


Abstract
We have investigated the effects of G protein-coupled receptor kinase (GRK) 3 and GRK6 on the phosphorylation and regulation of the M3 muscarinic acetylcholine receptor (mACh) endogenously expressed in SH-SY5Y cells. Overexpression of GRK3 or GRK6 enhanced M3 mACh receptor phosphorylation after high-concentration methacholine (100 μM, 1 min) addition. However, GRK6 was more potent, increasing receptor phosphorylation even after low (3 μM, 1 min) agonist stimulation. Compared with plasmid-transfected control cells expressing equivalent M3 mACh receptor number, GRK3- or GRK6-overexpressing cells exhibited a reduced phospholipase C activity reflected by a lower accumulation of total [3H]inositol phosphates and Ins(1,4,5)P3 mass. In addition, direct stimulation of G protein activation of phospholipase C (by AlF4 −) was inhibited in GRK3- but not GRK6-overexpressing cells. Guanosine-5′-O-(3-[35S]thio)triphosphate binding and immunoprecipitation of Gαq/11 indicated that acute methacholine-stimulated receptor/Gαq/11 coupling was unaffected by GRK overexpression. In contrast, agonist pretreatment of cells for 3 min caused M3 mACh receptor uncoupling from Gαq/11, which was markedly enhanced by GRK6 overexpression, particularly at lower agonist pretreatment concentrations. However, the increased M3 mACh receptor phosphorylation seen in clones overexpressing GRK3 was not accompanied by increased receptor-Gαq/11 uncoupling. Overall, these data suggest that GRK3 and GRK6 use different pathways to desensitize the M3 mACh receptor. GRK6 seems to act as a classical GRK, inducing increased receptor phosphorylation accompanied by an uncoupling of receptor and Gαq/11. Conversely, GRK3 may cause desensitization independently of receptor phosphorylation, possibly via Gβγ binding and/or direct Gαq binding via its regulator of G protein signaling domain to inhibit phospholipase C activity.
Desensitization of G protein-coupled receptors is a fundamental process that prevents excessive or inappropriate signaling. The process of rapid G protein-coupled receptor desensitization has been studied extensively in receptor systems that are coupled to increases in intracellular cAMP, primarily the β2-adrenergic receptor (Pippig et al., 1993; Kong et al., 1994). These studies implicate receptor phosphorylation as the primary desensitizing event, either via the action of second messenger kinases [protein kinase A (Benovic et al., 1985; Clark et al., 1989) or protein kinase C (Pitcher et al., 1992b)] or through the action of specific G protein-coupled receptor kinases (GRKs; reviewed in Krupnick and Benovic, 1998; Pitcher et al., 1998). At present, the GRK family consists of six members (Krupnick and Benovic, 1998). GRKs 2, 3, 5, and 6 are widely distributed in most tissues (Sterne-Marr and Benovic, 1996), whereas GRK1 (rhopodsin kinase) is found specifically in the retina (Somers and Klein, 1984) and GRK4 is found only in the testis and certain areas of the brain (Ambrose et al., 1992; Sallese et al., 2000b). GRKs cause homologous desensitization, phosphorylating only agonist-occupied receptors, leading to the binding of arrestins, which sterically suppress G protein interaction, thus terminating the signal (Lohse et al., 1990;Gurevich et al., 1995; Carman and Benovic, 1998).
Despite the abundance of data implicating GRK-mediated desensitization of adenylyl cyclase-linked receptor systems, relatively few studies have examined their role in the regulation of phospholipase C (PLC)-coupled receptors. These studies indicate the involvement of GRKs 2 and 3 in the desensitization of the substance P receptor (McConalogue et al., 1998; Barak et al., 1999), the α1B-adrenergic receptor (Diviani et al., 1996a,b), the type 1A angiotensin II receptor (Oppermann et al., 1996), and the endothelin A and B receptors (Freedman et al., 1997). Several studies have also shown that purified GRKs phosphorylate both M1 and M3 muscarinic acetylcholine (mACh) receptors (DebBurman et al., 1995; Haga et al., 1996). Furthermore, Tsuga et al. (1998) found that overexpression of both the M3 mACh receptor and GRK2 increased agonist-mediated receptor internalization. However, none of the studies involving the M3 mACh receptor have directly linked receptor phosphorylation with a measure of receptor desensitization, particularly in intact cellular preparations. This is important because GRKs may also inhibit receptor signaling through nonphosphorylation processes such as binding of free Gβγ subunits (which can themselves activate PLC; Haga and Haga, 1992; Pitcher et al., 1992a; Koch et al., 1993). In addition, two groups have recently shown that GRKs 2 and 3 are able to interact directly with activated GTP bound Gαq (Carman et al., 1999; Sallese et al., 2000a). This binding of GRKs 2 or 3 to Gαqthrough their N-terminal regulator of G protein signaling (RGS) domain is proposed to mediate their inhibitory effects on PLC signaling.
We have previously shown that recombinant human M1 and M3 mACh receptors overexpressed in CHO cells undergo agonist-stimulated phosphorylation and subsequent desensitization (Tobin, 1997; Tobin et al., 1997; Waugh et al., 1999). Although shown to enhance phosphorylation of the receptor, casein kinase 1α does not affect desensitization of the M3 mACh receptor (Budd et al., 2000). Thus, if phosphorylation is responsible for receptor desensitization, then other kinases, such as GRKs, should be considered. Therefore, we have examined the potential roles of GRK3 (which contains an RGS domain) and GRK6 (which has no RGS domain) in the desensitization of the endogenously expressed M3 mACh receptor population of SH-SY5Y cells. In this study both GRK3 and -6 enhance endogenous M3 mACh receptor phosphorylation and desensitization. Our data suggest that GRK6 acts as a classical GRK, through receptor phosphorylation accompanied by uncoupling of G protein interaction. In contrast, GRK3 seems to enhance M3 mACh receptor desensitization via a phosphorylation-independent pathway, possibly through direct binding of Gαq or free Gβγ.
Experimental Procedures
Cell Culture and Creation of Stably Transfected GRK Cell Lines.
SH-SY5Y human neuroblastoma cells were cultured in minimal essential medium containing 5% fetal calf and 5% newborn calf serums, 100 units/ml penicillin, 100 μg/ml streptomycin, and 2.5 μg/ml fungizone (Invitrogen, Paisley, UK). All cells were maintained at 37°C in humidified conditions under 5% CO2. Wild-type SH-SY5Y cells were transfected with either pcDNA3 alone, bovine GRK3, or human GRK6 cloned into pcDNA3, using FuGENE 6 according to the manufacturer's instructions. After 48 h, cells were subjected to geneticin (300 μg/ml) treatment. Surviving colonies were selected and expanded into cell lines.
Western Blotting.
Cells were lysed and subjected to electrophoretic separation exactly as described previously (Mundell et al., 1998). Separated protein was transferred to nitrocellulose and GRK expression was detected using anti-rabbit polyclonal IgG antibodies (1:1000 dilution) specific for GRKs 3 or 6 (Santa Cruz Biotechnology, CA). Gαq/11 expression was detected using an anti-rabbit polyclonal IgG (Santa Cruz Biotechnology, Santa Cruz, CA). Protein expression was determined by addition of enhanced chemiluminescence reagent (ECL; Amersham Pharmacia Biotech UK, Ltd., Little Chalfont, Buckinghamshire, UK), according to the manufacturer's instructions, and exposure to Hyperfilm (Amersham Pharmacia Biotech).
Determination of mACh Receptor Number and Receptor Internalization.
Cells were grown in 24-well plates until confluent. Cell monolayers were washed once with 1 ml of Krebs buffer (118.6 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 4.2 mM NaHCO3, 10 mM HEPES, 11.7 mM glucose, and 1.3 mM CaCl2), pH 7.4 before incubation with increasing concentrations of the nonmembrane permeable muscarinic antagonist, [3H]N-methylscopolamine (NMS; Amersham Pharmacia Biotech). To determine nonspecific binding, the mACh receptor antagonist atropine (20 μM) was included. After incubation at 37°C for 1 h, cell monolayers were washed twice with 1 ml of ice-cold Krebs buffer, pH 7.4, and bound [3H]NMS was extracted by addition of 0.5 ml of 0.1 M NaOH for 30 min. Samples were neutralized by addition of 0.5 ml of 0.1 M HCl and binding quantified by liquid scintillation counting.
