


Abstract
Previously, we reported that the time course for the rapid phosphorylation rate of μ-opioid receptor expressed in human embryonic kidney (HEK)293 cells did not correlate with the slow receptor desensitization rate induced by [d-Ala2,N-MePhe4,Gly-ol5]-enkephalin (DAMGO). However, others have suggested that receptor phosphorylation is the trigger for μ-opioid receptor desensitization. In this study, we demonstrated the relatively slow rate of receptor desensitization could be attributed partially to the recycling of internalized receptor as determined by fluorescence-activated cell-sorting analysis. However, the blockade of the endocytic and Golgi transport events in HEK293 cells with monensin and brefeldin A did not increase the initial rate of receptor desensitization. But the desensitization rate was increased by reduction of the μ-opioid receptor level with β-furnaltrexamine (βFNA). The reduction of the receptor level with 1 μM βFNA significantly increased the rate of etorphine-induced receptor desensitization. By blocking the ability of receptor to internalize with 0.4 M sucrose, a significant degree of receptor being rapidly desensitized was observed in HEK293 cells pretreated with βFNA. Hence, μ-opioid receptor is being resensitized during chronic agonist treatment. The significance of resensitization of the internalized receptor in affecting receptor desensitization was demonstrated further with human neuroblastoma SHSY5Y cells that expressed a low level of μ-opioid receptor. Although DAMGO could not induce a rapid desensitization in these cells, in the presence of monensin and brefeldin A, DAMGO desensitized the μ-opioid receptor's ability to regulate adenylyl cyclase with at 1/2 = 9.9 ± 2.1 min and a maximal desensitized level at 70 ± 4.7%. Furthermore, blockade of receptor internalization with 0.4 M sucrose enhanced the DAMGO-induced receptor desensitization, and the inclusion of monensin prevented the resensitization of the μ-opioid receptor after chronic agonist treatment in SHSY5Y cells. Thus, the ability of the μ-opioid receptor to resensitize and to recycle, and the relative efficiency of the receptor to regulate adenylyl cyclase activity, contributed to the observed slow rate of μ-opioid receptor desensitization in HEK293 cells.
Being a member of the subfamily of rhodopsin receptor within the superfamily of G protein-coupled receptor (GPCR), the molecular processes that lead to opioid receptor desensitization should be similar to those reported with the β2-adrenergic receptor (Lefkowitz, 1998). In this β2-adrenergic receptor model, agonist binding to the receptor results in the rapid phosphorylation of the receptor by protein kinases, including the G protein-coupled receptor kinases (GRKs), thereby promoting the association of arrestin with the receptor that leads to uncoupling of the signals. The trigger of this cellular adaptational process is the agonist-induced phosphorylation of the receptor.
There are many reports on opioid receptor phosphorylation as the mechanism for receptor desensitizaton. The concrete demonstration of opioid receptor phosphorylation was first demonstrated by Pei et al. (1995) with the δ-opioid receptor and by Arden et al. (1995) with the μ-opioid receptor. Agonist-induced phosphorylation of the κ-opioid receptor also was reported by Appleyard et al. (1997). In all cases, by immunoprecipitating the phosphorylated receptor with either the polyclonal antibodies to the receptor or the monoclonal antibodies to the epitope spliced to the N terminus of the receptor, a time and agonist concentration-dependent phosphorylation of the receptor was observed. Studies with the δ-opioid (Pei et al., 1995) and μ-opioid (Zhang et al., 1996; El-Kouhen et al., 1999) receptors suggested that agonist-induced phosphorylation is mediated via GRKs and not by protein kinase C. Basal phosphorylation of the μ-opioid receptor might involve other protein kinases such as Ca2+/calmodulin-dependent kinases (Wang et al., 1996). Predictably, the ability of opioid ligand to induce receptor phosphorylation is correlated to its efficacy (Yu et al., 1997).
There are studies that suggest agonist-induced receptor phosphorylation correlates with δ-opioid receptor desensitization. Pei et al. (1995)reported with the δ-opioid receptor expressed in human embryonic kidney (HEK)293 cells that the dominant negative mutants of GRKs could attenuate the [d-Pen2,d-Pen5]-enkephalin (DPDPE)-induced receptor phosphorylation and subsequently receptor desensitization. The overexpression of GRK3 and β-arrestin 2 in Xenopus oocytes could accelerate the rate of δ-opioid receptor desensitization with the voltage-dependent G protein-coupled inward rectifying potassium channels (GIRKs; Koover et al., 1997). Overexpression of GRK2 and β-arrestin 2 in HEK293 cells expressing the δ-opioid receptor also could increase the rate of desensitization as measured by receptor regulation of adenylyl cyclase activity (El Kouhen et al., 1999). The mutation of the last four Thr and Ser residues at the C terminus of the δ-opioid receptor would block GRK and arrestin-mediated desensitization (Koover et al., 1997). Thus, these data clearly established the relationship between δ-opioid receptor phosphorylation and desensitization. However, for μ-opioid receptor, the significance of the agonist-induced receptor phosphorylation on desensitization has not been established firmly.
There are reported studies that support the hypothesis in which μ-opioid receptor phosphorylation leads to receptor desensitization. By measuring μ-opioid agonist regulation of the GIRK1 channels expressed in Xenopus oocytes and agonist-induced receptor phosphorylation in Chinese hamster ovary cells, Zhang et al. (1996)suggested that μ-opioid receptor phosphorylation leads to rapid desensitization of the receptor. These findings were supported by the absence of [d-Ala2,N-MePhe4,Gly-ol5]-enkephalin (DAMGO)-induced receptor desensitization in cells expressing the μ-opioid receptors with the T394A mutation (Pak et al., 1997). Although actual receptor phosphorylation was not determined in this study, E393A mutation also eliminated DAMGO-induced receptor desensitization, suggesting the involvement of GRKs that are acidokinases (Fredericks et al., 1996). However, these findings could be partially explained by involvement of Thr394in recycling of the μ-opioid receptor. As demonstrated with MOR1B, a splice variant of MOR1, the rate of receptor desensitization could be enhanced with monensin (Koch et al., 1998). Mutation of the Thr394 of the wild-type μ-opioid receptor to Ala could increase the rates of receptor internalization and resensitization (Wolf et al., 1999). Because MOR1B is seven amino acids shorter than MOR1 (Zimprich et al.,1995), and therefore Thr394 is absent from the sequence, these data support the hypothesis Thr394 plays a role in the recycling of the μ-opioid receptor, and subsequently, the rate of receptor desensitization.
There are also studies that do not support μ-opioid agonist-induced receptor phosphorylation-triggered receptor desensitization. Coexpression of GRK3 and β-arrestin 2 with the μ-opioid receptor inXenopus oocytes did not alter the relatively slow rate of μ-opioid agonist-induced desensitization as measured by the GIRK regulation (Koover et al., 1997). Similarly, overexpression of GRK2 and β-arrestin 2 in HEK293 cells did not alter the rate of μ-opioid receptor but increased the rate of δ-opioid receptor desensitization as measured by regulation of adenylyl cyclase activity (El-Kouhen et al., 1999). El-Kouhen et al. (1999) also reported that the relative fast rate (minutes) of μ-opioid receptor phosphorylation did not correspond to the relative slow rate (hours) of receptor desensitization. Furthermore, DAMGO could desensitize the μ-opioid receptor with all Ser and Thr residues in the third intracellular loop and carboxyl tail being mutated to Ala (Capeyrou et al., 1997). These results are in agreement with studies in which the overexpression of β-arrestin 1 in HEK293 cells did not alter the μ-opioid receptor-mediated but attenuated the δ- and κ-opioid receptor-mediated inhibition of adenylyl cyclase activity (Cheng et al., 1998). All these studies suggested that mechanisms other than receptor phosphorylation and the subsequent interaction of arrestin are involved in the μ-opioid receptor desensitization.
