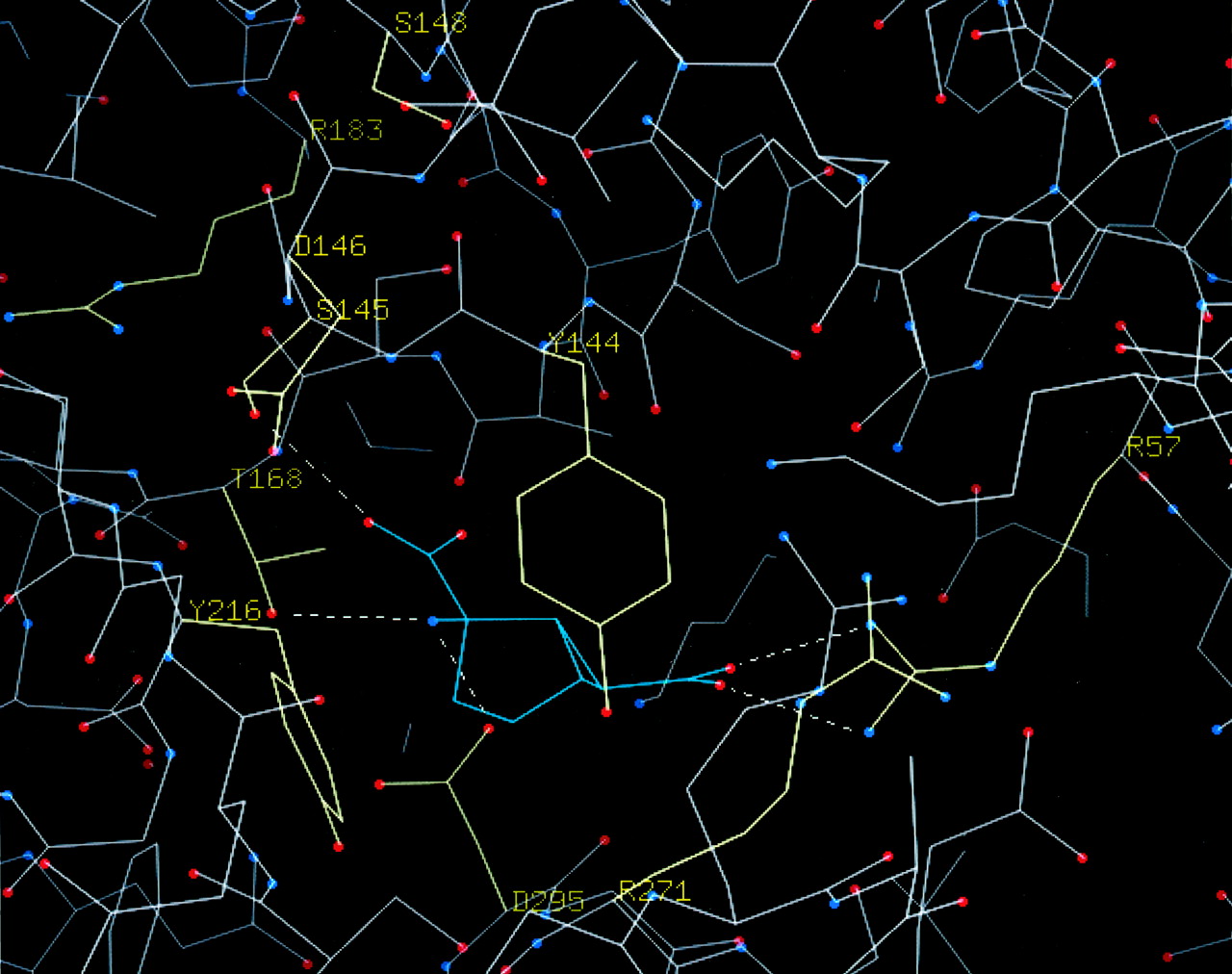
C-α representation of the binding site in the model of mGlu2 complexed with LY354740, based on the crystal structure of mGlu1 (Kunishima et al., 2000). The side chains of 10 crucial amino acids that were mutated in the current work are shown in yellow. Putative H-bonds between the mGlu2 and the ligand are shown as dashed lines.
Discussion
In the present work, we used site-directed mutagenesis and molecular modeling of group II mGlu receptors to elucidate critical residues involved in glutamate and LY354740 affinity and efficacy.