M3 mACh receptor internalization was assessed after treatment with either vehicle or methacholine (100 μM) for 3 or 30 min at 37°C, before intensive washing (4 × 1 ml of ice-cold Krebs buffer, pH 7.4). [3H]NMS binding sites were determined as above using a single saturating concentration (5 nM) of [3H]NMS, for 18 h at 4°C. Nonspecific binding was determined as described above. Receptor internalization was determined as the percentage loss of specific [3H]NMS binding sites after methacholine treatment compared with vehicle-treated control cells.
M3 mACh Receptor Phosphorylation.
The effects of GRK overexpression on the phosphorylation of endogenously expressed M3 muscarinic receptors was assessed by the method of Tobin and Nahorski (1993). Briefly, either plasmid control, or cells overexpressing either GRK3 or GRK6 were seeded into 6-well culture plates. Confluent cells were loaded with [32P]orthophosphate (specific activity, 5 μCi/ml; Amersham Pharmacia Biotech) in phosphate-free Krebs, pH 7.4, for 1 h before agonist challenge (methacholine 3 or 100 μM). After either 1 or 3 min, agonist was removed and the cells were solubilized by the addition of the following buffer: 10 mM Tris, 1 mM EDTA, 1% (v/v) NP40, 5 mg/ml deoxycholate, 100 μg/ml benzamidine, and 1 mM phenylmethylsulfonyl fluoride, pH 7.4, for 30 min on ice. Solubilized cell extracts were removed and centrifuged at 14,000 rpm for 5 min. The supernatant was added to a rabbit anti-M3 muscarinic receptor antibody (1:1000) raised against the third intracellular loop of the receptor (Tobin and Nahorski, 1993) to facilitate immunoprecipitation. After 1 h on ice, protein A Sepharose was added and the samples were placed on a roller for 30 min at 4°C. The protein A Sepharose beads were washed 3 times with the following buffer: 100 mM Tris, 1 to 5 M NaCl, 0.5% (v/v) Tween 20, and 3 times in 10 mM Tris, 1 mM EDTA, pH 7.4, before addition of 2× SDS-PAGE loading buffer. Samples were heated for 3 min at 85°C before separation by SDS-PAGE (8% acrylamide gel). Before gel loading, samples were equalized for receptor expression quantified by [3H]NMS binding. Gels were dried and the extent of phosphorylation was assessed by autoradiography, and subsequent densitometry using the Chemidoc gel documentation system (Bio-Rad, Hertfordshire, UK).
Casein Phosphorylation.
To determine whether overexpression of GRK3 or GRK6 produced functionally active kinases, GRK3 or GRK6 cells were lysed with the following buffer: 20 mM Tris/HCl, pH 7.6, 0.5% (v/v) NP40, 250 mM NaCl, 3 mM EDTA, 3 mM EGTA, 1 mM phenylmethylsulfonyl fluoride, 5 μg/ml benzamidine, and 1 mM dithiothreitol, for 15 min on ice. Lysates were then removed and centrifuged at 20,000g, at 4°C, for 5 min. The supernatant was removed and added to either GRK3 or GRK6 polyclonal antibodies (5 μg/ml). Samples were left on ice for 1 h before the addition of protein A Sepharose. After 30 min of rolling at 4°C, samples were washed twice with 10 mM Tris, pH 7.4, 1 mM EDTA buffer and once with kinase buffer (20 mM Tris, pH 7.5, 2 mM EDTA, and 5 mM MgCl2). After the final wash, the Sepharose beads were resuspended in reaction mix (5 μg of dephosphorylated α-casein, 10 μM unlabeled ATP, 5 μCi of [γ-32P]ATP, and kinase buffer to a total reaction volume of 20 μl) and left to incubate for 10 min at 37°C. The reaction was terminated by centrifugation (20,000g for 2 min) and 15 μl of the supernatant was removed and added to 2× SDS-PAGE loading buffer. Samples were separated by SDS-PAGE electrophoresis. Gels were dried and the extent of phosphorylation was assessed by autoradiography.
Measurement of Total [3H]Inositol Phosphate Accumulation and Ins(1,4,5)P3 Mass.
Either plasmid control or cells overexpressing GRK3 or GRK6 were seeded into 24-well plates at ∼50% confluence. After 24 h, cells were loaded with [3H]inositol (1 μCi/ml) in geneticin-free medium for a further 24 h. Confluent cell monolayers were then washed twice with 1 ml of Krebs buffer, pH 7.4, and incubated for 15 min at 37°C. LiCl (final concentration, 10 mM) was added to each well for 10 min, before addition of methacholine. The reaction was terminated via the addition of 0.5 M trichloroacetic acid and the samples were left on ice for 30 min. Total [3H]inositol phosphates were mixed with EDTA (50 μl, 10 mM) and extracted via addition of 0.6 ml of a 1:1 (v/v) mix of tri-n-octylamine and 1,1,2-trichlorotrifluoroethane. A 450-μl sample of the aqueous phase was removed and added to NaHCO3 (62.5 mM). The [3H]inositol phosphate fraction containing inositol mono-, bis- and tris-phosphates was recovered by ion-exchange chromatography on Dowex AG1-X8 (chloride form) columns as described previously (Challiss et al., 1993). A similar incubation/acid extraction protocol was used to assess Ins(1,4,5)P3 mass, except that cells were not prelabeled with [3H]inositol and LiCl was omitted. Ins(1,4,5)P3 mass levels were assessed as reported previously (Challiss et al., 1988).
Assessment of M3 mACh Receptor Desensitization by [35S]GTPγS Binding.
Plasmid control, GRK3- or GRK6-overexpressing cells were grown in 80-cm2cell culture flasks until confluent. Cells were then harvested in buffer [10 mM HEPES, pH 7.4, 0.2% (v/v) EDTA, 0.9% (v/v) NaCl]. After centrifugation, cells were resuspended in Krebs buffer, pH 7.4, at 37°C for 15 min before the addition of either vehicle or various concentrations of methacholine (0.01, 0.3, 3, or 100 μM). After 3 min, excess ice-cold Krebs buffer (50 ml) was added and the cells pelleted at 1000g for 5 min. The cell pellets were then resuspended in 30 ml of HEPES/EDTA buffer (20 mM HEPES, pH 7.4, 10 mM EDTA) and homogenized at maximum speed for 30 s, using a Polytron PT210 homogenizer (Kinematica, Basel, Switzerland). The resulting suspension was centrifuged at 20,000g for 15 min. The pellet was resuspended in 30 ml of a second HEPES/EDTA buffer (20 mM HEPES, pH 7.4, 0.1 mM EDTA) buffer and centrifuged for a further 15 min at 20,000g. The resultant pellet was resuspended at 1 mg/ml of protein and stored at −80°C until required.
[35S]GTPγS binding and immunoprecipitation were performed as described previously (Barr et al., 1998; Young et al., 2000; Akam et al., 2001). To each tube, the following was added: 1 μM GDP and either 100 μM methacholine, assay buffer (100 mM HEPES, 100 mM NaCl, and 10 mM MgCl2, pH 7.4), unlabeled 10 μM GTPγ S for determination of nonspecific binding, and 1 nM [35S]GTPγS (PerkinElmer Life Science Products, Boston, MA), in a volume of 50 μl. The tubes were warmed for 2 min at 30°C before the addition of 50 μg of membranes prepared as above. After 2 min of incubation, the reaction was terminated via the addition of 1 ml of ice-cold assay buffer and the membrane fraction was pelleted by centrifugation at 20,000gfor 5 min at 4°C. The pellets were resuspended in solubilization buffer [100 mM Tris/HCl, pH 7.4, 200 mM NaCl, 1 mM EDTA, 1.25% (v/v) NP40, and 0.2% (v/v) SDS]. Solubilized pellets were diluted further with 50 μl of solubilization buffer minus SDS and precleared via the addition of 1.3% (v/v) rabbit serum and 30 μl of protein A Sepharose, for 1 h at 4°C. The samples were centrifuged at 20,000g, 4°C, for 5 min. The supernatant (100 μl) was removed and added to anti-Gαq/11 antibody. Samples were incubated at 4°C overnight before addition of 70 μl of protein A Sepharose. After a further incubation at 4°C for 1.5 h the Sepharose beads were washed three times with 0.5 ml of solubilization buffer minus SDS. Finally all solubilization buffer was removed and scintillation fluid added before quantification by liquid scintillation counting. Desensitization was determined as a reduction in [35S]GTPγS binding after pretreatment with methacholine, and expressed as a percentage of the response found compared with a nonpretreated matched control cell. In addition to desensitization experiments, the kinetics of M3mACh receptor activation of Gαq/11 was assessed in nonpretreated cell membranes via generation of methacholine (300 nM to 1 mM) concentration-response curves, and time-course studies (0- to 15-min activation with 100 μM methacholine).