These apparent contradictory results were complicated further by studies in which the μ-opioid receptor regulation of the GIRK1/GIRK4 channels' activities in Xenopus oocytes can be desensitized within 20 min of DAMGO pretreatment in the presence of overexpressed GRK3 or GRK5 and β-arrestin 2 (Koover et al., 1998). This is in direct contrast with the earlier report in which overexpression of GRK3 and β-arrestin 2 did not alter the relative slow rate of μ-opioid receptor desensitization in the Xenopus oocytes (Koover et al., 1997). Because of such controversy, this study was carried out to address the basic issue of the causal relationship between μ-opioid receptor phosphorylation and desensitization. As demonstrated by our studies, the ability of the μ-opioid receptor to recycle rapidly and the relative efficiency of receptor signaling play critical roles in determining the rate of receptor desensitization. By limiting the ability of receptor to recycle with agents such as monensin and brefeldin A, and by controlling the level of the receptor level, we could demonstrate a rapid μ-opioid receptor desensitization at a rate corresponding to its phosphorylation.
Experimental Procedures
Culturing HEK293 Cells Stably Expressing the MORTAG.
The transfection of the HEK293 cells with pCDNA3 plasmids containing the hemagglutinin (HA) epitope tagged μ-opioid receptor, and the selection of the stable transfectants were carried out as described previously (El-Kouhen et al., 1999). The cells were then cultured in Eagle's minimal essential medium supplemented with 10% fetal calf serum, 100 μg/ml streptomycin, 100 I.U./ml penicillin, and 250 μg/ml geneticin (G418) under humidified atmosphere with 5% CO2.
Opioid Inhibition of Intracellular cAMP Level.
Forty-eight hours before the experiments, HEK293 cells were seeded into 24-well plates. On the day of experiments, the culture medium was removed and was replaced with 1.0 ml of minimal essential medium buffered with 10 mM HEPES at pH 7.1. In experiments in which the cells were pretreated with monensin or brefeldin A, the stock antibiotics in 95% ethanol solutions were added to individual wells in 10-μl aliquots 1 h before the addition of opioid agonists. Chronic treatment of HEK293 cells with etorphine was carried out by adding 100 μM etorphine solution to the individual wells at various time intervals to give the final etorphine concentration of 1 μM. After preincubating with etorphine, the medium was removed, the plates were placed on ice, and 0.5 ml of Krebs-Ringer-HEPES Buffer (110 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, 25 mM glucose, 55 mM sucrose, 10 mM HEPES, pH 7.4) containing 0.25 mM 3-isobutyl-1-methylxanthine and 10 μM forskolin with or without 1 μM etorphine was added. The plates were subsequently incubated at 37°C for 10 min and reaction was terminated by addition of 50 μl of 3.3 N perchloric acid. After neutralizing the perchloric acid in each well with 125 μl of 2 M KOH, 1 M Tris, and 60 mM EDTA, the amount of cAMP in each well was determined by comparing the ability of the diluted acetylated samples to compete for125I-cAMP binding to the antibodies with that of standard concentrations of acetylated cAMP as described previously (El-Kouhen et al., 1999). The degree of desensitization is calculated by comparing the percentage of forskolin-stimulated intracellular cAMP production being inhibited by 1 μM etorphine during the 10-min assay before and after etorphine pretreatment. The rate of desensitization was calculated with the single component exponential decay analysis with GrapPad. The values represent mean ± S.D. of the determinations from a minimum of three separate experiments.
Opioid Agonist-Induced Phosphorylation of μ-Opioid Receptor in HEK293 Cells.
Phosphorylation experiments with HEK293 cells were carried out as described previously (El-Kouhen et al., 1999). Cells from a 35-mm plate were combined for the wheat germ lectin column partial purification of the receptor and the subsequent immunoprecipitation of the receptor. The immunoprecipitated receptor was separated from other phosphoproteins with SDS-polyacrylamide gel electrophoresis. The degree of receptor phosphorylation was visualized and quantitated with the PhosphoImager Storm 840 system (Molecular Dynamics, Sunnyvale, CA).
μ-Opioid Receptor Internalization as Determined by Fluorescence-Activated Cell-Sorting Analysis (FACS) and Confocal Microscopy.
For the FACS experiments, the cell surface-located μ-opioid receptors in HEK293 cells were visualized with the high-affinity mouse monoclonal anti-HA antibody HA.11 clone 16B12 and the secondary antibodies goat anti-mouse IgG conjugated with Alexa 488. For the confocal microscopy experiment, the rat monoclonal anti-HA antibody 3F10, conjugated with fluorescein, was used to label the μ-opioid receptor. Two to 4 days before experiments, HEK293 cells were plated onto 35-mm culture dishes. In the confocal microscopy experiments, these dishes contained glass coverslips that were acid treated and polylysine coated. For FACS, the cells were treated with etorphine or control and then the medium was removed. The cells were incubated with 0.5 ml of HA.11 (1:500) at 4°C for 2 h. After removing the medium and washing with PBS, the cells were fixed with Lana's fixative, washed twice with PBS, and incubated with the secondary antibodies (1:1000) for 1 h at room temperature before FACS. For the confocal microscopy studies, the cells were incubated with the anti-HA antibodies, 3F10 conjugated with fluroescein, for 2 h at 4°C. Excess antibodies were removed by washing with growth medium. Then the cells were treated either with 1 μM etorphine or saline for 30 min at 37°C. Afterward, the cells were fixed with Lana's fixative, and the cellular location of the μ-opioid receptor was determined with confocal microscopy.
Materials.
Expression vector pCDNA3 was from Invitrogen (San Diego, CA). Dulbecco's modified Eagle's medium (DMEM), Met/Cys-free DMEM, and Geniticin (G-418) were purchased from Gibco Life Technologies (Grand Island, NY). [3H]Diprenorphine (58 Ci/mmol) was supplied by Amersham (Arlington Heights, IL). [32P]-Pi (>400 Ci/ml) was supplied by ICN (Costa Mesa, CA). [125I]-acetylated cAMP (2200 μCi/mmol) was purchased from Linco Research Inc. (St. Charles, MO). Polyclonal antibodies for the cAMP radioimmunoassay were developed by immunolizing rabbits with succinyl cAMP conjugated to keyhole limpet hemocyanin. Mouse monoclonal anti-HA 1.1 clone 16B12 was purchased from Babco (Richmond, CA). Rat monoclonal anti-HA 3F10 conjugated with fluorescein was from Roche Biochemicals (Indianapolis, IN). Forskolin was purchased from Calbiochem (San Diego, CA). Etorphine and other opioid ligands were supplied by National Institute on Drug Abuse. All other chemicals were purchased from Sigma Chemical Co. (St. Louis, MO).
Results
In previous studies, we reported that DAMGO-induced phosphorylation of the μ-opioid receptor stably expressed in HEK293 cells did not correlate with the loss of response, as measured by agonist inhibition of adenylyl cyclase activity (El-Kouhen et al., 1999). The HEK293 clone used in the studies expressed 14.4 ± 2.9 pmol/mg protein of μ-opioid receptor with the affinity for the opioid receptor universal ligand [3H]diprenorphine ofK d = 1.03 ± 0.1 nM. It is possible that the relatively high level of receptor being expressed could affect the potencies of the opioid agonists. When the IC50 values of several opioid agonists to inhibit the forskolin-stimulated adenylyl cyclase activity were measured, it was determined that the potency of etorphine was greatly enhanced, whereas the potency of DAMGO in this HEK293 clone remained similar to cell lines with lower receptor level (Table1). Because μ-opioid receptor phosphorylation is dependent on the potency and efficacy of the agonist (Yu et al., 1997), it is probable that the subsequent receptor desensitization induced by etorphine could be faster than that of DAMGO.
Affinities and potencies of various opioid agonists and antagonists for the μ-opioid receptor expressed in HEK293 cells
When the HEK293 cells expressing the HA-tagged μ-opioid receptor (MORTAG) were treated with 1 μM etorphine for up to 4 h, it was observed that the receptor also desensitized to etorphine slowly (Fig.1). Before etorphine treatment, 1 μM etorphine inhibited the 10 μM forskolin-stimulated intracellular cAMP production by 83.5 ± 5.4%. After pretreating the cells with etorphine for 4 h, 1 μM etorphine inhibited the cAMP production by 60.2 ± 10%, or 72% of the maximal activity was retained after chronic treatment. More importantly, after 15 or 30 min of pretreatment, 96.9 ± 3.7 and 95.2 ± 4.1% of the maximal etorphine activity remained, respectively. The rate of etorphine-induced receptor phosphorylation peaked at 5 min after the addition of the agonist to the HEK293 cells (Fig. 1). Thus, similar to the previous report with DAMGO as the agonist (El-Kouhen et al., 1999), the etorphine-induced receptor phosphorylation did not appear to correlate with the loss of agonist inhibition of adenylyl cyclase activity.