In general, all the mGlu2 receptor mutations resulted in a decrease of affinity for [3H]LY354740. However, three mGlu2 mutations, A166S, S167A, and S169A, which are all located in the vicinity of Thr-168, had no significant effect on either [3H]LY354740 affinity or glutamate affinity and efficacy in a functional assay. Previously, the mutation S181A in mGlu4a, which corresponds to Ser-167 of mGlu2, was reported to have only a moderate effect on l-AP4 affinity (Hampson et al., 1999).
Amino Acids Directly Involved in the Agonist Binding Site.
In an earlier study (O'Hara et al., 1993), it was demonstrated that the conversion of Ser-165 and Thr-188 of mGlu1 receptor (Ser-145 and Thr-168 in mGlu2) into alanine decreased the glutamate potency by 100- and 10,000-fold, respectively. In good agreement, we observed that both S145A and T168A mutations completely abolished glutamate- and LY354740-stimulated functional responses, although the T168A mutation decreased [3H]LY354740 affinity by only 21-fold. Recently, Hampson et al. (1999), probing the ligand-binding domain of the mGlu4a receptor, demonstrated that the two residues Ser-159 and Thr-182 (Ser-145 and Thr-168 in mGlu2) are also important molecular determinants for the binding of the agonistl-AP4. Similarly, the investigation of the agonist-binding domain of the other GPCR family 3 members has led to the conclusion that the Ser-147 and Ser-170 of Ca2+-sensing receptor (Brauner-Osborne et al., 1999) and the Ser-246 and Ser-269 of GABAB R1 (Ser-145 and Thr-168 in mGlu2) (Galvez et al., 1999) are key residues of the agonist-binding pocket. Indeed, this pocket is highly conserved and displays a similar structure among this subfamily of GPCRs. Our model of the mGlu2-LY354740 binding domain shows that the α-carboxyl group of LY354740 can form an H-bond with the hydroxyl group of Ser-145, whereas the α-amino group forms H-bonds with the side-chain hydroxyl group of Thr-168, both of which are amino acids that are conserved in all types of rat and human mGlu receptor.
Our mutational analysis also suggests an important role for Tyr-144 in the glutamate and LY354740 affinity at mGlu2 receptors. From the known crystal structure, we speculate that this tyrosine stabilizes the binding of LY354740 via C-H/π-interactions. In agreement with this hypothesis, the affinity of 2-amino-bicyclo-[3.1.0]hex-3-ene-2,6-dicarboxylate was only 2.4-fold less for the Y144S mutant than for the wild-type receptor whereas LY354740 affinity was 11-fold less for this mutant receptor, suggesting that the interaction with the tyrosine was already lost for the former compound. Indeed, replacing the tyrosine 144 with a phenylalanine had no effect on the affinity of [3H]LY354740. These observations indicate that the forces involved in the interaction of LY354740 with this tyrosine are direct C-H/π interactions rather than the indirect Van der Waals.
In our model, the γ-carboxyl group of LY354740 forms H-bonds to Arg-57, a residue that is not conserved in all rat mGlu receptors. mGlu2 and -3 receptors, which show the highest affinity for the LY354740 agonist, have an arginine at this position. mGlu8 and -4 receptors, which show a reduced affinity, have a lysine, whereas mGlu1 and -5 receptors, having the weakest affinity, have a tyrosine at this position. The mutations of Arg-57 to alanine, lysine, or tyrosine all led to the complete disappearance of [3H]LY354740 binding. As expected, the R57A mutation also resulted in 340-and 142-fold increased EC50 values for glutamate and LY354740, respectively, as assayed by electrophysiology.
When the closure of the venus flytrap around LY354740 was simulated, it became apparent that Asp-295 would come into sufficient proximity to have an important interaction with the agonist. It also showed that the α-amino group of LY354740 can form H-bonds to Asp-295, a residue conserved in all mGlu receptors. In agreement, the mutation of this residue (D295A or D295R) virtually eliminated the binding of the ligand to the receptor and led to a very large increase in EC50 values of glutamate (>1000 μM) and LY354740 (>10 μM) in the electrophysiological assay.
Two residues, Tyr-216 and Arg-271, which correspond to Tyr-236 and Glu-292 in mGlu1, respectively, were predicted by the mGlu1 3D structure to interact with glutamate (Kunishima et al., 2000). We found that the mutation Y216A had a detrimental effect on [3H]LY354740 affinity, whereas the mutation R271A had no significant effect. However, the conversion of tyrosine 216 (a residue conserved in all mGlus) to a phenylalanine also abolished the [3H]LY354740 binding. This may indicate that the forces involved in the interaction between Tyr-216 and ligand are not via directed C-H/π interactions. In the mGlu1 3D structure, Tyr-236 (Tyr-216 in mGlu2) makes an H-bond to Asp-318 (Asp-295 in mGlu2), which binds the α-amino group of the ligand. The binding partners of kainate at the iGlu2 receptor derived from X-ray crystal structure (PDB 1gr2) (Armstrong et al., 1998), namely Tyr-450, Arg-485, Thr-480, and Glu-705, each have a counterpart in our model of LY354740 binding (i.e., Tyr-216, Arg-57, Thr-168 and Asp-295, respectively).
