


Abstract
In the complex signal transduction networks involving G protein-coupled receptors there are numerous examples where Gi -linked receptors augment Gq-dependent signals. The mechanistic basis of such occurrences is thought to entail signal convergence at phospholipase Cβ (PLCβ) via the G protein βγ-dimers. Herein, we explored the possibility that augmentation by βγ-dimers requires preactivation of PLCβ. COS-7 cells were transiently cotransfected with cDNAs encoding various combinations of receptors and G protein subunits. The Gi-coupled δ- and κ-opioid receptors could not stimulate PLCβ unless they were coexpressed with Gα16. The opioid-induced response was dose-dependent and partially inhibited by pertussis toxin or coexpression with transducin, indicating the involvement of βγ-subunits released from the Giproteins. When PLCβ was preactivated by constitutively active mutants of Gα16, Gαq, or Gα14, opioids enhanced the activity by 80 to 300% and such responses were mostly pertussis toxin-sensitive. The opioid-induced enhancement was dose-dependent and could not be blocked by staurosporin, a protein kinase C inhibitor. Other Gi-coupled receptors that were ineffective on their own also acquired the ability to stimulate PLCβ in the presence of a constitutively active mutant of Gαq. Coactivation of endogenous or exogenous Gq-coupled receptors with the δ-opioid receptor produced strong stimulations of PLCβ and such responses could be partially blocked by pertussis toxin. These results show that enhancement of Gq-dependent signals by Gi-coupled receptors requires activated PLCβ and is mediated via the βγ-dimer.
In the nervous system, different extracellular signals are often required to coordinate complex neuronal activities such as neurotransmission and cognition. The multitude of extracellular signals is usually detected by a variety of cell surface receptors that use distinct yet overlapping signal transduction mechanisms. The ability to integrate and process incoming signals is an important characteristic of neurons. The superfamily of G protein-coupled receptors (GPCRs) constitutes a large array of cell surface detectors for neurotransmitters, hormones, lipids, pheromones, and photons. Multiple GPCRs are often coexpressed in any particular cell type, where they regulate the levels of intracellular second messengers independently, in synergism, or by antagonism. Of the two most widely studied effectors of GPCRs, adenylyl cyclase and phospholipase C (PLC), intricate regulatory mechanisms for the former have been discerned.
The mechanism by which signals generated from different GPCRs become integrated inside the cell is best exemplified by the type 2 adenylyl cyclase. Type 2 adenylyl cyclase can be stimulated by the G protein βγ-subunits only when it is already preactivated by either Gαs (Federman et al., 1992) or protein kinase C-mediated phosphorylation (Tsu and Wong, 1996). Hence, the βγ-subunits released on the activation of Gi-linked receptors can enhance the activity of type 2 adenylyl cyclase only if Gs- or Gq-linked receptors are simultaneously activated. This unique property of type 2 adenylyl cyclase allows it to integrate and process signals from various GPCRs, perhaps providing a temporal distinction of the different inputs (Lustig et al., 1993a).
Equally important for coordinating cellular functions is the regulation of PLC that generates diacylglycerol and inositol phosphate (IP)3, leading to the activation of protein kinase C and calcium mobilization. Many GPCRs stimulate PLCβ-isozymes through coupling to G proteins belonging to the Gq subfamily (Rhee and Bae, 1997). The regulation of PLCβ-isozymes by GPCRs bears some similarity to those of adenylyl cyclase. For instance, PLCβ can be stimulated by the α-subunits of all Gq subfamily members as well as by the βγ-dimers (Smrcka and Sternweis, 1993; Nakamura et al., 1995). The β2 and β3 isoforms of PLC are especially responsive to stimulation by βγ-subunits. However, most Gi-coupled receptors are incapable of activating PLCβ despite their ability to generate free βγ-subunits. In many biological systems such as the smooth muscles and cultured astrocytes, although activation of Gi-coupled receptors alone has no effect, it augments Gq-mediated responses (for review, seeSelbie and Hill, 1998). The augmentation produced by the stimulation of Gi-coupled receptors is presumably mediated by the βγ-dimers (Biber et al., 1997). These observations suggest that the βγ-subunits released on the activation of Gi are insufficient to stimulate PLCβ, and other signals or conditions may be required. A distinct possibility is that PLCβ can integrate signals from Gi-, Gs-, and Gq-linked receptors in a manner akin to the one used by the type 2 adenylyl cyclase. In the course of examining the coupling of opioid receptors to G16 (Lee et al., 1998), we noticed that although the opioid-induced response was mediated via G16, it was partially sensitive to pertussis toxin (PTX) treatment. In this report, we describe our efforts to decipher the molecular mechansim behind such PTX sensitivity. Our results suggest that when PLCβ is preactivated by the α-subunits of Gq, G14, or G16, it becomes responsive to stimulation by βγ-dimers. Such a precondition by which Gi-linked receptors can stimulate PLCβ may have important mechanistic implications on signal processing by PLCβ.
Materials and Methods
Reagents.
cDNAs encoding the formyl peptide (fMLP) and δ-opioid receptors were kindly provided by F. Boulay (LBIO/Laboratoire, France) and C. Evans (UCLA), respectively. The bombesin and κ-opioid receptors were gifts from J. Battey (National Institute of Neurological Disorders and Stroke) and G. Bell (University of Chicago, IL), respectively. The cDNA encoding the α-subunit of GL1 (the bovine homolog of G14; henceforth referred to as G14 for generality) was generously provided by Dr. T. Nukada (Tokyo Institute of Psychiatry, Japan). The origin and construction of other cDNAs have been described elsewhere (Wong et al., 1992; Lee et al., 1998). PTX and plasmid purification columns were purchased from List Biological Laboratories (Campbell, CA) and Qiagen (Hilden, Germany), respectively. COS-7 cells were obtained from the American Type Culture Collection (ATCC CRL-1651). [3H]Myo-inositol was obtained from DuPont-NEN (Boston, MA). Receptor agonists and staurosporin were purchased from Research Biochemicals (Natick, MA). Antisera against Gαq/11 (3A-180) and Gα14 (3A-195) were purchased from Gramsch Laboratories (Schwabhausen, Germany). Antiserum G51820 against Gαt1 was from Transduction Laboratories (Lexington, KY). Cell culture reagents were obtained from Life Technologies (Grand Island, NY) and all other chemicals were purchased from Sigma Chemical Co. (St. Louis, MO).
Construction of Gα16Q212L and Gα14R179C Mutants.
Polymerase chain reactions (PCRs) were used to generate the two mutants. The cDNAs subjected to site-directed mutagenesis were subcloned into pcDNAI (Invitrogen, San Diego, CA), which contained T7 and SP6 promoter sequences as flanking priming regions. Human Gα16 and bovine Gα14 were used to generate Gα16Q212L (Gα16QL) and Gα14R179C (Gα14RC). Primers encoding the desired mutations were listed below with the mismatch nucleotides underlined: 16-QL/S: GACGTCGGAGGCCTGAAGTCAGAGCGT; 16-QL/AS: ACGCTCTGACTTCAGGCCTCCGACGTC; 14-RC/S: GTGCTCCGTGTCTGCGTGCCCACCACT;14-RC/AS: AGTGGTGGGCACGCAGACACGGAGCAC. Two overlapping fragments that contained the mutation in their overlapping region were amplified separately with thermal cycling at 94°C (1 min)/50°C (1 min)/72°C (1 min) for 30 cycles with Robocycler 40 from Stratagene (La Jolla, CA). The PCR products were annealed together and the full-length fragments were made with the flanking primers. Extension time was increased to 1.5 min/cycle. Full-length Gα16QL was ligated into EcoRV-cut pcDNAI, whereas Gα14RC was subcloned into pcDNAI as a PstI/XbaI cassette. DNA sequences of the mutants were checked by dideoxynucleotide sequencing method with Sequenase V2.0 kit.
Transient Transfection and IP Assay.
COS-7 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (v/v), 50 U/ml penicillin, and 50 μg/ml streptomycin, and grown at 37°C in an environment of 5% CO2. Cells were seeded in 12-well plates at a density of ∼1 × 105 cells/ml and were transfected with the appropriate cDNAs 24 h later by means of the DEAE-dextran method (Wong, 1994). One day later, the transfected cells were labeled with [3H]myo-inositol (2.5 μCi/ml) in inositol-free DMEM (0.75 ml/well) containing 5% fetal calf serum (v/v) for 18 to 24 h. Where necessary, PTX (100 ng/ml) was added together with the radiolabel. Labeled cells were rinsed with 2 ml of assay medium (20 mM HEPES-buffered DMEM with 20 mM LiCl) followed by incubation at 37°C for 1 h in 1 ml of assay medium with the indicated drugs. The reaction was terminated by aspiration and addition of 0.75 ml of 20 mM formic acid. IP production was estimated by determining the ratio of [3H]IP to [3H]inositol plus [3H]IP as described previously (Tsu et al., 1995b).