Etorphine-induced μ-opioid receptor phosphorylation and desensitization in HEK293 cells. HEK293 cells stably expressing the μ-opioid receptors were treated with 1 μM etorphine for various periods of time. Top, ability of etorphine to induce phosphorylation was investigated as discussed in Experimental Procedures. Bottom, ability of 1 μM etorphine to inhibit 10 μM forskolin-stimulated intracellular cAMP production was determined after various times of etorphine pretreatment. The values represent mean ± S.D. from three to seven experiments.
The absence of rapid desensitization to etorphine pretreatment could be due to the rapid internalization and resensitization of the μ-opioid receptor. As suggested with the β2-adrenergic desensitization studies, agonist-induced receptor phosphorylation resulted in the rapid internalization of the protein where dephosphorylation occurred (Zhang et al., 1997). The dephosphorylated and resensitized receptor can be recycled back to the membrane surface. Thus, the lack of observed etorphine-induced receptor desensitization at maximal receptor phosphorylation could be due to the rapid internalization of the receptor. Rapid agonist-induced internalization of the μ-opioid receptor has been reported (Whistler and von Zastrow, 1998). The rapid internalization of the MORTAG expressed in HEK293 also was observed when the receptor was prelabeled with anti-HA monoclonal antibodies (Fig. 2A). The μ-opioid receptor was observed to localize mainly at the plasma membrane with some occasional immunofluorescence within the intracellular compartment of the cells (Fig. 2A, left). Incubation of the HEK293 cells at 37°C after the anti-HA binding did not induce the internalization of the antibodies (Fig. 2A, middle). In agreement with previously reported studies, within 30 min of 1 μM etorphine treatment, the majority of anti-HA immunofluorescence was detected intracellularly (Fig. 2A, right). However, when FACS was carried out to monitor the cell surface receptors, a much slower rate of receptor internalization was observed. These experiments were carried out by staining the μ-opioid receptor on the cell surface of HEK293 cells with the monoclonal anti-HA antibody after treating the cells with etorphine for various amounts of time. As shown in Fig. 2B, the rate of internalization was relatively slow, t 1/2 = 0.86 ± 0.19 h. Furthermore, the maximal decrease in the cell surface fluorescence, or the μ-opioid receptor, was 56.8 ± 4.2%. This maximal level of receptor internalized was not achieved until 4 h after the addition of etorphine (Fig. 2B). The relative slow rate of internalization could reflect the continued recycling of the internalized μ-opioid receptor to the cell surface of this HEK293 clone. If this is the scenario, the prevention of acidification of intracellular compartments by the sodium ionophore monensin should prevent the recycling and increase the rate of receptor internalization. When HEK293 cells stably expressing the MORTAG were pretreated with 50 μM monensin 1 h before etorphine treatment, the rate of μ-opioid receptor internalization was observed to increase (Fig. 2B). There were significant increases in the amount of receptor being internalized in the presence of monensin. Thet 1/2 of the internalization rate was determined to be 0.19 ± 0.036 h. Interestingly, although the maximal level of receptor being internalized was observed within 30 min of etorphine treatment, this level in the presence of monensin, 55.6 ± 2.6%, remained similar to that of control (Fig. 2B).

Rate of μ-opioid receptor internalization. The ability of 1 μM etorphine to induce endocytosis of the μ-opioid receptor was determined by confocal microscopy (A) and FACS (B) in the absence (○) or in the presence (●) of 50 μM monensin. A, HEK293 cells were prelabeled with anti-HA monoclonal antibody at 4°C as described in Experimental Procedures. Then the cells were either kept at 4°C (A) or at 37°C (B) for 30 min. Immunoflourescence of the anti-HA labeled under these conditions was compared with the cells treated with 1 μM etorphine for 30 min at 37°C (C). B, FACS was carried out with the anti-HA antibodies as described in Experimental Procedures. The values represent mean ± S.D. of FACS with three different passages of cells. ∗, statistical significance with Student's unpairedt test when the percentages of receptor being internalized in the presence of monensin were compared with that of control, P ≤ .005.
The inability of etorphine to internalize a higher percentage of receptor in the presence of monensin could be due to the transport of newly synthesized receptors to the cell surface during agonist treatment. Although monensin is known to be an inhibitor of the Golgi apparatus function (Mollenhauer et al., 1990), the concentration used has mainly the lysosmotic effects of the drug. Higher concentrations of monensin were not used because the antibiotic caused HEK293 cells to detach from the growing surface during etorphine treatment. Hence, brefeldin A was used to block the translocation of protein from endoplasmic reticulum to the Golgi without affecting endocytosis or lysosome function. Brefeldin A has been reported to cause the release of the coat protein β-COP from the apparatus (Duden et al., 1991) and to inhibit the GTP/GDP exchange of the ADP ribosylating factor involved in the vesicular transport (Donaldson et al., 1992;Helms and Rothman, 1992). Treatment of HEK293 cells with 1 μM etorphine for 30 min caused 38.1 ± 0.8% of the receptor being internalized. Pretreatment of HEK293 cells with 5 μM brefeldin A 1 h before etorphine addition resulted in 56.1 ± 7.3% of the receptor being internalized (Fig. 3). This apparent magnitude of receptor internalization in the presence of brefeldin A was similar to that in the presence of monensin alone, 53.2 ± 5.0%. When the etorphine-induced μ-opioid receptor internalization was measured in the presence of both monensin and brefeldin A, a further decrease in cell surface fluorescence was observed. An average of 75.6 ± 0.1% of the receptor was internalized after 30 min in the presence of these two drugs (Fig. 3). Increase in the brefeldin A concentration during the 1-h pretreatment did not decrease further the relative fluorescence or the μ-opioid receptor concentration in the cell surface. Hence, the ability of brefeldin A to augment the monensin effect suggests that a large fraction of the cell surface receptor in the HEK293 cells is contributed by the receptor pool being transported through the Golgi apparatus.
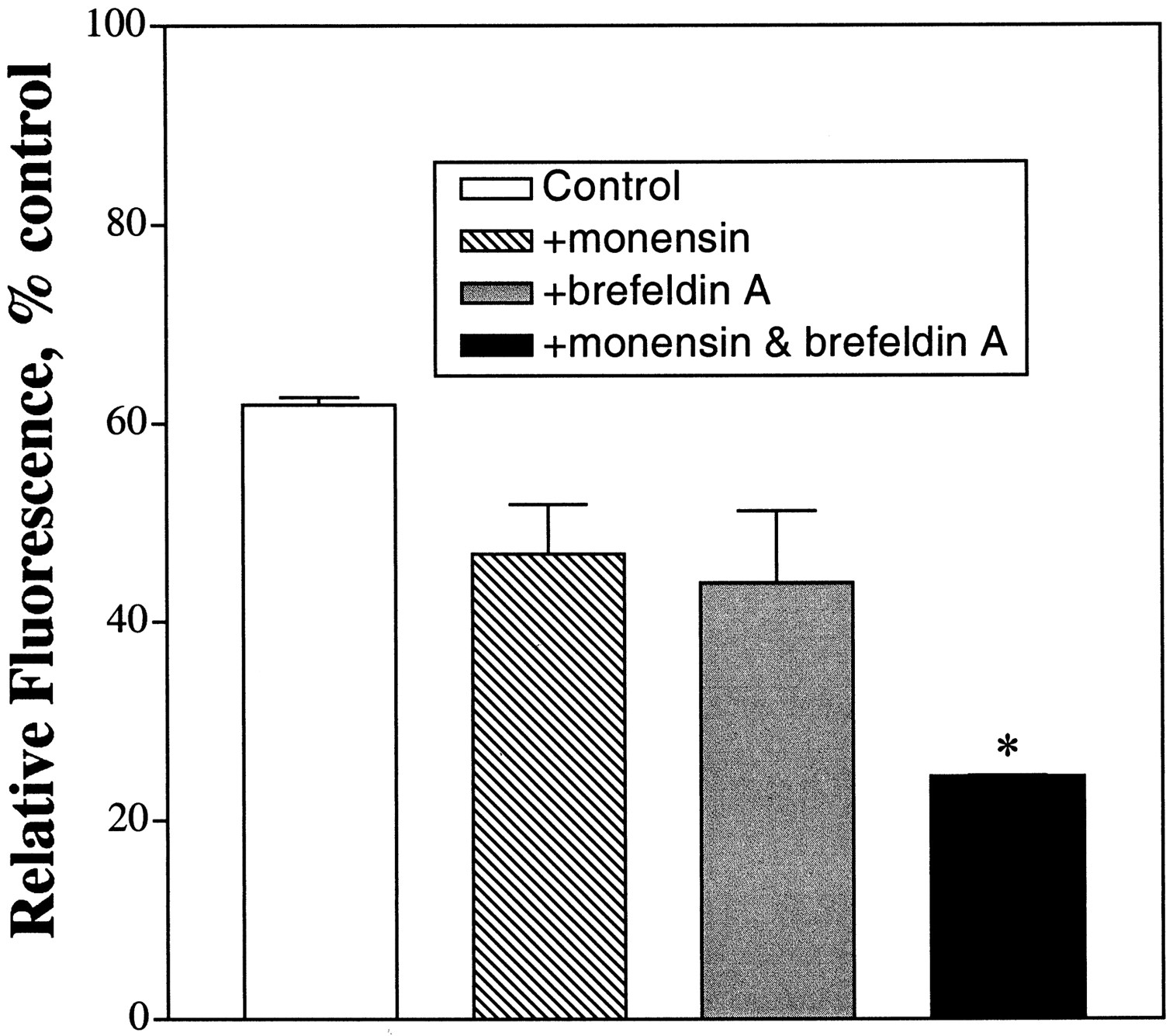
Effect of brefeldin A on μ-opioid receptor internalization. HEK293 cells were pretreated with 50 μM monensin (▧), 5 μM brefeldin A (░), or 50 μM monensin and 5 μM brefeldin A (▪) 1 h before the addition of etorphine as described in Experimental Procedures. The cells were then treated with 1 μM etorphine for 30 min. The amount of receptor being internalized in the presence of etorphine was determined by FACS. The values represent mean ± S.D. of three determinations. ∗, statistical significance with Student's unpaired t test when the amount of receptor being internalized was compared with that in the presence of monensin, P ≤ .005.