Amino Acids That Contribute to the Overall Stability of the 3D Structure of the Domain and/or Are Involved in the Closing Mechanism.
Ser-148 was identified as an important residue for the binding of [3H]LY354740 to mGlu2 receptor. Mutagenesis of Ser-148 to alanine drastically decreased the affinity of LY354740 for mGlu2 receptor. Surprisingly, in both models, Ser-148 is located too far from the binding pocket for any direct interaction with the ligand. According to the 3D model, Ser-148 (Ala-168 in mGlu1) is positioned near Ser-164 (Ala-184 in mGlu1) and Ser-167 (Ala-187 in mGlu1). However, further mutational analysis showed that the mutations S164A had no effect and S167A decreased [3H]LY354740 affinity by only 3-fold. We suggest that this Ser-148, which is positioned on a β-sheet structure, might act as a relay stabilizing the 3D-conformation of the domain and/or promoting its closure.
Arg-183, which is highly conserved among mGlu receptors LIVBP (Arg-116;Sack et al., 1989), GABAB R1 (Arg-284; Galvez et al., 1999), and Ca2+-sensing receptor (Arg-185;Brauner-Osborne et al., 1999), is another residue that seemed to play an important role in the effect of the agonist. The replacement of Arg-183 by alanine had a profound influence on both glutamate and LY354740 potency. Their EC50 values were increased 49- and 21-fold, respectively, in the electrophysiological assay, whereas the same mutation had only a moderate effect on the binding of [3H]LY354740; theK D value increased by 11-fold. Interestingly, the mutation R185Q in the human Ca2+-sensor causes a familial hypocalciuric hypercalcemia (Pollak et al., 1993). When the mutated protein was transiently expressed in HEK-293 cells, the response to extracellular polyvalent cations (Ca2+, Gd3+) was attenuated and their affinity was reduced (Bai et al., 1996). The 3D model shows that the Arg-183 is located near the hinge region. The potency, rather than the affinity, of both glutamate and LY354740 was affected by this mutation, which indicates that, as in the Ca2+-sensor, this amino acid may play a dynamic role in the agonist-induced conformational change of the mGlu2 receptor.
It has been shown that Ca2+ and other polycations have a modulatory effect on mGlu receptors (Saunders et al., 1998). Extracellular Ca2+ activates mGlu1, -3 and -5 receptors in the absence of glutamate and the site of this activation has been attributed to the serine residue at position 166 of mGlu1 (Ser-152 in mGlu3 and -5) (Kubo et al., 1998). In agreement, the mGlu2 receptor, which is the only mGlu receptor to have aspartate at this position (Asp-146), was not activated by extracellular Ca2+ (Kubo et al., 1998). In our study, we have observed that aspartic acid 146 of mGlu2 was largely accountable for the difference in [3H]LY354740 affinity observed between mGlu2 and -3 (K D values of 20 and 82 nM, respectively). The replacement of Ser-152 of mGlu3 by aspartate converted the mGlu3 affinity for [3H]LY354740 to a value comparable with that of mGlu2 (K D value of 27 nM for mGlu3 S152D), whereas the mGlu3 mutation S152H did not change the [3H]LY354740 affinity compared with wild-type receptor. Hampson et al. (1999) have reported that the mutation of Ser-160 in mGlu4a (Ser-152 in mGlu3) into alanine did not affect the binding of the agonistl-[3H]AP4. Obviously, it would be interesting to test the effect of an aspartic acid at this position in the mGlu4 receptor. Very recently, Galvez et al. (2000)reported that Ca2+ was required for the high-affinity GABA binding at GABAB receptors and that the Ser-269 of GABAB R1 subunit was involved in the Ca2+-effect. Interestingly, Ca2+ had no effect on the high-affinity binding of baclofen at GABAB receptor. In analogy to the model of baclofen binding (Galvez et al., 2000), the presence of Asp-146 in mGlu2 (Ser-166 in mGlu1), which is adjacent to the glutamate binding site (Ser-145), might exclude the binding of Ca2+ to the mGlu2 and might therefore be responsible for the lack of Ca2+ sensitivity of this receptor; it would also confer a higher affinity of mGlu2 for the agonist. In agreement with this, we have observed that CaCl2 at 2 mM had no effect on the affinity of the agonist for the wild type mGlu2 receptor, although it tends to enhance the agonist affinity for the mGlu2 with D146S mutation.