Preparation of Plasma Membranes and Immunodetection of Gα-Subunits.
COS-7 cells were grown on 150-mm dishes to 70 to 80% confluence and transfected as described for 12-well plates with proper adjustments to the volumes and amounts of the reagents used. Transfected cells were harvested 48 h later in PBS (Ca2+ and Mg2+ free) containing 10 mM EDTA. Cells were resuspended in lysis buffer (50 mM Tris-HCl containing 1 mM phenylmethylsulfonyl fluoride, 1 mM benzamidine-HCl, 1 mM EGTA, 5 mM MgCl2, and 1 mM dithiothreitol, pH 7.4) and lysed by one cycle of freeze-thawing followed by 10 passages through a 27-gauge needle. After removal of nuclei by centrifugation, membranes were collected, washed, and resuspended in lysis buffer. Protein concentrations were determined with the Bio-Rad protein assay kit. For each sample, 50 μg of membrane proteins was separated on a 12.5% polyacrylamide SDS gel and electrophorectically transferred to polyvinylidene difluoride membranes. Localization of protein markers on the polyvinylidene difluoride membrane was by Ponceau S staining. Antigen-antibody complexes were visualized by chemiluminescence with the enhanced chemiluminescence kit from Amersham (Arlington Heights, IL).
Data Analysis.
The IP levels were interpreted as the ratios of the counts per minute of [3H]IP fractions to those of the total labeled inositol fractions and expressed as the ratio of [3H]IP over total [3H]inositols. Absolute values for IP accumulations varied between experiments, but variability within a given experiment was in general <10%. Data shown in the figures are means ± S.D. of triplicates within one single experiment. At least three independent experiments yielded similar results. Bonferronit test with 95% confidence was adopted to verify the significance between different treatment groups within the experiments.
Results
Gα16-Mediated Stimulation of PLCβ by Gi-Coupled Receptors Is Partially Sensitive to PTX Treatment.
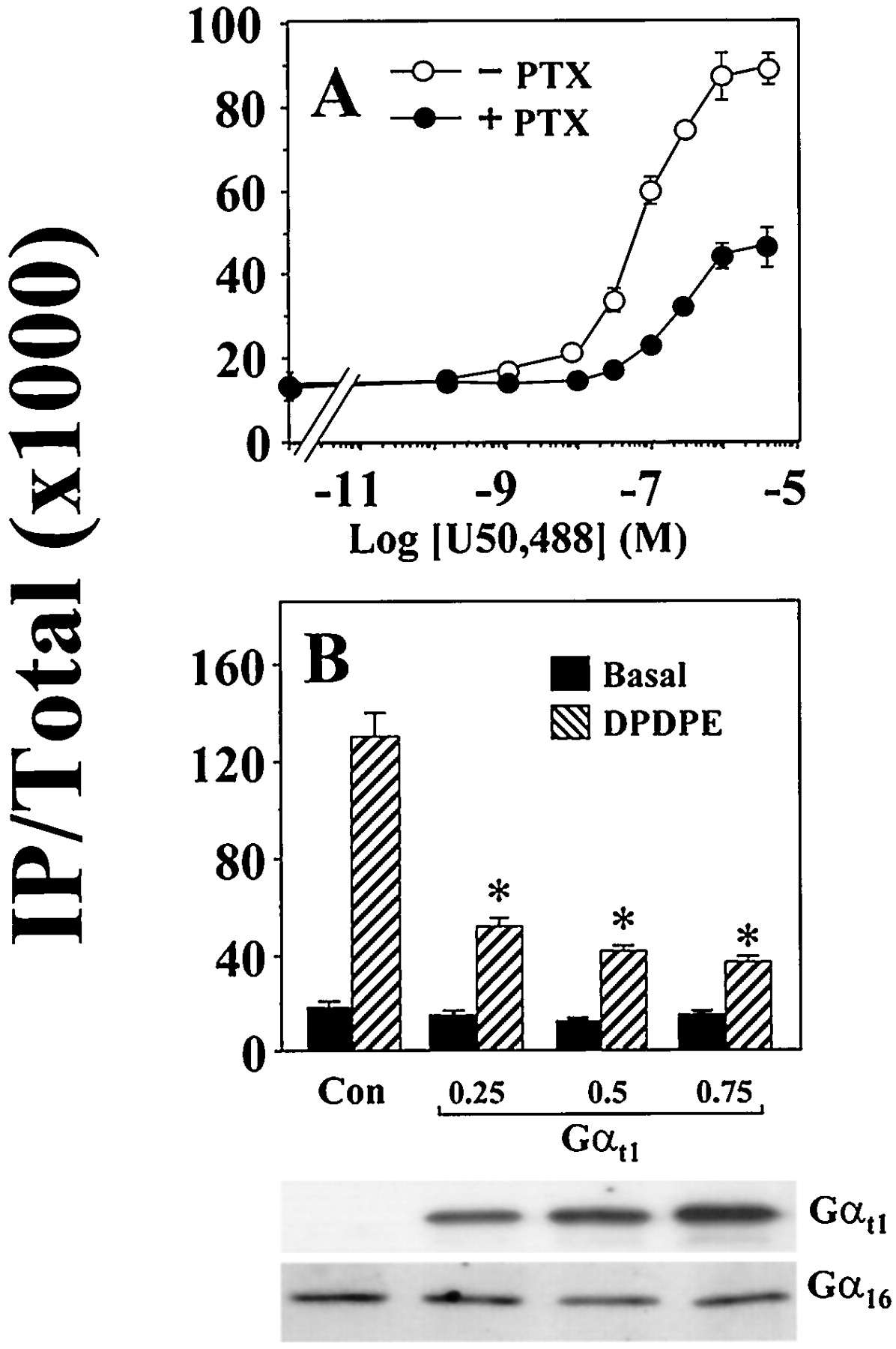
It has recently been shown that the δ-opioid receptor (DOR) can efficiently stimulate the formation of IP in COS-7 cells when it is coexpressed with Gα16 (Lee et al., 1998). As a member of the Gq subfamily, Gα16 lacks the ADP ribosylation site and is resistant to modification by PTX. Gα16-mediated stimulation of PLCβ should thus be PTX-insensitive. Surprisingly, when we examined the ability of DOR to stimulate PLCβ via Gα16 in transiently transfected COS-7 cells, the agonist-induced response was partially reduced by PTX. In the absence of PTX treatment, the δ-selective agonist [d-Pen2,5]enkephalin (DPDPE) stimulated IP formation by 9-fold (Fig.1). Pretreatment of transfected COS-7 cells with PTX suppressed the Gα16-mediated response by 55%. Three other Gi-coupled receptors that are incapable of activating PLCβ in the absence of Gα16 also were examined. COS-7 cells were cotransfected with cDNAs encoding Gα16 and the fMLP, opioid receptor-like (ORL1), or A1 adenosine receptor. Like DOR, the ability of these Gi-coupled receptors to stimulate PLCβ via Gα16 was attenuated by PTX (Fig. 1). Receptor-selective agonists induced 6- to 8-fold stimulation of PLCβ activity, but these Gα16-mediated responses were partially sensitive to PTX treatment. In COS-7 cells coexpressing the κ-opioid receptor (KOR) and Gα16, U50,488 (a κ-selective agonist) stimulated IP accumulation in a dose-dependent manner (Fig. 2A). Again, PTX treatment reduced the U50,488-induced IP formation by ∼60% and raised the EC50 of U50,488 for Gα16-mediated stimulation of PLCβ from ∼60 nM to ∼200 nM (Fig. 2A). A similar shift in EC50 values also was observed with DOR (J.W.M.L. and Y.H.W., unpublished data) and it may reflect the efficiency of coupling solely to the transfected Gα16. The potency with which PTX affects the opioid-induced stimulation of PLCβ and inhibition of adenylyl cyclase was approximately the same. The EC50 of PTX in suppressing the Gα16-mediated stimulation of PLCβ was 0.3 ng/ml, whereas PTX blocked the opioid-induced inhibition of adenylyl cyclase with an EC50 of 0.5 ng/ml (data not shown).