If the relative slow rate of etorphine-induced μ-opioid receptor desensitization is affected by the receptor recycling and the receptor transport from Golgi, then the blockade of these cellular events with monensin and brefeldin A should increase the rate of agonist-induced receptor desensitization. When HEK293 cells stably expressing the MORTAG were pretreated with 50 μM monensin or 5 μM brefeldin A for 1 h before the addition of 1 μM etorphine, there was no apparent increase in the initial rate of receptor desensitization (Fig.4). The degrees of receptor being desensitized in the presence of both drugs were similar to the control values. Only after 3 h of etorphine treatment was there an apparent difference in the magnitude of receptor being desensitized. In control cells, 3 h of etorphine pretreatment desensitized the ability of 1 μM etorphine to inhibit adenylyl cyclase activity by 13.5 ± 4.0% in control HEK293 cells. In cells pretreated with monensin or brefeldin A, etorphine could desensitize the system by 24.9 ± 3.4 and 17.7 ± 2.2%, respectively. Addition of both monensin and brefeldin A did not increase the percentage of receptor being desensitized. Regardless of whether HEK293 cells were treated with monensin, brefeldin, or both, the initial etorphine treatment, i.e., time less than 30 min, did not significantly desensitize the ability of the agonist to inhibit adenylyl cyclase activity (Fig. 4). Because the maximal level of receptor phosphorylation was observed 5 min after etorphine, the apparent lack of effect of the endocytic and Golgi function inhibitors on the receptor desensitization did not support receptor phosphorylation as the trigger for desensitization.

Receptor recycling and rate of μ-opioid receptor desensitization. The ability of the μ-opioid receptor in HEK293 cells to recycle was blocked by treating the cells with 50 μM monensin 1 h before the addition of 1 μM etorphine. The newly synthesized receptor was prevented from transport to the membrane with the addition of 5 μM brefeldin A. The ability of 1 μM etorphine to inhibit the forskolin-stimulated production of intracellular cAMP was determined after various times of etorphine pretreatment. These values were compared with the percentage of etorphine inhibtion without etorphine pretreatment but with the antibiotics treatment. The values represent mean ± S.D. from three separate experiments. The maximal inhibition level exhibited by 1 μM etorphine in control cells was 84.5 ± 4.1%, in monensin-treated cells 87.0 ± 1.1, and in brefeldin A-treated cells 83.2 ± 2.7%.
The relative high density of μ-opioid receptor expressed in HEK293 cells could attribute to the apparent lack of rapid desensitizaion of the receptor. As suggested by Pak et al. (1996), μ-opioid receptor activity is dependent on receptor density. Although in the presence of monensin and brefeldin A 75% of the cell surface receptor was internalized after 30 min (Fig. 3), with the receptor density of 14.4 pmol/mg protein, 3.6 pmol/mg protein of receptor remained on the cell surface. This represents a relatively high level of μ-opioid receptor considering that the level of μ-opioid receptor expressed endogenously in the human neuroblastoma SHSY5Y cells is only 43 fmol/mg protein (Prather et al., 1994). One method of reducing the surface receptor concentration is to use the μ-opioid receptor irreversible antagonist β-funaltrexamine (βFNA; Takemori et al., 1981). The covalent binding of βFNA to the receptor was shown to involve Lys233 of the second extracellular loop (Chen et al., 1996). Thus, by pretreating HEK293 cells with various concentrations of βFNA, the receptor level can be adjusted. When HEK293 cells were incubated with various βFNA concentrations for 2 h, a decrease in the level of receptor was observed to be βFNA dose-dependent as indicated by [3H]diprenorphine binding (Fig.5A). At the maximal concentration of βFNA, reduction of [3H]diprenorphine binding to intact cells was 79.5 ± 0.1%, with the calculated maximum 88 ± 9.8%. Although the concentration required to decrease the receptor level by 50%, 49 ± 26 nM, was higher than the reported affinity of βFNA for the μ-opioid receptor, this concentration reflected the covalent binding of the ligand to the receptor during the 2-h incubation. To evaluate the effect of such covalent labeling of the receptor by βFNA on the receptor function, the ability of etorphine to inhibit adenylyl cyclase was determined. As shown in Fig. 5B, even when ∼80% of the receptors were alkylated with βFNA, the maximal inhibition level exhibited was 73 ± 2.9% compared with 88 ± 2.3% in control cells. There was a 17% reduction in maximal activity of the agonist. However, there was a 45-fold decrease in the potency of etorphine to inhibit adenylyl cyclase activity, from 72 ± 16 pM in control cells to 3.2 ± 0.9 nM in HEK293 cells treated with 1 μM βFNA for 2 h. Thus, the potency of etorphine and not the maximal inhibitory level was affected greatly by receptor density on the cell surface.

Irreversible inhibition of μ-opioid receptor activity by βFNA. HEK293 cells stably expressing the μ-opioid receptor were pretreated with various concentrations of βFNA for 2 h at 37°C. A, after repeated washings with PBS at 37°C to remove excess βFNA, in 1 nM [3H]diprenorphine binding assays were carried out with the intact cells as described inExperimental Procedures. The control binding without βFNA was determined to be 1.87 pmol/mg protein. The competition curve was fitted by the GraphPad program. B, etorphine concentration-dependent inhibition of the forskolin-stimulated production of intracellular cAMP was determined in the control and in cells treated with 1 μM βFNA for 2 h. The dose-response curves also were fitted by the GraphPad program.