Data Analysis.
All concentration-response curves were fitted and EC50 values were determined using nonlinear regression analysis using Graphpad Prism II software. All data were analyzed using one or two-way analysis of variance (Excel 5.0; Microsoft, Redmond, WA). Significance was accepted whenp < 0.05.
Results
Creation of Stable GRK-Overexpressing SH-SY5Y Cell Lines.
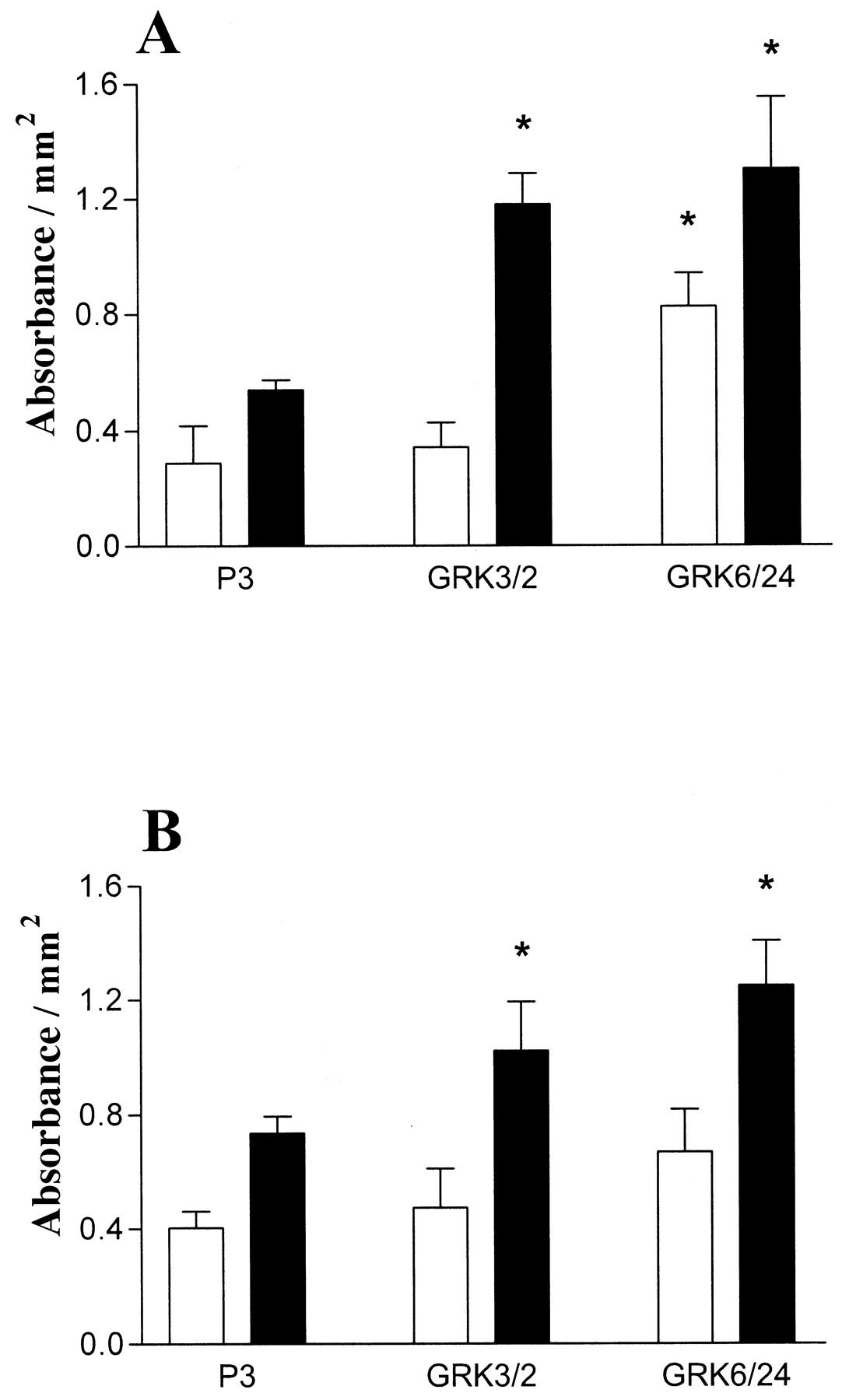
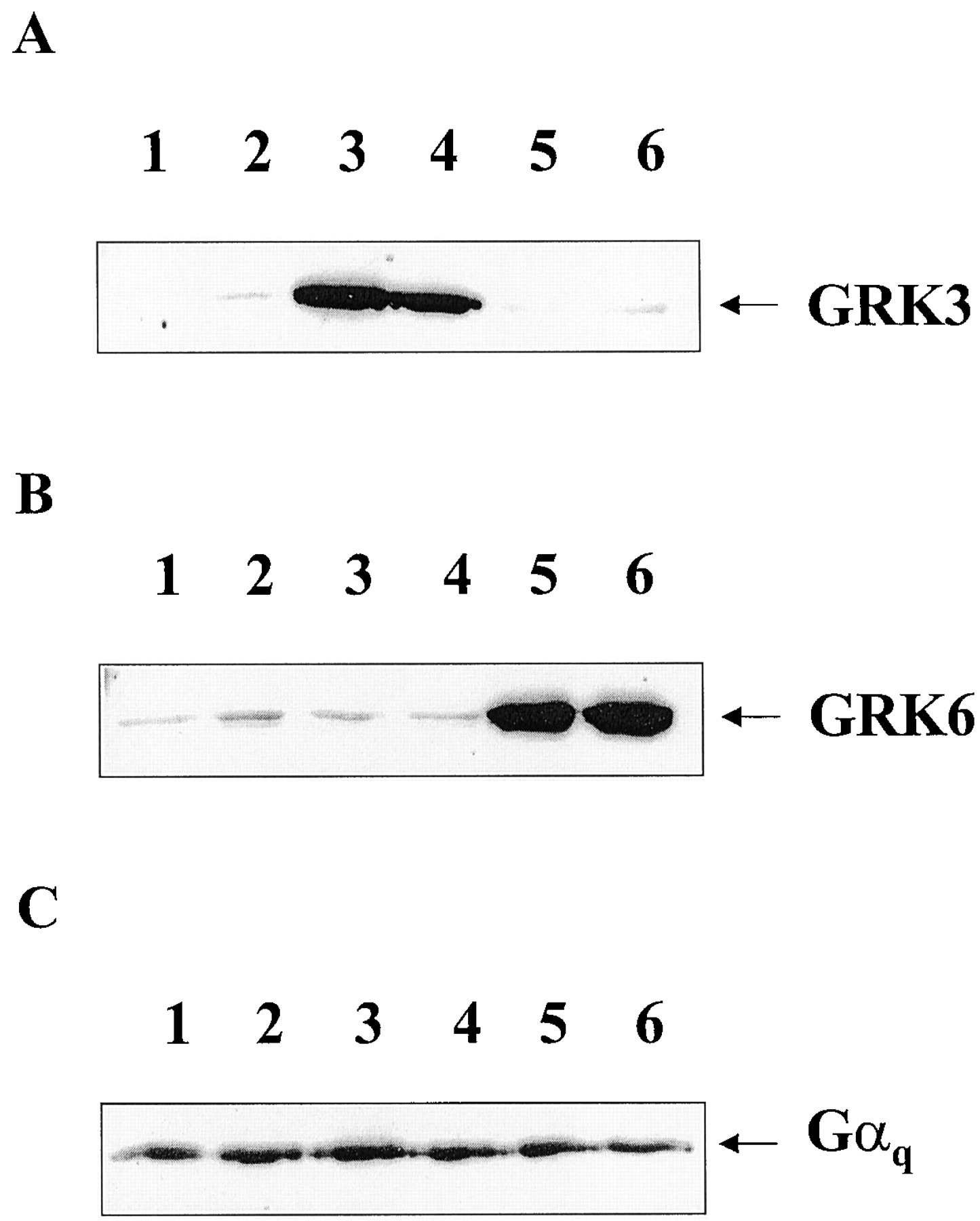
Wild-type SH-SY5Y neuroblastoma cells were transfected with either bovine GRK3 or human GRK6 in pcDNA3 or empty vector (pcDNA3). After culture with geneticin (300 μg/ml), surviving clones were isolated and expanded into cell lines. Several clones were identified by Western blot to express enhanced levels of either GRK3 or GRK6. From these clones at least two expressing varying amounts of either GRK3 (clones 2 and 12) or GRK6 (clones 6 and 24) were chosen for further study along with two plasmid-transfected control cell lines. Relative expression levels compared with endogenous GRK3 or -6 expression for the overexpressing clones are shown in Fig.1, A and B. To determine whether the overexpressed kinases were active, GRKs 3 or 6 were immunoprecipitated and dephosphorylated bovine α-casein was used as a substrate for phosphorylation with [γ-32P]ATP. In each cell line, overexpression of either GRK3 or GRK6 produced a greatly increased phosphorylation of casein compared with the endogenous level of kinase activity immunoprecipitated from plasmid control cells (Fig.1C). Enhanced phosphorylating activity was also found using rhodopsin from rod outer segments as substrate (data not shown), again indicating that both GRK3 and GRK6 overexpression produces active kinases. Determination of kinase overexpression levels as undertaken by Western blot comparison of sequentially diluted cell lysate with plasmid controls (40 μg of protein). GRK3 and GRK6 overexpression were estimated at 25- and 30-fold (for GRK3 clones 2 and 12) and 30- and 25-fold (for GRK6 clones 6 and 24) greater than plasmid control levels, respectively (data not shown). Gαq/11 levels were also shown to be similar in all clones examined (Fig. 1D).