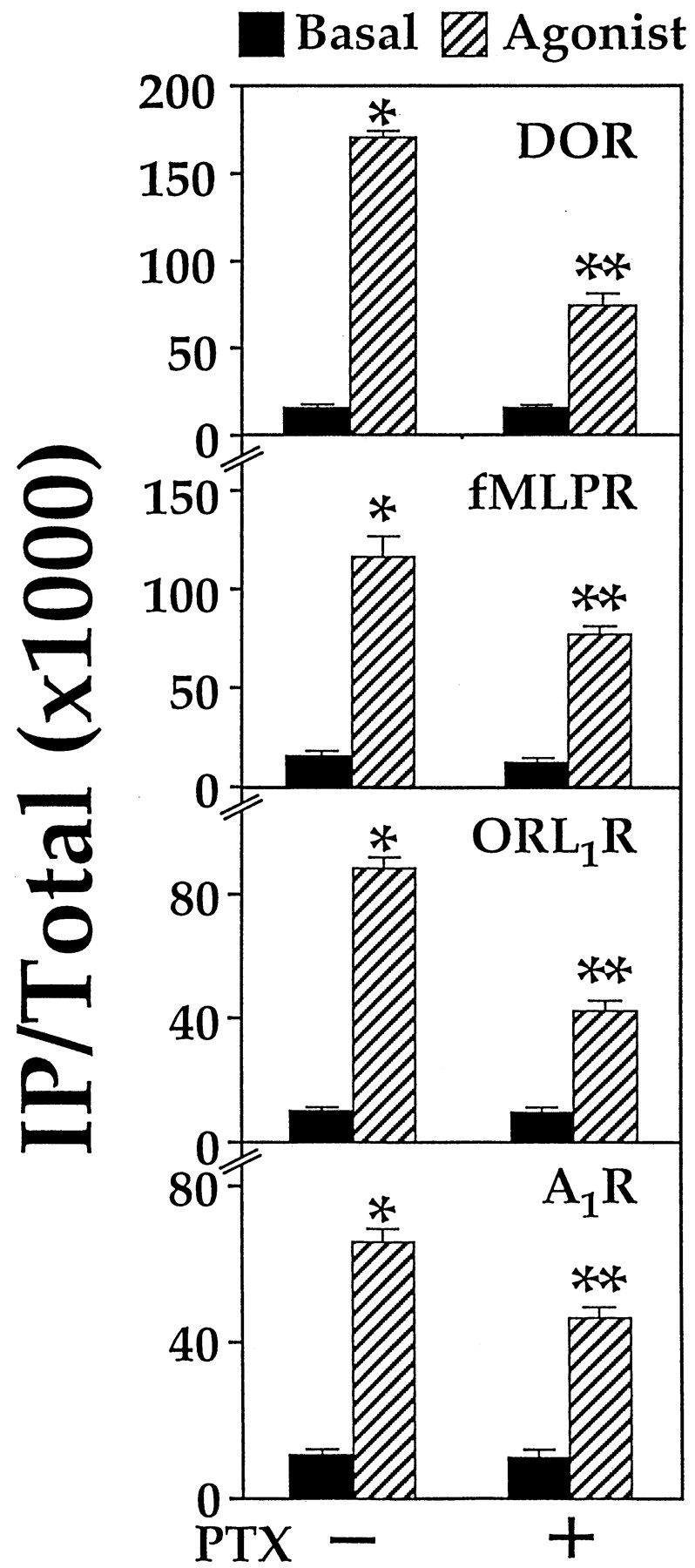
Gα16 -mediated stimulation of PLCβ by Gi-coupled receptors is PTX-sensitive. COS-7 cells were cotransfected with cDNAs encoding Gα16 and a Gi-coupled receptor: DOR, fMLPR, ORL1R, or A1R (each at 0.25 μg/ml). After 24 h, the cells were labeled overnight with 2.5 μCi/ml [3H]myo-inositol with or without 100 ng/ml PTX as indicated. IP production was assayed in the absence or presence of an agonist: 100 nM DPDPE, 200 nM fMLP, 100 nM nociceptin, or 10 μM (+)-N 6-(2-phenylisopropyl)-adenosine. ∗, agonist significantly increased IP accumulation over basal values. ∗∗, PTX significantly reduced the agonist-induced response;n = 3, Bonferroni t test,P < .05.
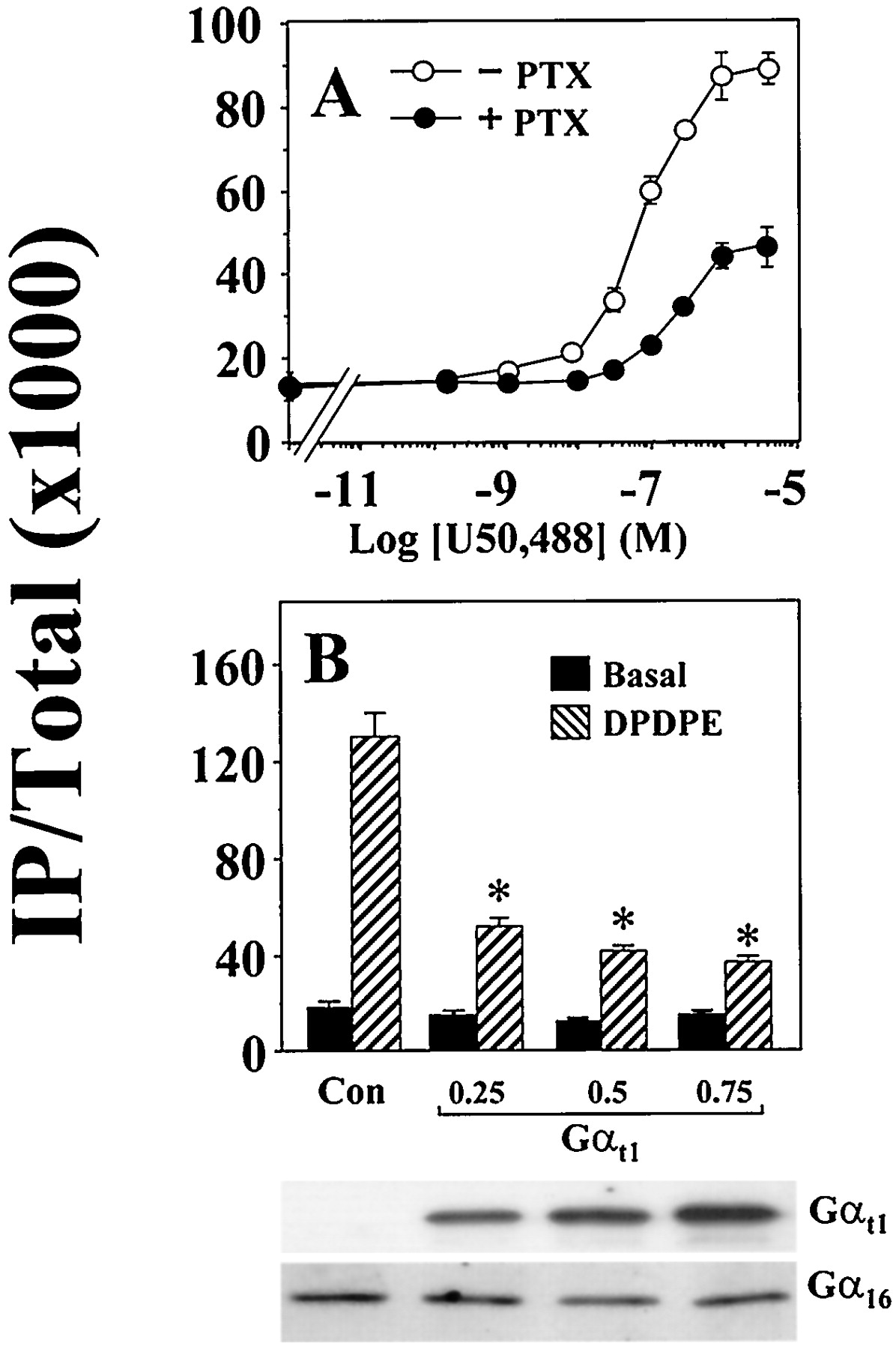
Permissive stimulation of PLCβ by opioid receptors through Gα16 is agonist dose-dependent and can be inhibited by Gαt1. A, COS-7 cells were transfected and labeled as in (A) but with KOR instead of the other receptors. IP production increased dose-dependently with increasing concentrations of the KOR agonist U50,488 (0–3 μM). B, COS-7 cells were cotransfected with the DOR and Gα16 cDNAs (both at 0.25 μg/ml) only or with varying concentrations of Gαt1 (0.25 to 0.75 μg/ml). Cells were assayed for IP production in the absence or presence of 100 nM DPDPE. ∗, all concentrations of Gαt1significantly reduced the DPDPE-induced IP accumulation;n = 3, Bonferroni t test,P < .05. Bottom, expression of Gαt1and Gα16 in the transfected cells as determined by immunodetection with antisera G51820 and 3A-180, respectively.
Involvement of βγ-Subunits.
The PTX sensitivity of the DPDPE response suggests the involvement of Giproteins. Activation of Gi proteins by DOR will invariably lead to the dissociation of the α- and βγ-subunits. Because none of the Gαi-subunits possess the ability to directly activate PLCβ, they are unlikely to stimulate the formation of IP. In contrast, the βγ-dimer is known to regulate a host of effectors, including different isoforms of PLCβ (for review, see Clapham and Neer, 1997). To test if βγ-subunits are involved in the Gα16-dependent stimulation of PLCβ by DOR, we attempted to block the agonist-induced response with a βγ-scavenger, the α-subunit of transducin (Gαt1). When Gαt1 was transiently coexpressed with DOR and Gα16, the DPDPE-induced response was suppressed by 60% (Fig. 2B). The extent of suppression by Gαt1 was similar to that produced by PTX. Increasing the concentration of Gαt1 cDNA used in the transfections from 0.25 μg/ml to 0.75 μg/ml did not further attenuate the DPDPE-induced response. The expression of Gαt1 was confirmed by immunodetection with a Gαt-specific antiserum (Fig. 2B). In our heterologous expression system, overexpression of Gαt1 did not affect the expression level of Gα16 (Fig. 2B). Coexpression of another βγ-scavenger, the carboxyl fragment of β-adrenergic receptor kinase (βARK495–690), with DOR and Gα16 also suppressed the DPDPE-induced response by ∼40% (data not shown). Such experiments implicate the involvement of βγ-subunits.