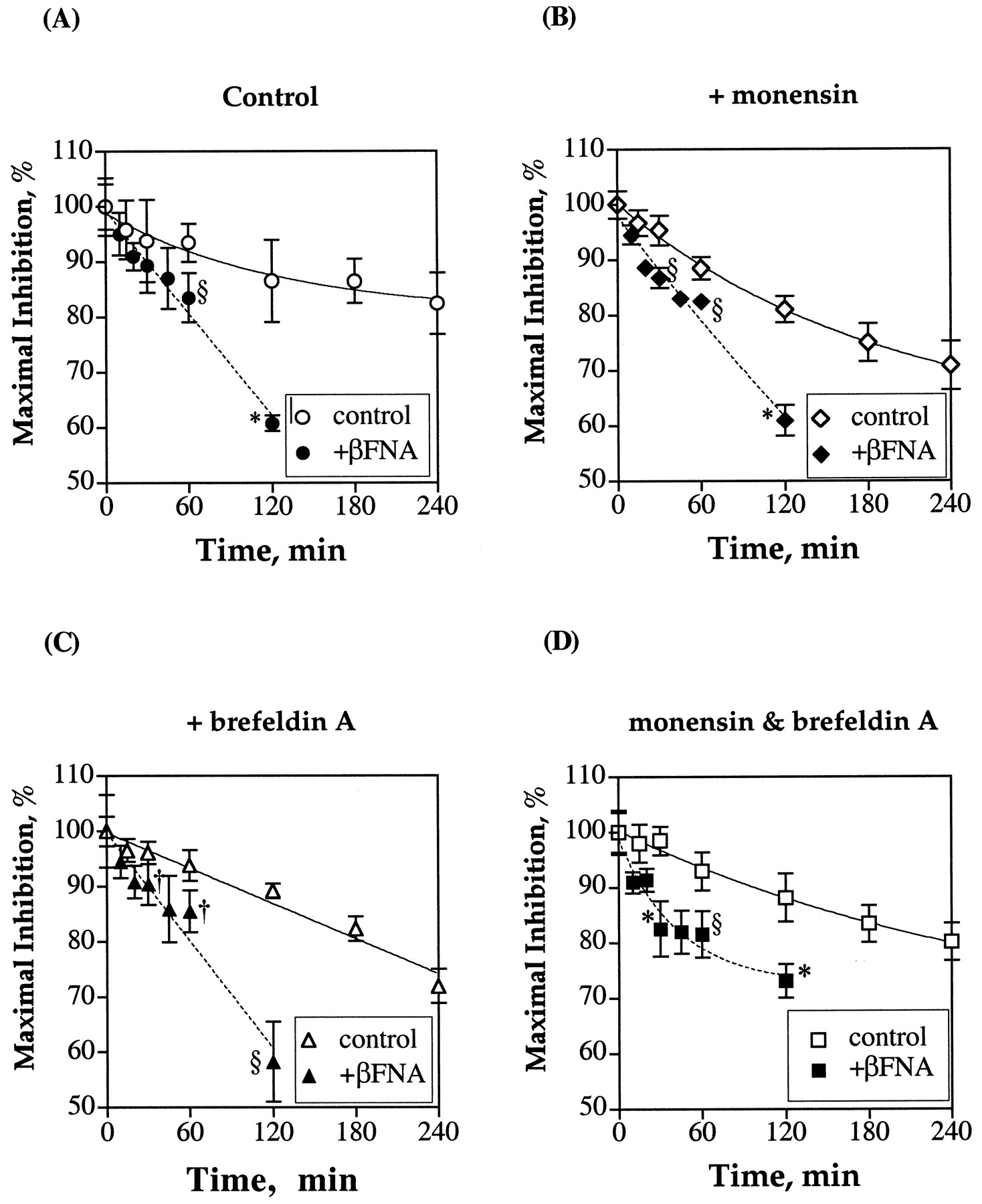
If the high receptor density expressed in HEK293 cells could affect the rate of agonist-induced receptor desensitization, then by inactivating a major percentage of the receptor with βFNA, there should be an increase in the rate of desensitization. When HEK293 cells expressing MORTAG were pretreated with 1 μM βFNA for 2 h before etorphine pretreatment, the rate of etorphine-induced desensitization of the μ-opioid was increased (Fig. 6A). However, even with >80% of the receptor being covalently labeled and inactivated by the antagonist, the rapid desensitization of the receptor was not observed. After 10 min of etorphine pretreatment, there was no statistically significant desensitization of the receptor. Only after 20 min of pretreatment was significant desensitization (9.0 ± 2.5%) of the μ-opioid receptor in HEK293 cells observed. At these two time intervals, it was demonstrated that etorphine-induced phosphorylation of the receptor had reached its maximal level (Fig. 1). Again, the phosphorylation of the receptor did not correlate with the agonist-induced receptor desensitization as measured by adenylyl cyclase activity. With the rapid recycling of the receptor and the contribution of the Golgi transport in maintaining the steady state of the membrane receptor pools, it is probable that the relative slow rate of desensitization after βFNA treatment is caused by resensitization of the receptor. Thus, the HEK293 cells were treated with monensin or brefeldin A or combination of the two antibiotics after βFNA alkylation of the receptor before etorphine pretreatment. Although the pretreatment of HEK293 cells with monensin and brefeldin A increased the rate of receptor internalization, these two drugs did not increase significantly the initial rates of etorphine-induced receptor desensitization in HEK293 cells compared with control (Fig. 6, B–D). The percentage of receptor being desensitized did not exceed 10% after 10 min of etorphine pretreatment in the presence of both monensin and brefeldin A (Fig. 6D). Nevertheless, the reduction in receptor density by βFNA pretreatment increased the rates of etorphine-induced receptor desensitization in the absence or presence of monensin and brefeldin A (Fig. 6).

Time-dependent decrease of μ-opioid receptor activity during chronic etorphine treatment. The effect of receptor density and receptor recycling was determined in HEK293 cells stably expressing the μ-opioid receptor and pretreated with 1 μM βFNA for 2 h at 37°C. The degree of desensitization was measured by comparing the ability of 1 μM etorphine to inhibit the 10 μM forskolin-stimulated intracellular cAMP production before and after various times of 1 μM etorphine treatment. Pretreatments of the cells with 50 μM monensin and 5 μM brefeldin A were carried out 1 h before the addition of etorphine. The maximal inhibition levels in control cells exhibited by 1 μM etorphine before pretreatment were as described in the legend of Fig. 4. For the βFNA-treated cells, levels were as follows: control = 69.6 ± 7.3%, +monensin = 77.2 ± 3.9%, +brefeldin A = 64.0 ± 8.4%, and +monensin + brefeldin A = 76.9 ± 6.7%. The values represent mean ± S.D. of four to six experiments. Statistical analyses with Student's unpaired t tests were carried out to determine significant differences between the percentages of desensitization in control cells and those in βFNA-treated cells under various conditions. †P ≤ .05; §P ≤ .01, and *P ≤ .001.