Determination of GRK overexpression and activity in SH-SY5Y cells stably transfected with either GRK3 or GRK6. Whole-cell lysates were subjected to SDS-PAGE followed by Western transfer and immunoblotting with either a polyclonal rabbit antibodies that recognize either GRK3 (A) or GRK6 (B). In A and B, lysates were corrected for protein and 40 μg was loaded per lane. A, lane 1, clone P1 (plasmid control); lane 2, clone 2 (GRK3-overexpressing); lane 3, clone P3 (plasmid control); and lane 4, clone 12 (GRK3-overexpressing) cells. B, lane 1, clone P1 (plasmid control); lane 2, clone 6 (GRK6-overexpressing); lane 3, clone P3 (plasmid control); and lane 4, clone 24 (GRK6-overexpressing) cells. C, GRKs 3 and 6 were immunoprecipitated from either P1 (lane 1), clone 12 (lane 2), clone 2 (lane 3), P3 (lane 4), clone 6 (lane 5), or clone 24 (lane 6) before addition of [32P]ATP and dephosphorylated α-casein for 10 min at 37°C. GRKs were removed from the reaction mix by centrifugation and the resultant supernatant separated by SDS-PAGE. α-Casein phosphorylation was determined by autoradiography. Data are representative of three separate experiments. D, representative blot showing Gαq/11 levels in either P3 (plasmid control, lane 1), clone 2 (GRK3-overexpressing, lane 2), or clone 24 (GRK6-overexpressing, lane 3) membranes. Protein levels were equilibrated and 30 μg was loaded per lane before SDS-PAGE gel electrophoresis followed by Western transfer and immunoblotting with a polyclonal rabbit anti-Gαq/11 antibody.
Determination of M3 mACh Receptor Number.
To determine whether overexpression of either GRK3 or GRK6 in SH-SY5Y cells had any effects on M3 mACh receptor number, we undertook whole-cell binding studies with [3H]NMS. The B maxand K D values shown in Table1 indicate that overexpression of either GRK3 or GRK6 had no significant effect on M3 mACh receptor expression. Furthermore, receptor expression did not alter with cell passage number (data not shown).
Whole cell [3H]NMS saturation binding to determine M3 mACh receptor expression in plasmid control, GRK3 and GRK6 overexpressing clones.
M3 mACh Receptor Phosphorylation.
The ability of GRKs 3 and 6 to increase the phosphorylation of the M3 mACh receptor after addition of methacholine (3 or 100 μM) was assessed in P3, GRK3 clone 2, and GRK6 clone 24, and these data are shown here; similar data were obtained for other clones (data not shown). Figure 2, A and B, show representative autoradiograms of M3 mACh receptor phosphorylation after 1 min of agonist exposure. Overexpression of GRK3 or GRK6 was accompanied by increased receptor phosphorylation after addition of a high methacholine (100 μM) concentration (Fig. 2B). In contrast, only GRK6 overexpression increased receptor phosphorylation at the lower methacholine concentration studied (Fig. 2A). Densitometric analysis confirmed that both GRK3 and GRK6 overexpression produced significantly increased (p < 0.05) M3 mACh receptor phosphorylation above that seen in the plasmid control, P3 (Fig.3, A and B). GRK3 overexpression produced greater M3 mACh receptor phosphorylation only after 100 μM methacholine, whereas GRK6 overexpression enhanced phosphorylation after 3 or 100 μM methacholine (Fig. 3A). Interestingly, the enhanced level of M3mACh receptor phosphorylation seen with GRK6 and GRK3 overexpression was most pronounced at 1 min, and was less evident after 3 min of methacholine stimulation because the level of receptor phosphorylation in the control cells begins to approach that observed in cells overexpressing GRK3 or GRK6 (Fig. 3B).