Comparison of the Agonist Binding Sites of mGlu1 and mGlu2 Receptors.
In the present study, the crucial residues were identified on the basis of a mGlu2 model built on the 3D structure of LIVBP. The sequence identity between the amino-terminal domain of the mGlu1 and mGlu2 receptors is 44%. Recently, the availability of atomic coordinates of the mGlu1 ATD complexed with glutamate (Kunishima et al., 2000) provided an alternative template for modeling of the mGlu2 ATD, allowing their comparison. Of the 21 residues located at a distance of 6 Å from glutamate in the binding pocket, 11 differ between mGlu1 and mGlu2 receptors. Among the residues shown in the mGlu1 crystal structure to interact directly and/or via a water molecule with glutamate, Arg-78, Ser-165, Thr-188, Asp-208, Tyr-236, and Asp-318 (Arg-61, Ser-145, Thr-168, Asp-188, Tyr-216, and Asp-295, respectively, in mGlu2) are identical in all mGlu receptors. That other residues, such as Tyr-74 (Arg-57 in mGlu2), Ser-164 (Tyr-144 in mGlu2), Ser-186 (Ala-166 in mGlu2), and Glu-292 (Arg-271 in mGlu2), are different might be critical for imparting ligand specificity to mGlu receptors.
The mGlu1 3D structure also demonstrates the pivotal role played by H2O-mediated H-bonds and salt bridges in the glutamate-binding pocket. The replacement of residues Tyr-74, Trp-110, Ser-186, Glu-292, and Gly-293 of mGlu1 with those of mGlu2, Arg-57, Ser-93, Ala-166, Arg-271, and Ser-272, respectively, would create a spatially different ligand-binding pocket in which many of the original H2O-mediated H-bonds have disappeared while new contacts were established. These can be clearly seen in the case of Ser-186 and Glu-292 in mGlu1, two residues that interact with the γ- and α-carboxyl groups of glutamate via H2O-46 and H2O-17 links, respectively. Interestingly, the mutations of corresponding residues Ala-166 and Arg-271 in mGlu2 had limited effect on agonist affinity. The new mGlu2 model also shows that Ser-148 (Ala-168 in mGlu1), an important residue for LY354740 binding, is positioned too remotely for a direct interaction with the ligand. Unfortunately, the low resolution in this vicinity prevents a clear understanding of how the ligand affinity and efficacy could be affected so dramatically by this Ser-148.
Although the crystal structure of mGlu1 confirms many of our findings, including the role of Arg-57 and Asp-295, a better appreciation of the relative importance of other critical residues identified in our study will not be feasible until the X-ray crystal structure of the mGlu2 receptor extracellular domain complexed with agonist molecules has been determined.
Acknowledgments
We thank Prof. S. Nakanishi (Kyoto University, Kyoto, Japan) for the mGlu2 and -3 receptor cDNA clones, Prof. F. Diederich for invaluable advice and discussion on the model, Drs. J. G. Richards and J. N. C. Kew for their critical reading of the manuscript. We are grateful to Rachel Fimbel, Agnès Nilly, Sylvie Chaboz, Daniele Buchy, Veit Metzler, and Klaus Christensen for their excellent technical assistance.
Footnotes
- Received December 19, 2000.
- Accepted August 6, 2001.
Abbreviations
- NMDA
- N-methyl-d-aspartate
- AMPA
- (S)-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- mGlu
- metabotropic glutamate
- l-AP4
- l-(+)-2-amino-4-phosphono butyric acid
- GPCR
- G-protein-coupled receptor
- ATD
- amino-terminal domain
- LIVBP
- leucine/isoleucine/valine-binding protein
- iGlu
- ionotropic glutamate
- GABAB
- γ-aminobutyric acid, type B
- LY354740
- (+)-2-aminobicyclo-[3.1.0]-hexane-2,6-dicarboxylate
- GIRK
- G-protein coupled inwardly rectifying potassium channel
- GTPγS
- guanosine-5′-O-(3-thiotriphosphate)
- HEK
- human embryonic kidney
- CHO
- Chinese hamster ovary
- BAPTA
- 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
- 3D
- three-dimensional
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}