Constitutively Active Gα-Mutants Permit Gi-Linked Receptors to Stimulate PLCβ.
Because DOR is incapable of stimulating endogenous PLCβ in the absence of Gα16 (Lee et al., 1998), somehow the expression of Gα16 allowed the endogenous PTX-sensitive G proteins to participate in the activation of PLCβ. Interestingly, mechanisms exist for permissive activation of effectors. The type 2 adenylyl cyclase can be stimulated by G protein βγ-dimers when it is preactivated by Gαs·GTP (Federman et al., 1992) or by protein kinase C-mediated phosphorylation (Tsu and Wong, 1996). It is conceivable that similar mechanisms exist for the regulation of PLCβ. This might explain why βγ-complexes can participate in the stimulation of PLCβ when Gα16 was coexpressed with DOR, but cannot do so in the absence of Gα16. By drawing an analogy to the type 2 adenylyl cyclase system, we tested whether preactivation of PLCβ allows Gi-linked receptors to subsequently stimulate PLCβ. To induce preactivation of PLCβ, COS-7 cells were cotransfected with cDNAs encoding DOR and Gα16QL, a constitutively activated mutant of Gα16 (Heasley et al., 1996). Because Gα16QL is “locked” in the GTP-bound state, it is relatively unresponsive to activation by DOR compared with Gα16 wild-type. In the absence of any opioid agonist, Gα16QL-expressing cells exhibited higher basal PLCβ activity, which is indicative of the constitutive activity of Gα16QL (Fig.3). Application of 100 nM DPDPE to the transfected cells further enhanced the IP formation by ∼90%. The DPDPE-induced enhancement was completely PTX-sensitive (Fig. 3), indicating the involvement of Gi proteins instead of G16. These results imply that when PLCβ is activated by Gα16QL, it may then become responsive to stimulation by Gi-linked receptors through a Gi-mediated mechanism. This mechanism might involve the βγ-subunits because coexpression of β1γ2 with Gα16QL significantly increased the basal PLCβ activity by 25.7 ± 2.9% (n = 4;P < .05 by paired t test) compared with that obtained with the expression of Gα16QL alone. No enhancement of Gα16QL activity was observed by coexpressing the nonfunctional β3γ2-complex.

Constitutively active Gα-mutants permit stimulation of PLCβ by DOR. COS-7 cells were cotransfected with 0.25 μg/ml DOR and one of the following cDNAs: Gα16QL, GαqRC, or Gα14RC (all at 0.15 μg/ml). After labeling with or without 100 ng/ml PTX, IP accumulation was assayed in the absence (basal) or presence of 100 nM DPDPE. ∗, DPDPE significantly increased IP production beyond basal levels;n = 3, Bonferroni t test,P < .05.
Because DOR can use Gα16 to stimulate PLCβ (Lee et al., 1998), it is conceivable that the DPDPE-induced stimulation was due to improved maintenance of Gα16QL in the active state. To eliminate this possibility, we repeated the experiment with a constitutively activated mutant of Gαq (GαqRC;Conklin et al., 1992), which should not interact with DOR. As shown in Fig. 4, coexpression of DOR and wild-type Gαq in COS-7 cells did not permit DPDPE to stimulate IP accumulation. In contrast, coexpression of GαqRC raised the basal accumulation of IP by 8-fold, and activation of DOR further increased the IP formation (Fig.4). Again, the DPDPE-induced stimulation of PLCβ in the presence of GαqRC was PTX-sensitive (Fig. 3). In contrast, replacement of GαqRC by the constitutively active mutant of Gαs(GαsRC) did not allow DPDPE to stimulate the PLCβ activity (Fig. 4). Additional experiments using the constitutively active mutant of Gα14(Gα14RC) yielded similar results, except that the DPDPE-induced response was not completely abolished by PTX (Fig.3). The reason for the incomplete blockade of the DPDPE-induced response by PTX is unclear. Nevertheless, these data suggest that preactivation of PLCβ by constitutively active mutants of Gαq, Gα14, or Gα16 permits DOR to stimulate PLCβ in a PTX-sensitive manner.

DPDPE-induced PLCβ-activation is protein kinase C-independent. COS-7 cells were transfected with 0.25 μg/ml DOR alone or together with one of the following: GαsRC (0.25 μg/ml), GαqWT (0.25 μg/ml), or GαqRC (0.1 μg/ml). After labeling with [3H]myo-inositol, cells transfected with DOR alone were treated with 100 nM PMA. Where indicated, DOR/GαqRC-transfected cells were pretreated with 500 nM staurosporin (stauro). After 15 min of pretreatment, cells were assayed for IP accumulation in the absence (basal) or presence of 100 nM DPDPE. ∗, DPDPE significantly enhanced IP production over basal values;n = 3, Bonferroni t test,P < .05.
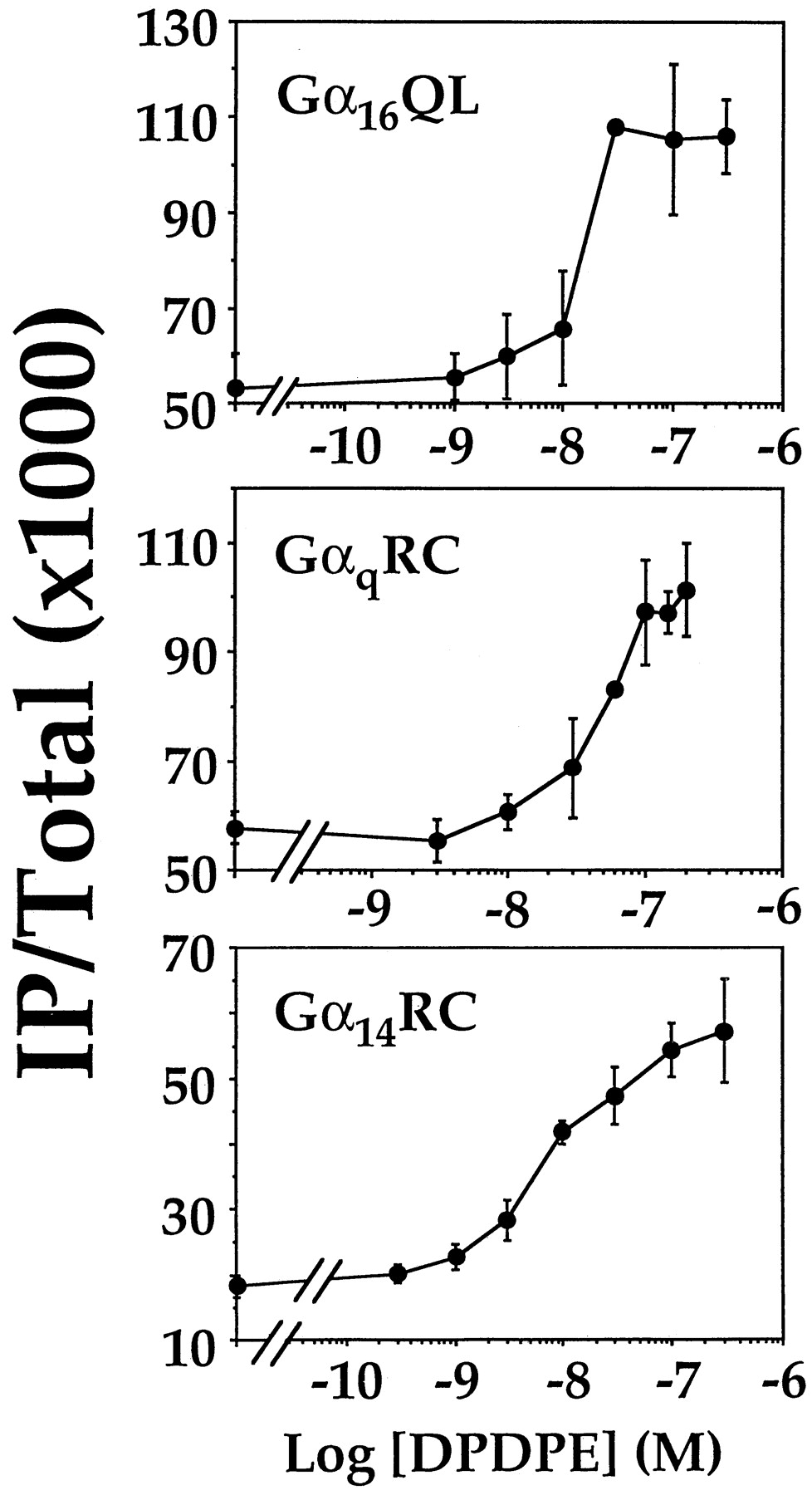
Next, we asked if such permissive stimulation of PLCβ by DOR is dependent on the extent of preactivation. COS-7 cells were transiently cotransfected with cDNAs encoding DOR (0.25 μg/ml) and varying amounts of Gα14RC cDNA up to 5 μg/ml. Transfected cells were then assayed for IP formation in the absence or presence of 100 nM DPDPE. No direct correlation was observed between the magnitude of the DPDPE-induced stimulations and the extent of preactivation of PLCβ by the mutationally activated Gα-subunits; the magnitude of the DPDPE-induced enhancement did not correspond with the level of expression of Gα14RC (Fig.5). Replacement of Gα14RC by GαqRC or Gα16QL produced similar results (data not shown). To examine if these DPDPE-induced enhancements exhibit agonist dose-dependence, we cotransfected COS-7 cells with cDNAs encoding DOR and one of the three constitutively active mutants. The cDNA concentration for the constitutively active mutants was lowered to 0.1 μg/ml to enhance the signal-to-noise ratio. The transfected cells were stimulated with varying concentrations of DPDPE (ranging from 0.3 to 300 nM). DPDPE dose-dependently stimulated IP formation in all three cases (Fig. 6). In the presence GαqRC, the EC50 for the DPDPE-induced response was ∼50 nM. The EC50values for the DPDPE-induced response were ∼10 to 20 nM for Gα16QL and Gα14RC transfected cells, and were slightly lower than that obtained with their wild-type counterparts (∼40 nM; Lee et al., 1998). Such studies demonstrate that agonist-dependent activation of DOR in the presence of constitutively active Gq subfamily mutants can potentiate PLCβ-activity.