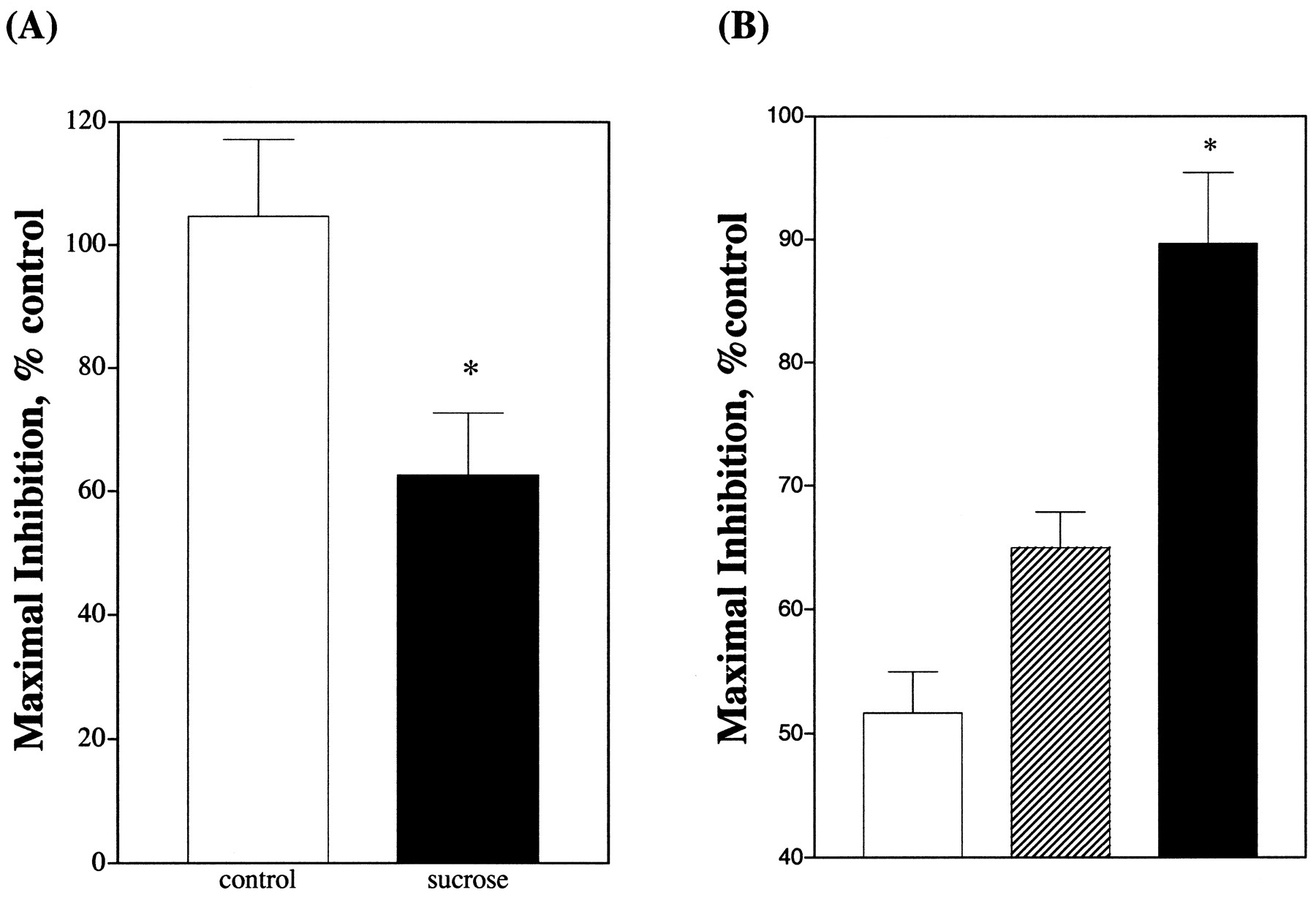
The role of resensitization of the μ-opioid receptor after internalization can be demonstrated further by examining the etorphine-induced receptor desensitization under conditions when receptors do not internalize. It has been well established that opioid receptor endocytosed via the clathrin-coated pit pathway (Chu et al., 1997). This endocytotic pathway can be blocked by the dominant negative mutants of β-arrestin and dynamin (Chu et al., 1997; Whistler and von Zastrow, 1998; Zhang et al., 1998). Additionally, the addition of 0.4 M sucrose to the medium during chronic agonist treatment also blocked the endocytosis of the opioid receptor (Keith et al., 1996). To evaluate the effect of receptor internalization and recycling on the rate of desensitization, etorphine pretreatment of HEK293 cells was carried out in the presence of 0.4 M sucrose. Before the desensitization experiments, the effect of 0.4 M sucrose on internalization of the receptor was examined. HEK293 cells were pretreated with 50 μM monensin to prevent the receptor recycling, and then were treated with 1 μM etorphine in the presence or absence of 0.4 M sucrose. As shown Fig. 7A, in the presence of monensin, 1 μM etorphine induced 42 ± 2.4% of the cell surface μ-opioid receptor to internalize. This agonist-induced receptor internalization was completely blocked by 0.4 M sucrose. Similar inhibition of the decrease in cell surface fluorescence was observed when experiments were carried out in the presence of brefeldin A. When the ability of etorphine to desensitize the receptor was determined under identical conditions, the μ-opioid receptor could be desensitized within the 30 min of etorphine pretreatment. Only the conditions in the presence of brefeldin A were examined because 0.4 M sucrose completely blocked internalization of the receptor (Fig. 7A), and thus the recycling of the receptor should not occur. Longer time periods were not examined due to shrinkage of HEK293 cells in the presence of 0.4 M sucrose and the gradual decline of adenylyl cyclase activity in the presence of sucrose. Nevertheless, pretreatment of HEK293 cells expressing the μ-opioid receptor with etorphine showed a time-dependent desensitization of the receptor (Fig. 7B). Thirty minutes of etorphine pretreatment could desensitize the μ-opioid receptor by 61 ± 0.3%, as measured by adenylyl cyclase activity. In the absence of 0.4 M sucrose, under the conditions in which the receptor can be internalized and resensitized, there was no significant level of desensitization (Fig. 1B). Intriguingly, the inclusion of brefeldin A during the etorphine pretreatment did not increase the rate or magnitude of etorphine-induced receptor desensitization in the presence of 0.4 M sucrose. Instead, there is an apparent decrease in the magnitude of desensitization in the presence of brefeldin A (Fig.7B).

Blockade of receptor endocytosis and μ-opioid receptor desensitization. A, HEK293 cells were treated with 50 μM monensin for 1 h before the addition of 1 μM etorphine. The percentage of the receptor that remained on the cell surface after 30 min of etorphine treatment in the absence or presence of 0.4 M sucrose was then determined by FACS. The values represent mean ± S.D. of three determinations. ∗, P values ≤ .001. B, abilities of 1 μM etorphine to inhibit intracellular cAMP production were determined in HEK293 cells treated with 1 μM βFNA for 2 h at 37°C and then treated with 1 μM etorphine for various times in the presence of 0.4 M sucrose. Cells that were not treated with etorphine also were exposed to 0.4 M sucrose for the same duration. Control represents cells that were not pretreated with brefeldin A and were pretreated with etorphine in the presence of 0.4 M sucrose. The maximal inhibition level of etorphine inhibition in control cells was determined to be 66.0 ± 6.8% and in brefeldin A-treated cells 68.3 ± 1.6%. The values represent mean ± S.D. of three separate experiments. ∗, statistically significant,P ≤ .001 compared with the cells not treated with etorphine. †statistical significance,P ≤ .001 compared with cells treated with brefeldin A.
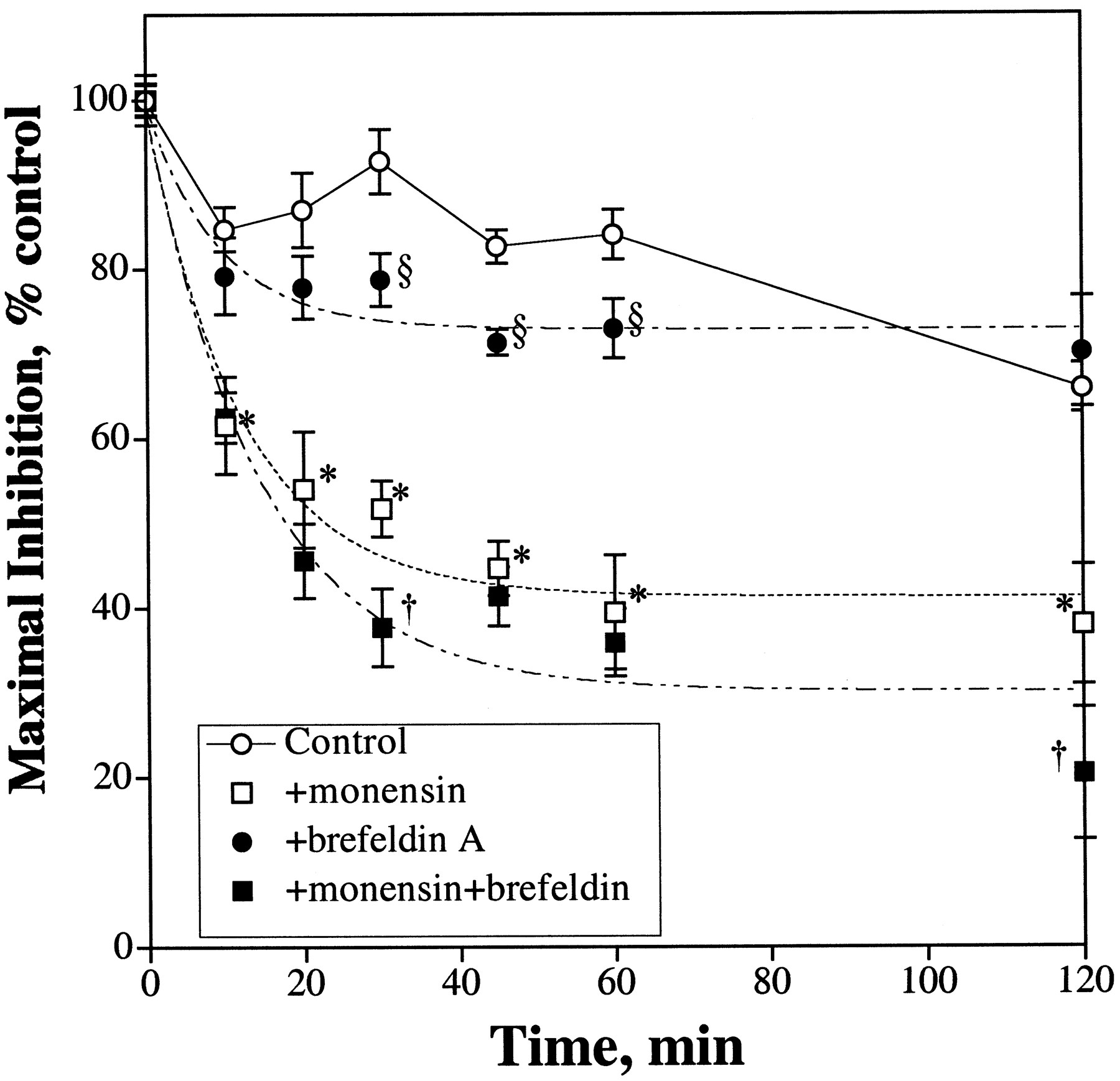
The significance of receptor internalization and resensitization on the rate of μ-opioid receptor desensitization also could be demonstrated with human neuroblastoma SHSY5Y cells. As discussed above, this cell line expressed 43 fmol/mg protein of μ-opioid receptor at the cell surface (Prather et al., 1994). As expected, 1 μM DAMGO induced a time-dependent phosphorylation of the μ-opioid receptor (data not shown). DAMGO was used instead of etorphine so as to confine the agonist activation at the μ- but not at the δ-opioid receptor expressed in these cells. Even with this relatively low level of μ-opioid receptor expressed, DAMGO could inhibit the forskolin-stimulated intracellular cAMP production by 57.9 ± 4.0%. There appeared to be an initial rapid desensitization of the μ-opioid receptor followed by a gradual resensitization (Fig.8). Ten minutes after DAMGO pretreatment, the receptor was desensitized by 15 ± 2.6%. However, after pretreating the SHSY5Y cells with DAMGO for 30 min, 93 ± 3.8% of the maximal activity remained. After 2 h of DAMGO pretreatment, the μ-opioid receptor was only 34% desensitized (Fig. 8). Because the μ-opioid receptor in this cell line could be phosphorylated rapidly in the presence of agonist (data not shown), again, there appears to be a discrepancy between the rate of μ-opioid receptor desensitization and the phosphorylation