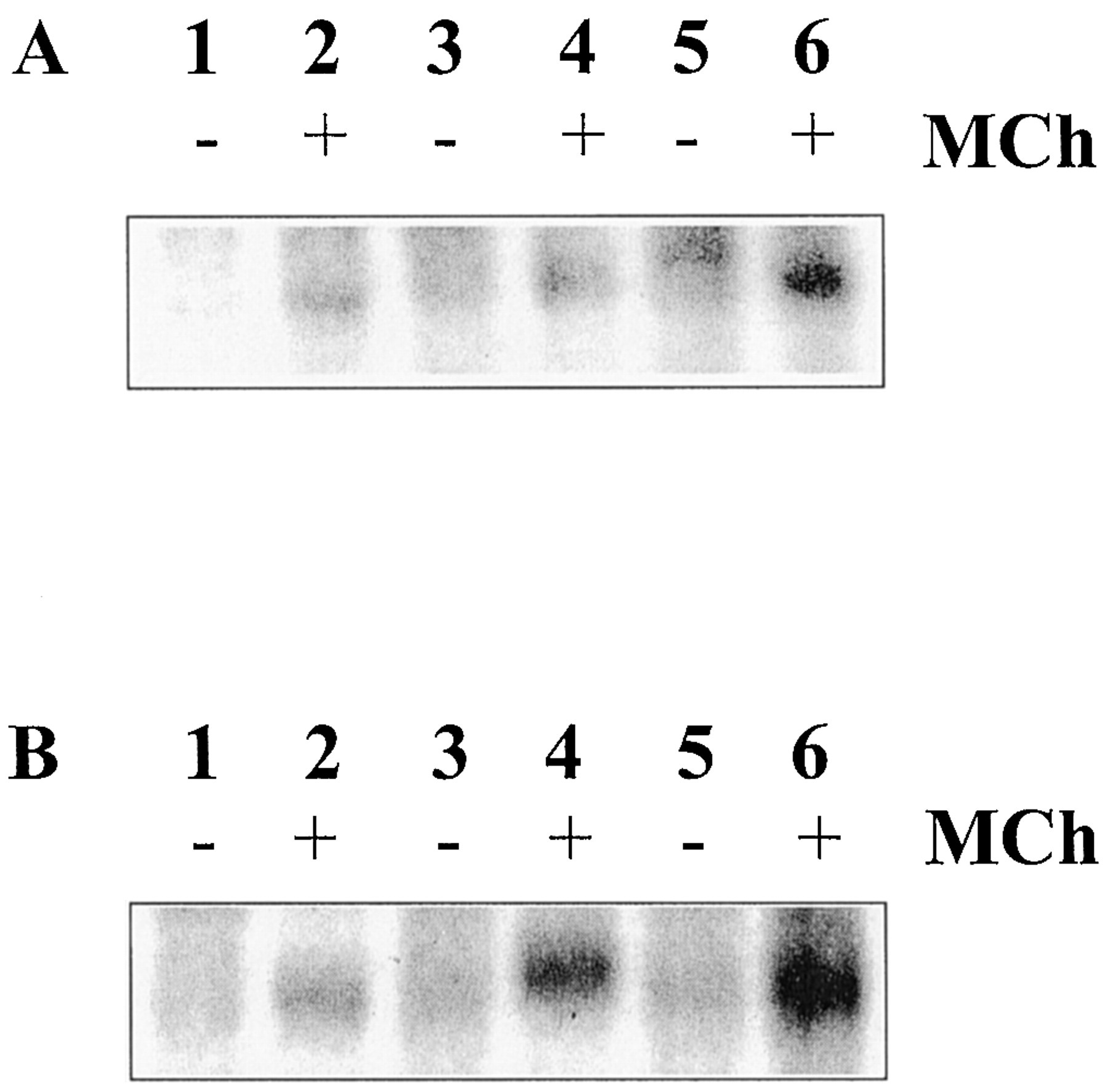
Effects of GRK overexpression on agonist-induced M3 mACh receptor phosphorylation. Confluent P3, clone 2 (GRK3-overexpressing), or clone 24 (GRK6-overexpressing) cells were loaded with [32P]orthophosphate (5 μCi/ml) in phosphate-free Krebs buffer, pH 7.4. After 1 h, cells were challenged with methacholine (3 or 100 μM) for 1 min. Next, the cell monolayers were solubilized and the M3 mACh receptors immunoprecipitated with a specific M3 mACh receptor antibody. Samples were process as described under Experimental Procedures and corrected for receptor expression, before quantification of [32P]orthophosphate incorporation by autoradiography. Representative autoradiograms are shown for M3 mACh receptor phosphorylation after 3- (A) and 100- (B) min methacholine stimulation. A and B, lanes 1 and 2, P3 cells; lanes 3 and 4, GRK3 clone 2; and lanes 5 and 6, GRK6 clone 24.

Analysis of the effects of GRK overexpression on methacholine stimulated M3 mACh receptor phosphorylation. Samples prepared as described under Experimental Procedures and in Fig. 2 were subjected to densitometric analysis using the Chemidoc image analysis system and software (Bio-Rad). The level of M3 mACh receptor phosphorylation after either 1 or 3 min of methacholine stimulation (■, 3 μM; ▪, 100 μM) is shown in A and B, respectively. Data are expressed as mean ± S.E.M. of three separate experiments after subtraction of basal phosphorylation. Basal phosphorylation levels were as follows: P3, 0.31 ± 0.08; GRK3 clone 2, 0.33 ± 0.06; GRK6 clone 24, 0.29 ± 0.07 (mean ± S.E.M., absorbance units/mm2). *p < 0.05; significantly greater M3 mACh receptor phosphorylation compared with plasmid control cells as measured by Student's ttest.
Total [3H]Inositol Phosphate Accumulation Studies.
Accumulation of total [3H]inositol phosphates was measured to assess agonist-stimulated PLC activity. In all cases, clones overexpressing either GRK3 or GRK6 produced significantly less total [3H]inositol phosphates after methacholine stimulation than plasmid control cells (Fig.4). To examine in greater detail the effects of GRK3 and GRK6 on M3 mACh receptor desensitization, concentration-response curves to methacholine were produced at 3 min (Fig. 5). The data show that both GRK3 and GRK6 overexpression significantly reduced the ability of the M3 mACh receptor to stimulate [3H]inositol phosphate accumulation. Moreover, the degree of inhibition correlated with overexpression of kinase, especially in the case of GRK6. Although maximal accumulation of [3H]inositol phosphates was decreased in clones overexpressing GRKs 3 and 6, the EC50 values were not significantly altered (data not shown). These data were confirmed and extended by measuring agonist-stimulated Ins(1,4,5)P3 accumulation in the P3, GRK3-overexpressing (clone 2) and GRK6-overexpressing (clone 24) clones. Basal Ins(1,4,5)P3 levels were similar in all cell lines (P3, 23.9 ± 4.8; GRK3 clone 2, 23.7 ± 6.4; and GRK6 clone 24, 20.1 ± 4.9 pmol/mg protein, n= 4). Time course studies clearly demonstrated that although in GRK6 cells, the initial peak Ins(1,4,5)P3 accumulation was similar to that seen in P3 cells (P3, 85.1 ± 6.7; GRK6 clone 24, 86.4 ± 13.6 pmol/mg protein, n = 4), the response in GRK3 cells was acutely ablated (GRK3 clone 2, 36.3 ± 5.0 pmol/mg protein; n = 4; p < 0.01). In agreement with the total [3H]inositol phosphate data, the concentration-dependencies for Ins(1,4,5)P3 accumulation after methacholine stimulation for 3 min were similar (EC50 values of 11.1, 17.9, and 11.4 μM for P3, GRK3 clone 2, and GRK6 clone 24 cells lines, respectively), whereas maximal responses were reduced consistently with the data reported in Fig. 5B.

Effects of GRK overexpression on methacholine (100 μM) stimulated total [3H]inositol phosphate accumulation in either: A, P1 (control, ▪) or clone 12 (GRK3-overexpressing, ■) cells; B, P3 (control, ▪) or clone 2 (GRK3-overexpressing, ■) cells; C, P1 (▪) or clone 6 (GRK6-overexpressing cells, ■); and D, P3 (▪) or clone 24 (GRK6-overexpressing, ■) cells. Total [3H]inositol phosphate accumulation was determined as described underExperimental Procedures. Data are expressed as the mean ± S.E.M. for three separate experiments. Total [3H]inositol phosphate accumulation was significantly lower (p < 0.001) for clones overexpressing GRKs 3 or 6 compared with plasmid control cells.