cDNA dose-dependence of Gα14RC-mediated permissive activation of PLCβ by DOR. COS-7 cells were cotransfected with DOR (0.25 μg/ml), and varying concentrations of Gα14RC cDNAs (3 ng/ml to 5 μg/ml). Cells were then labeled and assayed with or without 100 nM DPDPE. Inset shows the expression of Gα14RC in cells transfected with the seven different doses of cDNA was determined by Western blot analysis with a Gα14-specific antiserum 3A-195.

DPDPE dose-dependently increases IP formation in cells coexpressing DOR and mutants of Gαq ,Gα16, or Gα14. COS-7 cells were cotransfected with cDNAs encoding DOR (0.25 μg/ml) and GαqRC, Gα16QL (both at 0.1 μg/ml), or Gα14RC (0.25 μg/ml). The cells were labeled and stimulated with varying concentrations of DPDPE (0–200 nM for GαqRC and 0–300 nM for Gα16QL or Gα14RC).
In the activation of type 2 adenylyl cyclase, phosphorylation by protein kinase C is one of the permissive conditions under which βγ-dimers can stimulate the production of cAMP (Tsu and Wong, 1996). Because the constitutive activity of GαqRC (as well as Gα16QL and Gα14RC) leads to the activation of PLCβ and subsequently protein kinase C, we asked if this permits βγ-dimers to further stimulate PLCβ in a manner similar to that observed for type 2 adenylyl cyclase. We began by substituting GαqRC with phorbol-12-myristate-13-acetate (PMA), a direct activator of protein kinase C. In COS-7 cells expressing DOR alone, prior treatment with 100 nM PMA for 15 min did not allow DPDPE to stimulate PLCβ (Fig. 4). Moreover, 500 nM staurosporin (a protein kinase C inhibitor) did not prevent DPDPE from stimulating PLCβ in cells coexpressing DOR and GαqRC (Fig. 4). Hence, the DOR-induced, GαqRC-dependent stimulation of PLCβ did not seem to require the activation of protein kinase C.
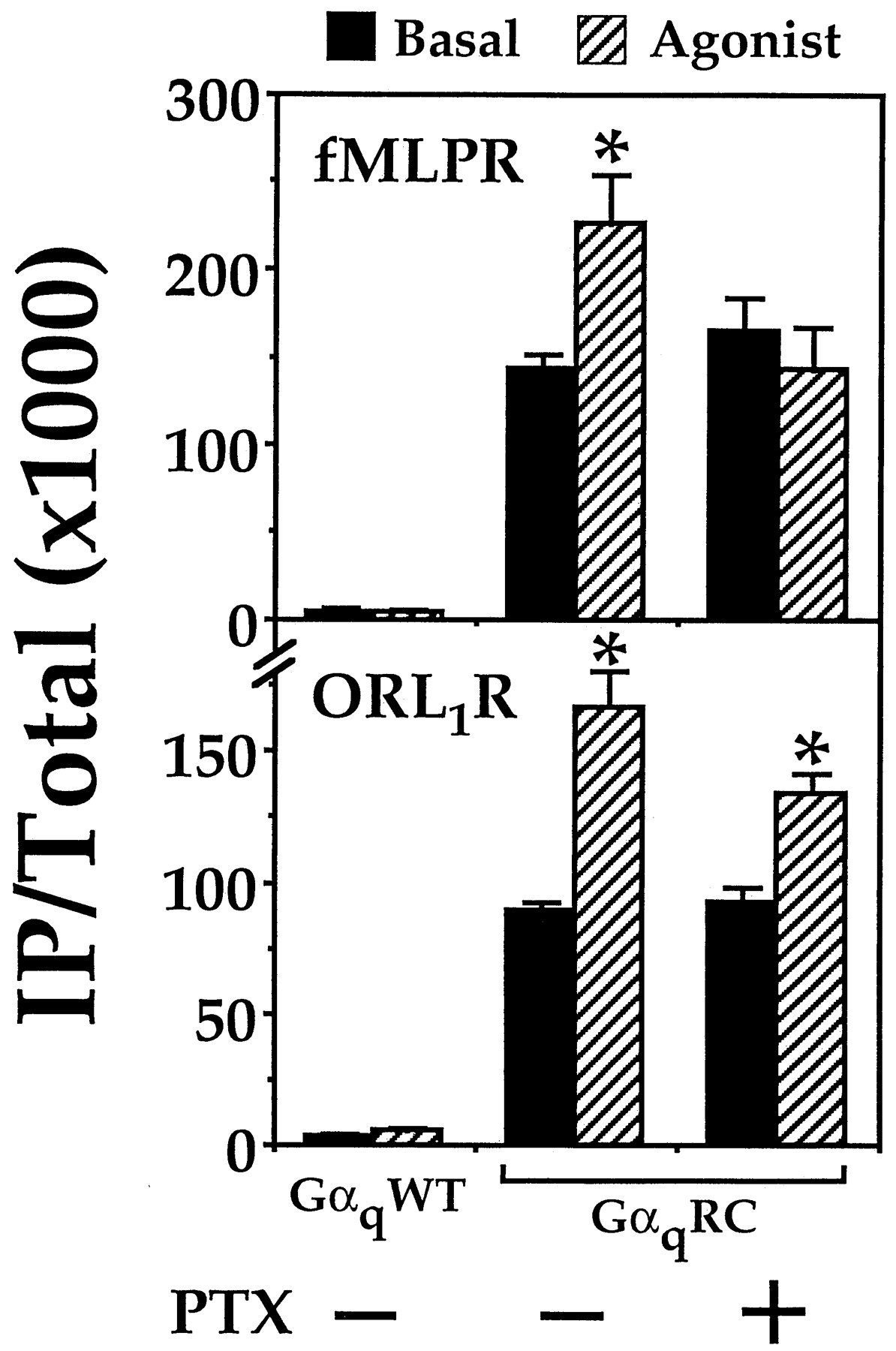
To extend our findings beyond DOR, we assayed for Gi-mediated stimulation of PLCβ by two other receptors in the presence of GαqRC. The fMLP and ORL1 receptors are incapable of coupling to Gαq (Tsu et al., 1995a; Yung et al., 1999). Each receptor was coexpressed with either Gαqor GαqRC in COS-7 cells and then assayed for agonist-induced stimulation of IP formation. In cells coexpressing the fMLP receptor and GαqRC, 200 nM fMLP stimulated the production of IP in a PTX-sensitive manner (Fig.7). Replacement of GαqRC with the wild-type Gαq effectively abolished the fMLP-induced stimulation of IP accumulation. The presence of GαqRC also was required for the ORL1 receptor-mediated stimulation of PLCβ (Fig. 7). Interestingly, the stimulatory response induced by 100 nM nociceptin was insensitive to PTX treatment (Fig. 7). This result indicated that the ORL1 receptor might use PTX-insensitive G proteins to release βγ-dimers and mediate the GαqRC-dependent stimulation of PLCβ. Indeed, there is indirect evidence to implicate the association of the ORL1 receptor to the PTX-insensitive Gα12 (Yung et al., 1999), which is present in COS-7 cells. It also should be noted that the apparent insensitivity of the nociceptin response to PTX treatment was partly due to the high basal PLCβ-activity induced by GαqRC. Irrespective of their PTX sensitivity, the GαqRC-dependent stimulation of PLCβ was not limited to DOR, and could be extended to include other Gi-linked receptors.