of the receptor. However, when the DAMGO pretreatment was carried out in the presence of monensin, the rate of μ-opioid receptor desensitization was greatly increased. After 10 min of DAMGO pretreatment, the μ-opioid receptor was desensitized by 38 ± 5.7%. There was a continued time-dependent decrease in DAMGO inhibition of the intracellular cAMP production with agonist pretreatment (Fig. 8). The maximal level of desensitization, 59 ± 2.7%, was reached within 60 min of pretreatment, with a calculated t 1/2 = 8.4 ± 1.4 min. Continued incubation of the SHSY5Y cells with DAMGO for up to 2 h did not increase the magnitude of desensitization. Addition of brefeldin A alone also increased the initial rate of desensitization. During the same time period of DAMGO pretreatment, in the presence of brefeldin A, the agonist could desensitize the μ-opioid receptor inhibition of adenylyl cyclase by 27 ± 1.8% with a calculatedt 1/2 = 6.5 ± 1.7 min (Fig. 8). Inclusion of brefeldin A with monensin during the DAMGO treatment further enhanced the receptor desensitization. Brefedlin A did not increase the initial rate of receptor desensitization when added to cells treated with monensin (t 1/2 = 9.9 ± 2.1 min), but appeared to increase the magnitude of desensitization significantly (70 ± 4.7%). Hence, in the SHSY5Y cells where the μ-opioid receptor density is low, the attenuation of receptor resensitization with monension and brefeldin A greatly increased the rate of receptor desensitization to a degree similar to the rate of agonist-induced receptor phosphorylation.

Receptor recycling and μ-opioid receptor desensitization in human neuroblastoma SHSY5Y cells. Human neuroblastoma SHSY5Y cells were pretreated with 1 μM DAMGO for various amounts of time and the ability of 1 μM DAMGO to inhibit 10 μM forskolin-stimulated intracellular cAMP production was then determined. When the effects of 50 μM monensin and 5 μM brefeldin were measured, the cells were pretreated with these antibiotics 1 h before the addition of DAMGO. The maximal inhibtion levels exhibited by DAMGO in control = 57.9 ± 4.0%, +monensin = 60.0 ± 2.0%, +brefeldin A = 55.9 ± 6.7%, and +monensin + brefeldin A = 61.6 ± 4.5%. The values represent mean ± S.D. of four separate experiments. Statistical significance compared with control cells, *P ≤ .001 and §P ≤ .005, respectively. †, statistically significant compared with cells treated with monensin,P ≤ .005.
The impact of the μ-opioid receptor's resensitization in SHSY5Y cells on the rate of agonist-induced receptor desensitization could be demonstrated further by blocking the agonist-induced receptor internalization with 0.4 M sucrose. Similar to our experiments with HEK293 cells, the blockade of clathrin-coated vesicle-mediated receptor internalization with hypertonic sucrose resulted in an increase in agonist-induced receptor desensitization. As summarized in Fig.9A, treatment of SHSY5Y cells with 1 μM DAMGO for 60 min resulted in minimal loss of activity. However, when these cells were treated with DAMGO in the presence of 0.4 M sucrose, a 38 ± 10% desensitization was observed. These results clearly suggest that the μ-opioid receptors are rapidly recylced and resensitized in SHSY5Y cells during chronic DAMGO treatment. If this is the case, and if monensin is used to prevent the recycling, then the SHSY5Y cells should remain desensitized to DAMGO after the removal of the agonist. When SHSY5Y cells were treated with 1 μM DAMGO for 30 min in the presence of 50 μM monensin, rapid desensitization was observed [48 ± 3.3% desensitized (Fig. 9B)]. After the repeated washing of the cells to remove DAMGO, the ability of DAMGO to inhibit adenylyl cyclase exhibited minimal recovery when the cells were incubated at 37°C for 30 min in the presence of 50 μM monensin. A 35 ± 2.9% desensitization remained (Fig. 9B). However, when the SHSY5Y cells were incubated at 37°C for 30 min in the absence of monensin, the ability of DAMGO to inhibit adenylyl cyclase was similar to the DAMGO-nontreated cells (Fig. 9B). These data indicate that the removal of monensin allows the recycling of the internalized receptor to the cell surface of SHSY5Y cells and the restoration of the ability of DAMGO to regulate adenylyl cyclase activity.

Blockade of receptor internalization or recycling on the desensitization and resensitization of μ-opioid receptor in SHSY5Y cells. A, SHSY5Y cells were pretreated with 1 μM DAMGO for 60 min in the presence (▪) or in the absence (■) of 0.4 M sucrose. Cells that were not treated with DAMGO were exposed to sucrose for the same period of time. Then the ability of 1 μM DAMGO to inhibit the 10 μM forskolin-stimulated intracellular cAMP production was determined. B, SHSY5Y cells were pretreated with 50 μM monensin for 1 h followed by 1 μM DAMGO for 30 min. Then the cells were washed three times with 1 ml of PBS to remove DAMGO and monensin. One milliliter of DMEM was added to each well and the cells were incubated at 37°C for an additional 30 min. The ability of 1 μM DAMGO to inhibit 10 μM forskolin-stimulated intracellular cAMP production was then measured. (■) represents cells that were treated with DAMGO and were not washed. (▧) represents cells that were washed and 50 μM monensin was added to DMEM during the 30-min recovery period. (▪) represents cells that were washed and monensin was not added back during the recovery period. The values represent averages of three independent experiments. ∗, statistically significant compared with the control values in A and to the nonwashed cells in B, P < .005.