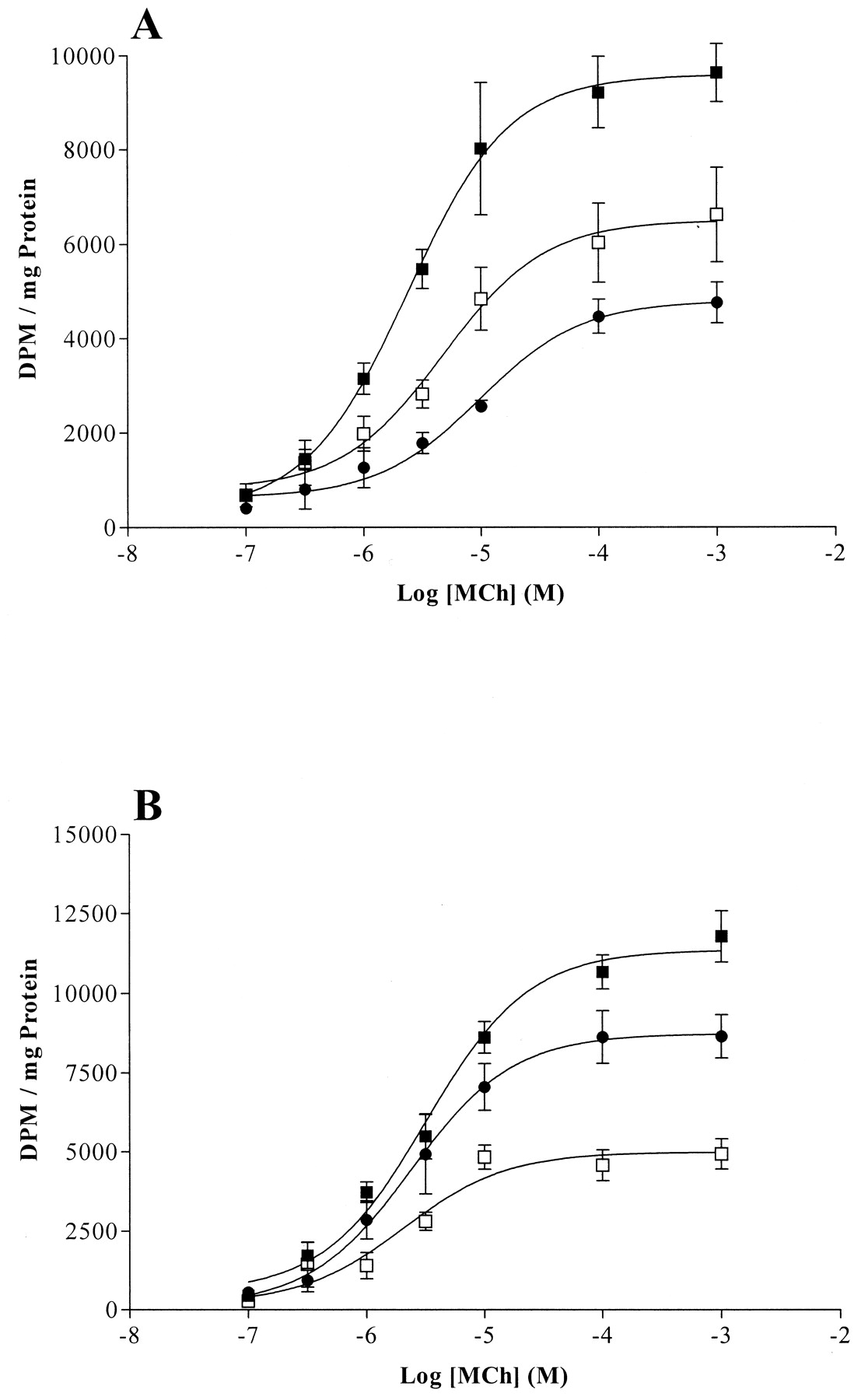
Methacholine concentration-response curves in plasmid control or GRK-overexpressing cells. Either plasmid control cells or GRK3- or GRK6-overexpressing cells were subjected to increasing concentrations of methacholine (100 nM to 1 mM) for 3 min under lithium block. Total [3H]inositol phosphate accumulation was determined as described under Experimental Procedures. Data are expressed as the mean ± S.E.M. for three separate experiments. A shows P1 (control, ▪), clone 12 (GRK3-overexpressing, ■), and clone 6 (GRK6-overexpressing, ●) cells. B shows P3 (control, ▪), clone 2 (GRK3-overexpressing, ■), and clone 24 (GRK6-overexpressing, ●). Total [3H]inositol phosphate accumulation was significantly lower (p < 0.001) for clones-overexpressing GRKs 3 or 6 compared with plasmid control cells.
The effects of GRK3 and GRK6 overexpression on receptor independent stimulation of PLC activity were assessed by the direct activation of G proteins via addition of AlF4 −for 20 min. Whereas GRK6 had no effect on AlF4 −-stimulated total [3H]inositol phosphate accumulation (P3, 79 ± 16.5; GRK6 clone 24, 75 ± 11.6% increase over basal;n = 6), the response in GRK3 cells was almost completely inhibited (14 ± 9.3% over basal; n = 6; p < 0.05). These data indicate that GRK3, but not GRK6, seems to block PLC signaling via a receptor-independent pathway.
Assessment of M3 mACh Receptor Desensitization by [35S]GTPγS Binding.
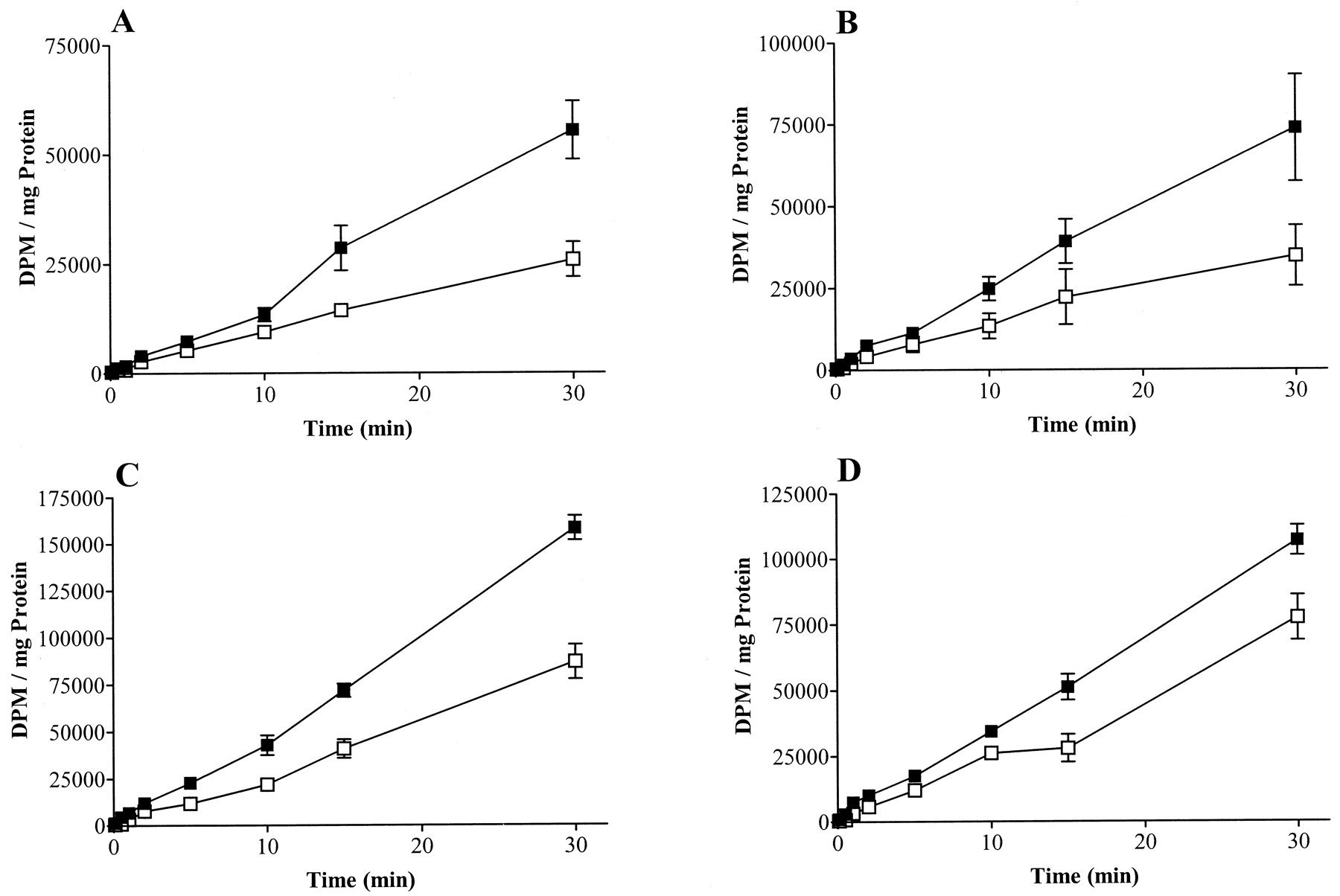
The above data were consistent with a role for receptor phosphorylation by GRKs 3 and 6 in desensitization of the M3 mACh receptor, as measured by the different indices of PLC activity. However, GRK6 or GRK3 may affect other components of the PLC signaling pathway, via either binding of activated Gαq (Carman et al., 1999; Sallese et al., 2000a) or free Gβγ (Haga and Haga, 1992; Pitcher et al., 1992a; Koch et al., 1993). Therefore, we have used agonist-stimulated [35S]GTPγS binding and immunoprecipitation of Gαq/11directly to assess receptor desensitization at the point of receptor catalyzed GTP/GDP exchange. The time course of [35S]GTPγS binding was determined in membranes from plasmid control P3, GRK3 clone 2-, and GRK6 clone 24-overexpressing cells (Fig. 6A). No significant differences were noted in the [35S]GTPγS binding to Gαq/11 on receptor activation in the different clones. Indeed, the data are consistent with our previous findings in CHO cells expressing M3 mACh receptors, showing that Gαq/11 activation peaks at 2 min (Akam et al., 2001). Therefore, all further experiments were carried out at 2 min to allow optimal Gαq/11/[35S]GTPγS binding. In addition, concentration-response curves to methacholine (Fig. 6B) showed identical binding of [35S]GTPγS to Gαq/11, indicating that M3 mACh receptor coupling in SH-SY5Y membranes is unaffected by overexpression of either GRK3 or GRK6.