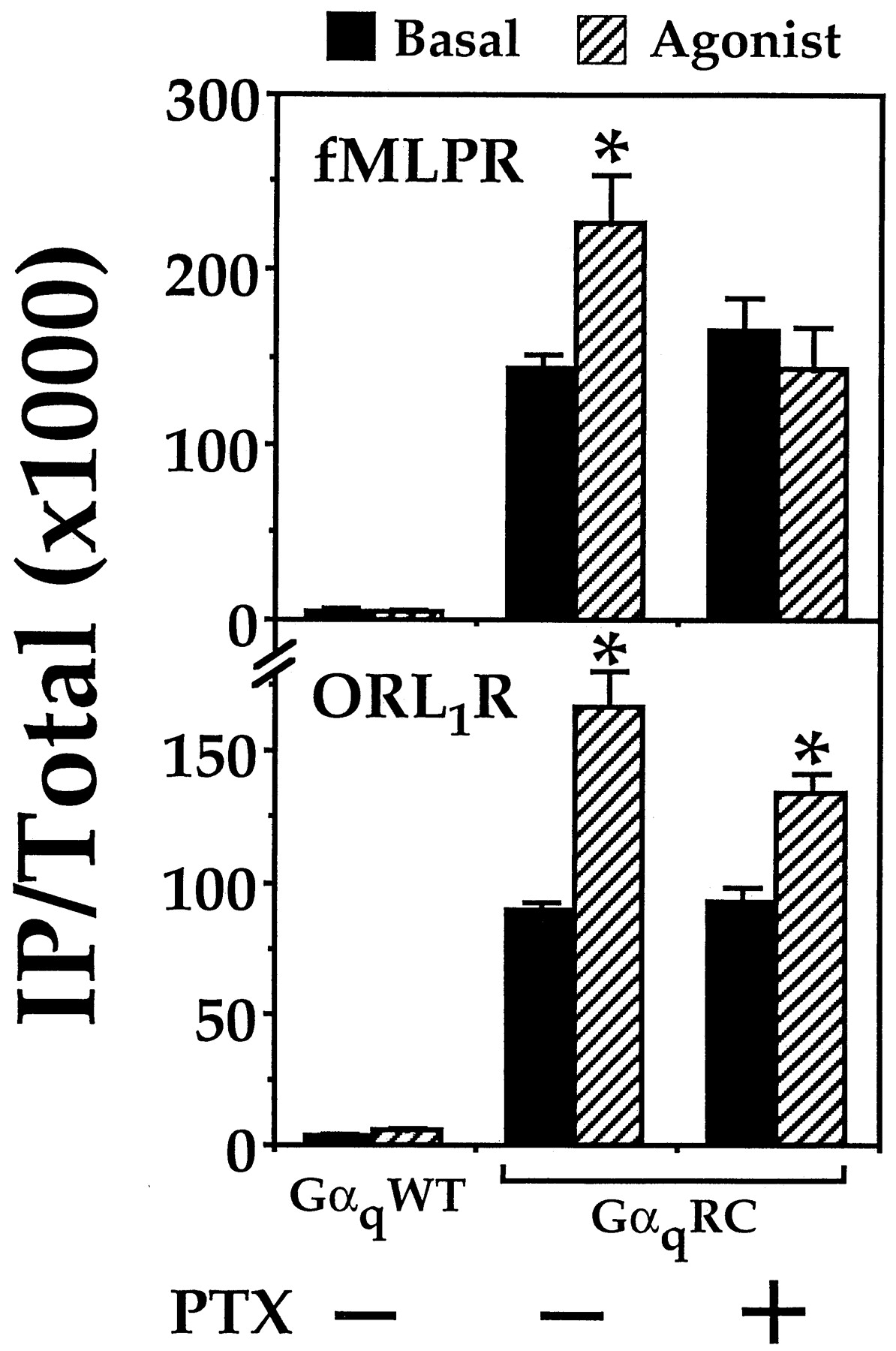
Permissive activation of PLCβ by other Gi -coupled receptors in the presence of GαqRC. COS-7 cells were cotransfected either with GαqWT (0.25 μg/ml) or GαqRC (0.1 μg/ml) and either the fMLP or ORL1 receptor cDNAs (both at 0.25 μg/ml). After transfection, the cells were labeled in the absence or presence of 100 ng/ml PTX as indicated. IP accumulation was assayed in the absence (basal) or presence of agonist (200 nM fMLP or 100 nM nociceptin). ∗, agonist-induced responses were significantly higher than the corresponding basal values; n = 3, Bonferroni t test, P < .05.
Synergism between Gi- and Gq-Linked Receptors on PLCβ-Activity.
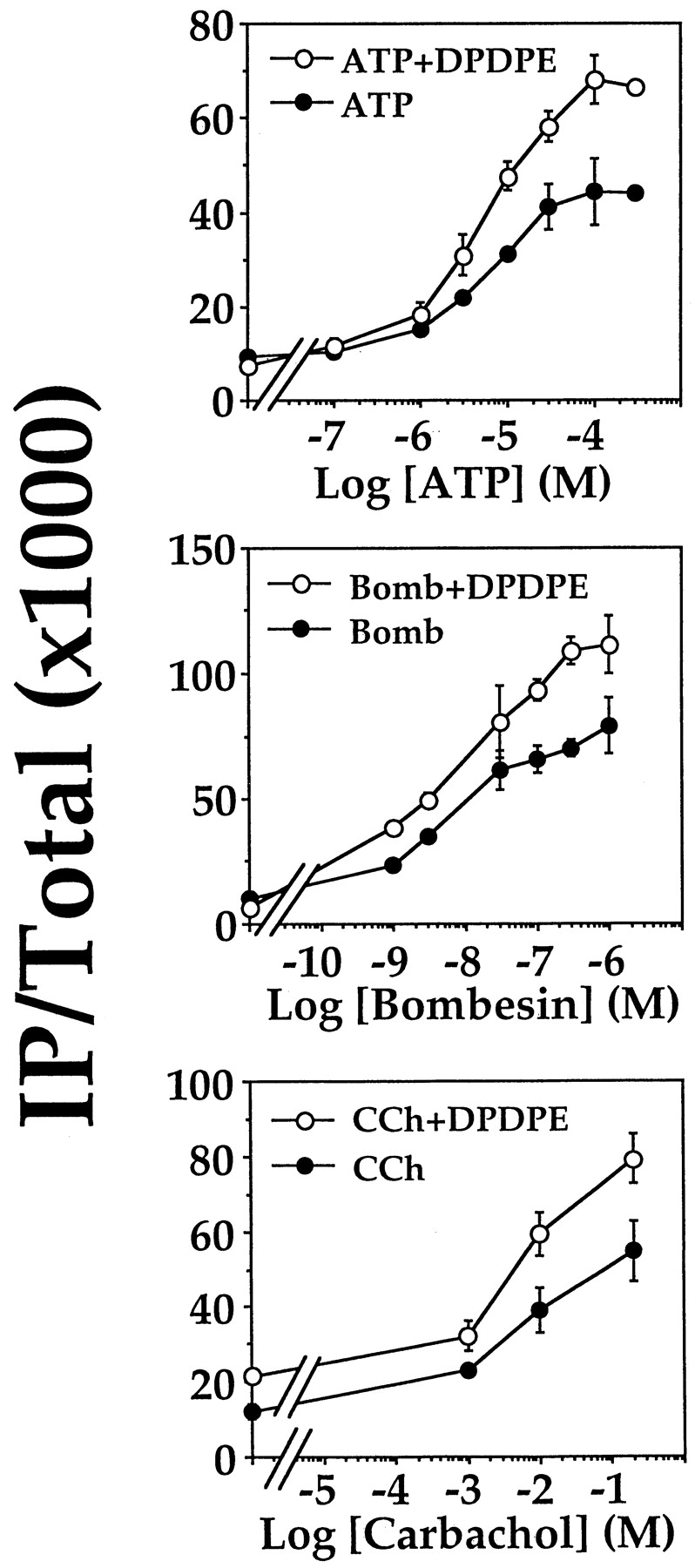
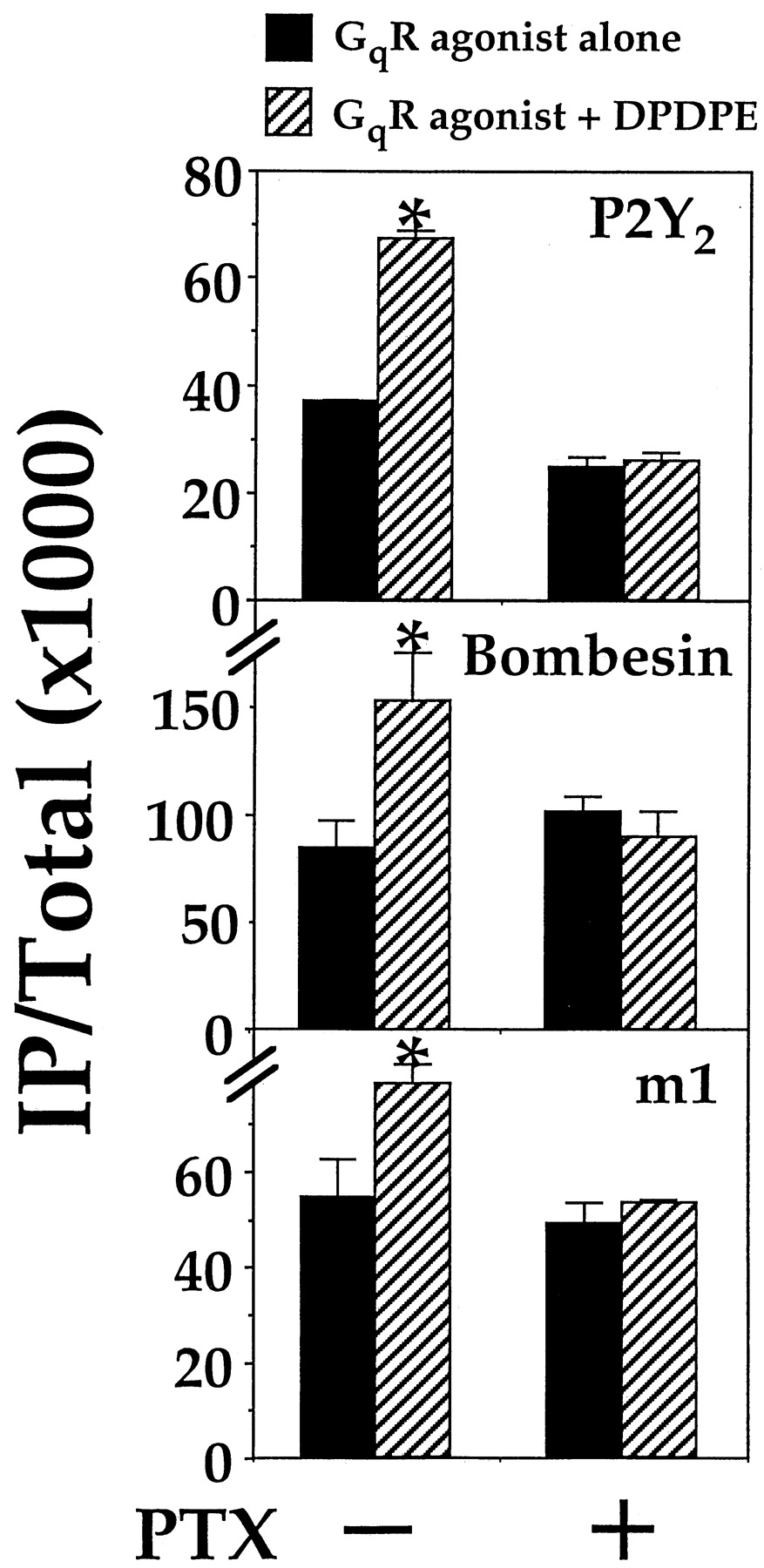
Given that Gi-linked receptors can stimulate PLCβ-activity in the presence of GαqRC, they should be able to enhance signals derived from the activation of Gq-linked receptors. We tested this hypothesis by substituting GαqRC with either an endogenously or recombinantly expressed Gq-linked receptor. COS-7 cells were found to endogenously express a purinergic P2Y receptor. Over a concentration range from 0.1 to 300 μM, ATP dose-dependently stimulated the formation of IP (data not shown but they were similar to those presented in Fig.8). Preliminary characterization with selective antagonists indicated that this purinergic receptor belongs to the P2Y2 class (North and Barnard, 1997); suramin hexasodium blocked the ATP-induced stimulation of IP formation, whereas pyridoxal-phosphate-6-azophenyl-2′,4′-disulphonic acid tetrasodium was ineffective (data not shown). The P2Y2 receptor is known to stimulate PLCβ via Gq/11 proteins and is expressed in the kidney (Lustig et al., 1993b), which is the tissue origin of COS-7 cells. In COS-7 cells transiently expressing DOR, DPDPE alone did not stimulate IP formation (Lee et al., 1998), whereas ATP dose-dependently elevated the IP levels by ∼4-fold with an EC50 of ∼6 μM (Fig. 8). When both δ-opioid and P2Y2receptors were simultaneously activated, the resultant IP accumulation was significantly greater than that obtained by activating P2Y2 receptors alone (Fig. 8). Addition of 100 nM DPDPE to the ATP dose-response curve raised the maximal stimulation by ∼65% with no change on the EC50 value (∼5 μM; Fig. 8). The DPDPE-induced enhancement of the P2Y2 receptor-mediated stimulation of PLCβ was completely inhibited by PTX (Fig. 9).

Synergistic activation of PLCβ by Gq - and Gi-coupled receptors. COS-7 cells were transfected with DOR alone (top) or together with either the bombesin receptor or muscarinic m1 receptor (all cDNAs at 0.25 μg/ml). After transfection, the cells were labeled overnight and assayed for IP accumulation with varying concentrations of ATP (0–300 μM; the agonist for endogenous P2Y2 receptors), bombesin (0–1 μM), or carbachol (0–200 μM; the m1 receptor agonist), with or without 100 nM DPDPE. Agonist stimulation of PLC with or without DPDPE was in all cases dose-dependent. DPDPE increased the maximal response of Gq-coupled receptor agonists alone.