Discussion
The working hypothesis of the receptor phosphorylation as the trigger for desensitization of these receptors has been demonstrated conclusively for many GPCRs. Phosphorylation of receptors such as the β2-adrenergic (Lefkowitz, 1998), muscarinic (Pals-Rylaarsdam et al., 1995), prostaglandin E EP2 and EP4 (Bastepe and Ashby, 1996), adenosine A1 and A3 (Palmer et al., 1996), somatostatin (Roth et al., 1997, Hipkin et al., 1997), neurokinin-2 (Alblas et al., 1995), and secretin (Holtmann et al., 1996) receptors have been shown to be critical for receptor desensitization. However, there are exceptions. Phosphorylation is not involved in desensitization of the β3-adrenergic receptor (Liggett et al., 1993, Nantel et al., 1993; Chaudhry and Granneman, 1994), chemoattractant (Kim et al., 1997), cholecystokinin (Pohl et al., 1997), follitorphin (Hipkin et al., 1995), and the adenosine A2 (Palmer and Stiles, 1997) receptors. Sequestration of the receptors has been proposed to be the mechanism for somatostatin receptor desensitization (Beaumont et al., 1998). Hence, it is possible that the GPCR can be desensitized without being phosphorylated.
The μ-opioid receptor could be one GPCR for which phosphorylation might not be the obligatory event for receptor desensitization. This conclusion is based mainly on the following observations: 1) receptor phosphorylate rate did not correlate with the desensitization rate (El-Kouhen et al., 1999); 2) μ-opioid receptor desensitized slowly in the Xenopus oocyte and the expression of GRK and β-arrestin did not increase the rate of receptor desensitization (Koover et al., 1997); and 3) DAMGO could desensitize a mutant receptor in which all the Ser/Thr residues within the third intracellular loop and carboxyl tail have been substituted with Ala (Capeyrou et al., 1997). Because the desensitization rates observed in those studies are relatively slow, it is still possible that the rapid desensitization of the receptor involved the phosphorylation of the receptor. Such a notion is supported by the observation that mutation of Thr394, a putative GRK site, could block the agonist-induced μ-opioid receptor desensitization (Pak et al., 1997). The receptor phosphorylation hypothesis also is supported by the observation that DAMGO inhibition of adenylyl cyclase activity was not affected by pretreatment of HEK293 cells expressing the μ-opioid receptor with morphine for 5 min but was attenuated greatly if the cells were pretreated with DAMGO for 5 min (Whistler and von Zastrow, 1998). Because morphine could not induce the phosphorylation of the receptor and DAMGO could (Arden et al., 1995), these data suggested the phosphorylation of the μ-opioid receptor is the key for receptor desensitization. However, in the same study, the overexpression of β-arrestin resulted in the internalization of the μ-opioid receptor and the subsequent rapid desensitization of agonist inhibition of adenylyl cyclase activity (Whistler and von Zastrow, 1998). The cell surface receptor level has been proposed to be critical for the overall activities of the μ-opioid receptor (Pak et al., 1996). Thus, the loss in activities of the μ-opioid receptor can involve both the phosphorylation and the internalization of the receptor.
This study indicates that the μ-opioid receptor does internalize and recycle to the cell surface of HEK293 cells. The increase in the rate of disappearance of cell surface fluoresence in the presence of monensin supports such a life cycle of the μ-opioid receptor (Fig.2). This increase in the internalization rate of MOR1 in the presence of monensin was in direct contrast with that reported by Koch et al. (1998). Such discrepancy could stem from the different agonists that were used in the two studies. Nevertheless, with the ability of the μ-opioid to recycle, it is probable that the receptor is being resensitized after internalization. With the β2-adrenergic receptor studies, blockade of receptor internalization with the dominant negative β-arrestin 1(V53D) mutant prevented the resensitization of the receptor (Zhang et al., 1997). Dephosphorylation of the receptor occurred intracellularly. Likewise, blockade of the clathrin-coated pit-mediated receptor endocytosis with sucrose accelerated the μ-opioid receptor desensitization (Fig. 7). These data supported the hypothesis that the opioid receptor can be resensitized after internalization.
If resensitization does occur, then the rate of μ-opioid receptor desensitization should be accelerated if the recycling of the receptor is prevented. As indicated in this study, addition of monensin during agonist treatment did not increase the receptor desensitization rate in HEK293 cells. Similar observations were reported by Koch et al. (1998)with HEK293 cells. The inclusion of brefeldin A to block the transport of the receptor from Golgi did not increase the rate of receptor desensitization either. The lack of monensin and brefeldin A effects argued against the resensitization process. However, it is apparent that the μ-opioid receptor, similar to other GPCRs, is very efficiently coupled to adenylyl cyclase. Our βFNA irreversible antagonist experiments indicate there is minimal alteration in the maximal inhibition level even with >80% of the receptor alkylated (Fig. 5). This is best illustrated with SHSY5Y cells that express 49 fmol/mg protein of μ-opioid receptor, or 0.3% of the receptor level expressed in the HEK293 cell line that is used in this study. DAMGO could inhibit the production of intracellular cAMP by 58%, or 66% of the maximal opioid agonist activity observed in HEK293 cells. Thus, probably, the failure to observe rapid desensitization in HEK293 cells under the receptor recycling blockade conditions is due to the relatively high density of receptor expressed in these cells. The increase in the desensitization rate after reduction of the receptor density by βFNA (Fig. 6) supported this hypothesis. The direct correlation between the receptor phosphorylation rate and the receptor desensitization rate is illustrated with the SHSY5Y cells when the receptor recycling is abolished (Fig. 8). The internalized and endosomal located receptors in the SHSY5Y cells can be resensitized after the removal of the agonist DAMGO (Fig. 9). It is important to point out that even with the low receptor level expressed in SHSY5Y cells, in the absence of monensin or brefeldin A, the desensitization of the receptor was slow. Thus, the ability of μ-opioid receptor to recycle affects the ability of the agonist to desensitize the system.
As a model, we propose that the dynamic cycle between receptor activation, phosphorylation, internalization, dephosphorylation, and recycling determines the activity of the μ-opioid receptor on the cell surface. The loss of response occurs only if one part of this dynamic cycle predominates. For example, if the activation of the receptor results in the phosphorylation of the receptor that leads to its internalization, the ability to dephosphorylate and recycle the internalized receptor will result in the activity of the receptor remaining the same. Only if the recycling and hence the resensitization of the receptor is prevented would the phosphorylation and internalization of the receptor dominate and lead to subsequent loss of response. However, when the receptor level is in excess of the level needed for maximal activation, internalization or sequestration of the receptor could not alter the μ-opioid receptor activity. Hence, the dynamic nature of the receptor level on the cell surface will greatly affect the ability of the agonist to regulate the μ-opioid receptor activity. If this model is correct, then extra precaution has to be taken in the interpretation of the receptor mutation studies. An example is the Thr394 mutation of the μ-opioid receptor. This mutation has been reported to attenuate DAMGO-induced receptor desensitization (Pak et al., 1997). The interpretation is that the phosphorylation of Thr394 by GRK is the trigger for receptor desensitization. However, the same mutation has been reported to increase the recycling of the receptor (Wolf et al., 1999). Hence, the attenuation of the agonist-induced μ-opioid receptor desensitization by the T394A mutation could be due simply to the increase in the recycling rate of the receptor. Thus, in addition to the role of the receptor density, future identification of the involvement of putative receptor phosphorylation sites in the μ-opioid receptor desensitization must consider ability of the μ-opioid receptor to resensitize and recycle.
Footnotes
- Received December 10, 1999.
- Accepted May 17, 2000.
-
Send reprint requests to: Dr. Ping-Yee Law, Department of Pharmacology, University of Minnesota Medical School, 6-120 Jackson Hall, 321 Church St. SE, Minneapolis, MN 55455. E-mail:Ping{at}mail.ahc.umn.edu
-
This study was supported in parts by National Institutes of Health Grants DA07339, DA11806, and DA00564 and F. Stark Fund of Minnesota Medical Foundation.
Abbreviations
- GPCR
- G protein-coupled receptor
- GRK
- G protein-coupled receptor kinase
- HEK
- human embryonic kidney
- GIRK
- G protein-coupled inward rectifying potassium channel
- DAMGO
- [d-Ala2,N-MePhe4,Gly-ol5]-enkephalin
- HA
- hemagglutinin
- FACS
- flourescence-activated cell-sorting analysis
- DMEM
- Dulbecco's modified Eagle's medium
- βFNA
- β-funaltrexamine
- DPDPE
- [d-Pen2,d-Pen5]-enkephalin
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}