Time course and concentration-response to methacholine stimulated M3 mACh Gαq/11recruitment in cells overexpressing GRKs 3 and 6, as measured by [35S]GTPγS binding after Gαq/11immunoprecipitation. A, time course of methacholine (100 μM) stimulated Gαq/11 activation in nonpretreated P3 (control, ▪), clone 2 (GRK3-overexpressing, ■), and clone 24 (GRK6-overexpressing, ●) cell membranes. Data are expressed as mean ± S.E.M. of fold increase over basal, and are representative of three to four separate experiments. B, dose response curves to increasing concentrations of methacholine (300 nM to 1 mM) after 2 min of stimulation, in membranes prepared from P3 (control, ▪), 2 (GRK3-overexpressing, ■), and 24 (GRK6-overexpressing, ●) cell membranes. Data are expressed as mean cpm ± S.E.M. minus basal values and are representative of three separate experiments. Basal values were as follows: P3, 336 ± 64; GRK3 clone 2, 317 ± 24; GRK6 clone 24, 359 ± 43; mean cpm ± S.E.M. for seven separate experiments.
However, pretreatment of intact plasmid control cells with methacholine (>3 μM) resulted in reduced agonist-stimulated [35S]GTPγS binding in the subsequent membrane assay (Fig. 7A). SH-SY5Y cells overexpressing GRK3 displayed an identical desensitization profile to the control cells, but GRK6-overexpressing cells displayed a substantially greater degree of uncoupling (Fig. 7, A and B). This was particularly evident at submaximal methacholine pretreatment doses (4-fold greater than plasmid control or GRK3-overexpressing cells). Similar data were obtained after 1-min methacholine pretreatment (data not shown). To assess whether the lack of any enhanced M3 mACh receptor/Gαq/11uncoupling in GRK3-overexpressing cells was caused by the absence of the kinase, membranes from clones either pretreated with methacholine (100 μM, 3 min) or vehicle were probed for GRK3 or GRK6. In GRK3-overexpressing cells (clone 2), a substantial amount of kinase associated with the membrane (Fig. 8A). Membrane-associated GRK6 (clone 24) levels seemed consistent, even after methacholine pretreatment (Fig. 8B). Furthermore, in all clones examined, methacholine pretreatment (3 min) had no effect on the level of membrane-associated Gαq/11 (Fig. 8C).

Effects of GRK overexpression on M3 mACh receptor desensitization as measured by [35S]GTPγS binding. A, plasmid control (P3, ▪), GRK3-overexpressing (clone 2, ■), and GRK6-overxpressing (clone 24, ●) cells were pretreated with increasing concentrations of methacholine (10 nM to 100 μM) for 3 min at 37°C. After pretreatment, cells were harvested and converted into membranes before a second challenge with methacholine (100 μM) for a further 2 min at 30°C in the presence of [35S]GTPγS. Methacholine-stimulated [35S]GTPγS binding to Gαq/11 was determined after immunoprecipitation of Gαq/11 as described under Experimental Procedures. Data were expressed as fold increase over basal. Desensitization was determined as the decrease in [35S]GTPγS binding to Gαq/11 after pretreatment with methacholine compared with that achieved in a nonpretreated control (i.e., 0 = no desensitization and 100% = full receptor desensitization). Data are shown as the mean ± S.E.M. of three separate experiments. GRK6 overexpression significantly (p < 0.001) increased M3 mACh receptor desensitization. Basal values were as follows and are expressed as the mean cpm ± S.E.M. for 12 separate experiments: P3, 422 ± 25; GRK3 clone 2, 456 ± 47; and GRK6 clone 24, 472 ± 34. B, M3 mACh receptor desensitization was examined in either control (P1), GRK3-overexpressing clone 12, or GRK6-overexpressing clone 6 cells as described in A. However, only one pretreatment concentration of methacholine (3 μM) was applied for 3 min. Data was expressed as above and displayed as mean ± S.E.M. of three separate experiments. **p < 0.01; GRK6 overexpression significantly increased M3 mACh receptor desensitization. Basal values were as follows and are expressed as the mean cpm ± S.E.M. for three separate experiments: P3, 447 ± 18; GRK3 clone 2, 460 ± 41; and GRK6 clone 24, 461 ± 22.

Western blot analysis of GRK3, GRK6, or Gαq/11 membrane association after methacholine pretreatment. Intact cells were pretreated with either methacholine (100 μM, 3 min) or vehicle before conversion into membranes. In all cases, 30 μg of membrane protein was added to each well. Lanes 1, 3, and 5 represent vehicle-pretreated controls for P3, GRK3 clone 2, and GRK6 clone 24, respectively. Lanes 2, 4, and 6 represent methacholine-pretreated P3, GRK3 clone 2, and GRK6 clone 24, respectively. A, GRK3 membrane association; B, GRK6 membrane association; and C, Gαq/11 membrane association. Data are representative of three separate experiments.
Assessment of M3 mACh Receptor Internalization.
A possible explanation for the differential effects of GRK3 and GRK6 on [35S]GTPγS binding may be that overexpression of GRK6 increases receptor-mediated internalization, whereas GRK3 overexpression does not. Assessment of M3 mACh receptor internalization via [3H]NMS equilibrium binding after prior exposure of cells to methacholine for 3 min (100 μM) indicated negligible loss of cell surface mACh receptor binding sites compared with vehicle-treated control cells (Fig.9). However, after 30 min, methacholine treatment M3 mACh receptor internalization was evident in all cell lines, with slight but significant enhancement with GRK3 overexpression.

Effects of GRK3 or GRK6 overexpression on methacholine-stimulated M3 mACh receptor internalization. Confluent plasmid control (P3), GRK3 clone 2, or GRK6 clone 24 cells were challenged with either vehicle or methacholine (100 μM) for 3 (■) or 30 (▪) min at 37°C. Cell monolayers were then washed four times with ice-cold Krebs buffer. Specific [3H]NMS binding was assessed after incubation of cells at 4°C for 18 h with a saturating concentration of [3H]NMS (5 nM). Receptor internalization is expressed as the percentage loss of specific [3H]NMS binding sites after methacholine treatment compared with vehicle-treated controls. Data are shown as means ± S.E.M. for three separate experiments. *p < 0.05; significant increase in M3mACh receptor internalization in GRK3-overexpressing cells compared with P3 controls.
Discussion
In this study, we have examined the potential roles of GRK3 and -6 in the phosphorylation and desensitization of M3 mACh receptors endogenously expressed in human SH-SY5Y neuroblastoma cells. Previous studies have established that not only the M2 and M4adenylyl cyclase-linked, but also the Gαq/11PLC-coupled M1/M3 mACh receptors are in vitro substrates for GRK2 and -3 (DebBurman et al., 1995; Haga et al., 1996). However, in the absence of direct evidence for such a role of these GRKs in intact cells, this area remains controversial. Indeed, in a series of studies from these laboratories, recombinant M3 mACh receptors expressed in CHO cells have been shown to undergo rapid agonist-dependent phosphorylation on serines in the third intracellular loop and furthermore that recombinant casein kinase 1α can enhance this phosphorylation in membrane preparations (Tobin et al., 1997). More recent studies have revealed that a dominant negative mutant of this kinase can partially suppress agonist-mediated phosphorylation of the M3 mACh receptor; importantly, this does not significantly suppress desensitization of PLC activity (Budd et al., 2000).