The synergistic effect produced by DPDPE is abolished by PTX. COS-7 cells were transfected as in Fig. 8 but treated with or without 100 ng/ml PTX as indicated. IP production was assayed with 100 μM ATP, 100 nM bombesin, or 200 μM carbachol with or without 100 nM DPDPE. ∗, DPDPE significantly enhanced IP production;n = 3, Bonferroni t test,P < .05. The DPDPE induced increase in IP accumulation was in all cases abolished by PTX.
The observed synergism between the P2Y2 and DOR suggest that DOR also may synergise with other Gq-linked receptors. Thus, we cotransfected COS-7 cells with cDNAs encoding DOR and the Gq-linked bombesin receptor. Bombesin alone stimulated IP formation in a dose-dependent manner with an EC50 of ∼1 nM (Fig. 8). Similar to the endogenous P2Y2 system, inclusion of 100 nM DPDPE elevated the maximal bombesin response by ∼50% but it did not alter the EC50 value (∼1 nM; Fig. 8). Replacement of the bombesin receptor with another Gq-linked receptor, the m1 muscarinic receptor, produced similar results. DPDPE enhanced the carbachol-induced stimulation of IP formation (Fig. 8). In both cases, the DPDPE-induced enhancement was totally abolished by PTX (Fig. 9). Collectively, these results show that simultaneous activation of Gq- and Gi-linked receptors can produce a more efficient stimulatory control on PLCβ.
Discussion
The complex network of signal transduction pathways regulated by GPCRs must possess critical loci for signal integration and processing. Synergistic cross talk interactions between Gi/Gs- and Gq-coupled receptors may provide a mechanism for the fine-tuning of signals generated from GPCRs. For example, activation of the Gi-linked adenosine A1 receptor augments the stimulation of PLCβ evoked by Gq-linked receptors such as α1-adrenergic, bradykinin, histamine H1, and muscarinic receptors (for review, seeSelbie and Hill, 1998). Often, stimulation of Gi-coupled receptors alone has no effect, but augments Gq-mediated responses when both are stimulated concurrently. The present study provides a mechanistic basis for synergistic cross talk between Gq- and Gi-coupled receptors because preactivation of PLCβ by the α-subunit of Gq subfamily members apparently allows βγ-dimers to further stimulate PLCβ.
Several observations support our notion that preactivation of PLCβ permits subsequent stimulation by Gi-coupled receptors. First, DOR could not stimulate PLCβ in COS-7 cells unless activated Gα-subunits of the Gq subfamily are available. Active Gα-subunits can be provided in the form of constitutively active mutants (GαqRC, Gα16QL, and Gα14RC) or generated through receptor coupling to the promiscuous G16. Second, the constitutively active Gαs-mutant did not permit DPDPE to stimulate PLCβ. Third, in the presence of a mutationally activated α-subunit of the Gq subfamily, DPDPE-induced enhancement of IP production occurs in a dose-dependent and PTX-sensitive manner. Fourth, concurrent activation of Gq- and Gi-linked receptors resulted in significantly higher PLCβ activities compared with stimulating a Gq-linked receptor alone. And fifth, the G16-dependent stimulation of PLCβ by DOR was partially sensitive to PTX, suggesting dual mechanisms of activation of PLCβ. Collectively, these results indicate that PLCβ can integrate coincident signals in much the same way as the type 2 adenylyl cyclase, where prior activation by one type of signal allows subsequent detection of other signals.
The mechanism by which Gi-linked receptors synergise with Gαq-mediated stimulation of PLCβ appears to involve the βγ-dimer. It is well established that the βγ-dimer can directly activate PLCβ1- to β3-isozymes and the sensitivity of PLCβ-isozymes to βγ-subunits decreases in the order PLCβ3 > β2 > β1 (for review, see Rhee and Bae, 1997). In the present study, there are strong indications for the involvement of βγ-subunits in mediating the synergistic effects of Gi- and Gq-coupled receptors on PLCβ. The synergistic effect can be potently inhibited by Gαt1, a known scavenger of βγ-subunits. Moreover, the EC50 for DPDPE to stimulate PLCβ in the presence of activated Gαq-subunits (10–50 nM; Fig. 6) is much higher than that required for its inhibitory effect on adenylyl cyclase (∼1 nM; Tsu et al., 1995b). This is in agreement with the concept that the EC50 for a βγ-mediated response is considerably higher than responses mediated through the corresponding α-subunits (Iñiguez-Lluhi et al., 1993). Likewise, adenosine A1 receptor-mediated potentiation of PLCβ activity in cultured astrocytes requires higher concentrations of agonist than for adenylyl cyclase inhibition (Biber et al., 1997).
It is not clear by which mechanism preactivation of PLCβ allows subsequent stimulation by βγ-subunits. Activation of PLCβ by Gαq requires the C-terminal extension unique to the β-isozymes (Park et al., 1993; Wu et al., 1993), whereas βγ interacts with the pleckstrin homology and EF-hand domains (Kuang et al., 1996). Reconstitution experiments have demonstrated that the effects of Gαq and βγ on PLCβ3 are additive (Smrcka and Sternweis, 1993). The present study provides evidence that, in intact cells, regulation of PLCβ by activated Gαq may modulate its responsiveness to βγ-dimers. Structural information on the PLCβ-isozymes in the future will hopefully resolve how the binding of Gαq might modulate the βγ-binding site.
Unlike the type 2 adenylyl cyclase, phosphorylation of PLCβ by protein kinase C does not permit subsequent stimulation by βγ-dimers. Although avian PLCβ is phosphorylated by protein kinase C in vivo, it is accompanied by a concomitant loss of enzyme activity (Filtz et al., 1999). Because the role of GαqRC in permitting DOR to stimulate PLCβ could not be substituted by phorbol ester treatment, and that the response was not suppressed by staurosporin, the Gi-mediated enhancement did not seem to require the activation of protein kinase C. However, these studies do not exclude the possibility that protein kinase C can regulate long-term potentiation of PLCβ-activity (Schmidt et al., 1998) or suppress receptor-induced stimulation of PLCβ mediated via Gα16 (Aragay and Quick, 1999).
It is well documented that in many cells and tissues, Gi- and Gs-coupled receptors rarely stimulate PLCβ on their own. COS-7 cells endogenously express the PLCβ1 and PLCβ3, but not PLCβ2 (Katz et al., 1992). Both β1 and β3 isoforms can be efficiently stimulated by Gαq, but only PLCβ3 can exhibit augmentation by βγ-dimers (Smrcka and Sternweis, 1993). With a 10-fold greater potency, only Gαq-mediated signals can efficiently stimulate PLCβ in COS-7 cells. If preactivation facilitates the βγ-mediated stimulation of PLCβ, then those Gi-coupled receptors that possess a weak ability to activate Gq will be able to induce a PLCβ-response. An example of such an occurrence is the α2-adrenergic receptor (Conklin et al., 1992). Efficient stimulation of PLCβ2 by Gi-linked receptors in some cell types (e.g., HL-60; Camps et al., 1992; Katz et al., 1992) suggests that this isoform probably does not require preactivation for βγ-mediated stimulation. Supportive evidence from fluorescence spectroscopy indicates that βγ-dimers bind to PLCβ2 more tightly than to β1 or β3 (Runnels and Scarlata, 1999). Moreover, it has been shown that recombinant Gα16 and Gαq do not change the sensitivity of PLCβ2 to stimulation by βγ-dimers (Kozasa et al., 1993). Whether preactivation-dependent, βγ-mediated stimulation of PLCβ is generally applicable to β1–3 isoforms, or if these isozymes are indeed differentially regulated, would require further studies.
The need of preactivation for βγ-dimers to efficiently stimulate PLCβ in intact cells has major mechanistic implications on signal processing via the PLCβ-pathway. In the absence of stimulation by Gαq, PLCβ (perhaps except the β2 subtype) is relatively nonresponsive to free βγ-dimers, hence activation of Gi-coupled receptors will only lead to the regulation of adenylyl cyclase (Fig.10A). This might explain why many Gi-coupled receptors require the coexpression of PLCβ2 in COS-7 cells to manifest a βγ-mediated stimulation of IP formation (Lee et al., 1993). Activation of a Gq-coupled receptor will generate two signaling components, Gαq- and the βγ-dimer, that exhibit differential abilities to stimulate PLCβ (Fig. 10B). Costimulation of Gi- and Gq-linked receptors will produce a stronger stimulation of PLCβ because the βγ-subunits released from Gi activation can now augment the Gq-derived signal (Fig. 10C). The Gi-linked adenosine A1receptor can certainly augment IP signals generated from a variety of Gq-linked receptors (Selbie and Hill, 1998). The present study shows that DOR can augment the PLCβ-activities evoked by purinoceptor, muscarinic, or bombesin receptors. In the central nervous system, cholecystokinin has been reported to enhance the analgesic potentials of opioid peptides (Noble et al., 1993). Because the cholecystokinin receptors are typically coupled to Gq, and opioid receptors are associated with Gi proteins, it is conceivable that PLCβ may act as the point of signal convergence in neurons where both receptors are colocalized. Last, a single receptor that can activate both Gi and G16 should be able to stimulate the PLCβ-activity efficiently (Fig. 10D). Because part of the signal is derived from Gi, the overall response should be partially sensitive to PTX treatment. Examples of such observations can be readily found. Gα16-dependent signaling by the P2Y1 purinoceptor (Baltensperger and Porzig, 1997) and leukotriene B4 receptor (Gaudreau et al., 1998) are indeed partially sensitive to PTX. These results are in good agreement with our findings on the PTX sensitivity of the G16-mediated stimulation of PLCβ by DOR. Although G16 is not expressed in the central nervous system, it colocalizes with neuropeptide receptors, such as the opioid receptors, in a number of hematopoietic cells. The mechanism depicted in Fig. 10D may in fact be applicable to neuropeptides involved in the modulation of immune and endocrine responses.

Mechanistic models in which Gi -coupled receptors can activate PLCβ in the absence or presence of costimulation by Gq-coupled receptors. Four models (A-D) for the activation of PLCβ by Gi-released βγ-subunits are depicted. For simplicity, the adenylyl cyclase (AC) isoforms are assumed to be nonresponsive to βγ-dimers. H, hormone; Ri, Gi-coupled receptor; Rq, Gq-coupled receptor. Arrows indicate activation of signaling pathways. Inactive PLCβ is shown as a rectangle, whereas activated isoforms are illustrated as ovals.
In conclusion, this study provides evidence that augmentation of Gq-stimulated PLCβ-activity by Gi-linked receptors requires preactivation. The proposed mechanism resembles the one used by type 2 adenylyl cyclase. Signal integration by cells or neurons is a complex and delicate process that often requires fine-tuning to discern an array of incoming signals. Temporal summation of various signals allows a cell to reinforce critical inputs and perhaps to establish itself as part of a distinct neural circuit. We envisage that many Gi- and Gs-coupled receptors when costimulated with Gq-coupled receptors can produce synergistic actions on PLCβ in neurons and other target cells.
Acknowledgments
We thank Drs. J. Battey, G. Bell, F. Boulay, H. Bourne, C. Evans, R. Lefkowitz, T. Nukada, and M. Simon for generously providing the various receptor and G protein constructs used in this study.
Footnotes
- Received July 23, 1999.
- Accepted December 20, 1999.
-
Send reprint requests to: Yung H. Wong, Department of Biology and the Biotechnology Research Institute, Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China. E-mail: boyung{at}ust.hk
-
This work was supported by the Research Grants Council of Hong Kong (HKUST 653/96M and HKUST 6096/98M) and the Hong Kong Jockey Club.
Abbreviations
- GPCR
- G protein-coupled receptor
- PLCβ
- phospholipase Cβ
- IP
- inositol phosphate
- PTX
- pertussis toxin
- fMLP
- formyl peptide
- PCR
- polymerase chain reaction
- DMEM
- Dulbecco's modified Eagle's medium
- DOR
- δ-opioid receptor
- DPDPE
- [d-Pen2,5]enkephalin
- ORL
- opioid receptor-like
- KOR
- κ-opioid receptor
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}