In view of these data, we have chosen to investigate the phosphorylation and regulation of endogenous M3mACh receptors, expressed at relatively low levels in SH-SY5Y cells, stably transfected to overexpress GRK3 or GRK6. Our results provide the first evidence in intact cells that overexpression of GRK3 and GRK6 is accompanied by enhanced agonist-mediated phosphorylation of a Gαq/11-linked mACh receptor. DebBurman et al. (1995) reported an agonist and Gβγ-dependent phosphorylation of M3 mACh receptors in urea-treated Sf9 membranes by GRK2 and -3, but not by GRK5 or -6. It is difficult to directly compare in vivo results from intact SH-SY5Y cells with those in which recombinant GRKs are added in vitro to already overexpressed M3 mACh receptors in Sf9 cells. However, it is noteworthy that purified GRK6 displays low activity toward rhodopsin outer segments that can be increased via palmitoylation and increased membrane association of the kinase (Loudon and Benovic, 1997). Our findings indicate that, in SH-SY5Y cells, both endogenous and overexpressed GRK6 are exclusively membrane-bound and presumably palmitoylated and active. Furthermore, GRK6 seems to promote greater maximal receptor phosphorylation than GRK3 even at submaximal agonist occupation of the M3 mACh receptor. Moreover, assessment of kinase overexpression via blotting of multiple cell lysate dilutions, and activity by α-casein phosphorylation, suggests similar levels of GRK overexpression. Overall therefore, these data provide strong evidence that the M3 mACh receptor is a substrate for GRK3 and particularly GRK6 under agonist-stimulation in vivo.
Agonist-dependent phosphorylation of the endogenous M3 mACh receptors in SH-SY5Y cells might be anticipated to be associated with desensitization of transmembrane signaling (Tobin et al., 1997). Indeed, evaluation of total [3H]inositol phosphate accumulation (1–30 min) and inositol 1,4,5-trisphosphate mass (at 10–300 s) provided evidence of a blunted PLC response in cells overexpressing GRK3 or GRK6. This reduced responsiveness could not be attributed to a reduced M3 receptor or Gαq/11expression and it is tempting to associate it with GRK-mediated receptor phosphorylation. However, GRKs 2 and 3 are known to bind free Gβγ subunits via a C-terminal pleckstrin homology domain and it has been suggested that this may play a direct role in regulating G protein-mediated signaling (Haga and Haga, 1992; Pitcher et al., 1992b;Koch et al., 1993). In addition, recent evidence suggests that GRK2 and -3 (but not GRK5 or -6) can selectively bind activated Gαq/11 and potentially sequester it from PLCβ (Carman et al., 1999; Sallese et al., 2000a). Indeed, there is now substantial evidence that GRK2 and -3 overexpression can severely blunt agonist-mediated PLC activity and that this may be caused in part by such phosphorylation-independent events downstream of the receptor- Gαq/11 interaction (Diviani et al., 1996b;Oppermann et al., 1996; Freedman et al., 1997; Dicker et al., 1999). In the present study, GRK3 overexpression severely blunts AlF4 − activation of PLC, which would be consistent with an action independent of receptor phosphorylation.
In view of these complications, we have also assessed M3 mACh receptor-Gαq/11coupling directly using a [35S]GTPγS immunoprecipitation strategy well established in these and other laboratories (Barr et al., 1998; Young et al., 2000; Akam et al., 2001). The results from these approaches revealed striking differences between cells overexpressing GRK3 and GRK6. Membranes prepared from all non–agonist-pretreated cells displayed similar extents of [35S]GTPγS binding to Gαq/11 over 2 min. However, in contrast, when intact cells were pretreated with agonist and prepared membranes rechallenged with agonist, a concentration-dependent uncoupling of the M3 mACh receptor was detected as a marked decrease in binding of [35S]GTPγS to immunoprecipitated Gαq/11. Over-expression of GRK6 markedly enhanced the degree of M3 mACh receptor uncoupling from Gαq/11 above that seen in plasmid control or GRK3-overexpressing cells. Indeed, the most striking differences were seen at agonist pretreatment concentrations, which produced little or no desensitization in either plasmid or GRK3-overexpressing cells. The inability of GRK3 overexpression to enhance M3 mACh receptor-Gαq/11 uncoupling, despite evidence of increased phosphorylation of the receptor, was surprising. Perhaps the phosphorylation evoked by GRK6 but not GRK3, particularly at low agonist occupation, allowed the detection of a greater uncoupling than plasmid-transfected cells. Alternatively, because GRK3 is predominantly cytosolic and only recruited to the membrane upon receptor stimulation, any enhanced inhibition of [35S]GTPγS binding caused by Gαq binding may be partially lost during membrane preparation. However, it should be noted that substantial levels of both GRK6 and GRK3 were detected in membrane preparations before and after methacholine pretreatment. Moreover, because [35S]GTPγS binding is performed in the absence of ATP, the presence or absence of any GRK in the membrane preparations is likely to have minimal effect upon receptor phosphorylation during the second methacholine challenge.
The data obtained from experiments involving GRK3 and GRK6 overexpression leads to the question of whether these kinases, endogenously expressed in SH-SY5Y cells, are responsible for M3 mACh receptor desensitization. We have preliminary data that suggest that a kinase-dead GRK6 dominant negative (mutated at K215R), when stably expressed in SH-SY5Y cells at levels comparable with the overexpression of the wild-type GRK6, inhibits methacholine-stimulated (100 μM, 3 min, 37°C) M3 mACh receptor/Gαq/11uncoupling (P3 control, 44.6 ± 2.4%; GRK6 dominant negative, 22.4 ± 2.3%, n = 3, p < 0.01). However, assessing the role of GRK3 using a dominant negative strategy remains problematic. The presence of the N-terminal RGS domain and the C-terminal Gβγ binding domain would suggest that even if the dominant negative GRK3 could inhibit M3 mACh receptor phosphorylation, it could prove to be impossible to dissect this role from its potential to inhibit PLC signaling. With this in mind, we are attempting to deplete the effects of endogenously expressed GRK3 with the application of antisense technology.
One further potential mechanism that may explain the differential effects of GRK3 and GRK6 on [35S]GTPγS binding could involve GRK6-mediated enhancement of M3 mACh receptor internalization. However, this seems not to be the case because after 3 min, methacholine treatment receptor internalization was minimal and no significant difference between either vector controls or GRK3- or GRK6-overexpressing clones was observed. Although both GRK3 and GRK6 overexpression slightly enhanced M3 mACh receptor internalization after 30 min of methacholine exposure, this should have little bearing on the [35S]GTPγS binding, which was undertaken after 3 min of methacholine pretreatment.
In conclusion, we have examined the potential roles of GRK3 and GRK6 in the desensitization of the M3 mACh receptor endogenously expressed in SH-SY5Y cells. Our data show that whereas overexpression of both kinases lead to reduced agonist-activated PLC activity, only GRK6 overexpression results in a greater uncoupling of the M3 receptor from Gαq/11. Although these differences may relate to different extents or patterns of receptor phosphorylation, it seems more likely at this time that the ability of activated GRK3, but not GRK6, to sequester Gαq/11 and/or Gβγ may provide additional phosphorylation-independent regulation of PLC activity. Our data suggest that in contrast, however, GRK6 may uncouple the M3 mACh receptor from Gαq/11 via a classical agonist-dependent receptor phosphorylation. This study has highlighted clear effects of GRK6 on agonist-sensitive M3 mACh receptor phosphorylation, coupling to Gαq/11, and consequent suppression of PLC in intact cells suggests that further investigation of this kinase is warranted.
Acknowledgments
We would like to thank Mr. Raj Mistry and Mr. Neil Johnston for their technical assistance.
Footnotes
- Received February 28, 2001.
- Accepted May 15, 2001.
-
This work was supported by Wellcome Trust Grant 0168895.
Abbreviations
- GRK
- G protein-coupled receptor kinase
- PLC
- phospholipase C
- mACh
- muscarinic acetylcholine receptor
- RGS
- regulator of G protein signaling
- NMS
- N-methylscopolamine
- NP40
- Nonidet P40
- PAGE
- polyacrylamide gel electrophoresis
- Ins(1,4,5)P3
- inositol 1,4,5-trisphosphate
- CHO
- Chinese hamster ovary
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}