


Abstract
The binding free energy for the interaction between serines 204 and 207 of the fifth transmembrane helix of the β2-adrenergic receptor (β2-AR) and catecholic hydroxyl (OH) groups of adrenergic agonists was analyzed using double mutant cycles. Binding affinities for catecholic and noncatecholic agonists were measured in wild-type and mutant receptors, carrying alanine replacement of the two serines (S204A, S207A β2-AR), a constitutive activating mutation, or both. The free energy coupling between the losses of binding energy attributable to OH deletion from the ligand and from the receptor indicates a strong interaction (nonadditivity) as expected for a direct binding between the two sets of groups. However, we also measured a significant interaction between the deletion of OH groups from the receptor and the constitutive activating mutation. This suggests that a fraction of the decrease in agonist affinity caused by serine mutagenesis may involve a shift in the conformational equilibrium of the receptor toward the inactive state. Direct measurements using a transient transfection assay confirm this prediction. The constitutive activity of the (S204A, S207A) β2-AR mutant is 50 to 60% lower than that of the wild-type β2-AR. We conclude that S204 and S207 do not only provide a docking site for the agonist, but also control the equilibrium of the receptor between active (R*) and inactive (R) forms.
The phenomenon of agonist-mediated activation of a G protein-coupled receptor (i.e., seven transmembrane domain, or 7TM, receptors) can be described conveniently through the formalism of a two-state allosteric transition. The receptor exists in equilibrium between the active and inactive conformations, R and R*. In the absence of a bound agonist, constraints (that are structural in nature, perhaps) maintain the equilibrium shifted toward the inactive form, thus, little or no ligand-independent signaling occurs. Agonist binding, but also, remarkably, some mutations that cause constitutive activity (Kjelsberg et al., 1992; Parma et al., 1993; Samama et al., 1993; Shenker et al., 1993; Scheer et al., 1996, 1997), remove such constraints and make the active receptor form R*predominant. Consequently, the free energy change that we measure for the agonist-binding process (e.g., as equilibrium affinity) must reflect at least two contributions. One is the total sum of energies related to the intermolecular forces that hold ligands and receptors together. The other results from the perturbation of intramolecular forces that the bound receptor endures in the transition to the bioactive form. Both make up experimentally measured ligand binding constants. Mutations that alter ligand affinity, therefore, can do so by changing either component, or both. How to dissect or measure each contribution?
We sought to present this question to the case of β2-adrenergic receptors (β2-AR), where a number of sites that are crucial for agonist binding were identified early in a series of ingenious mutagenesis studies (Strader et al., 1987, 1988, 1989). One fundamental interaction in the binding of catecholamine agonists (Strader et al., 1989) is considered to be hydrogen bonding between catecholic hydroxyl groups and two serine residues (S204 and S207 for human β2-AR) located in the fifth putative transmembrane domain (TM5). The position of such residues, especially that of S207, is highly conserved among all members of the catecholamine receptor family, and their modification by site-directed mutagenesis decreases agonist-, but not antagonist-, binding affinity in all types of catecholamine receptors (Wang et al., 1991; Link et al., 1992; Cavalli et al., 1996; Hwa and Perez, 1996).
The magnitude of binding energy that is lost to elimination of each serine residue was consistent with what may be expected for the deletion of one hydrogen bond (Strader et al., 1989). However, the modification of S204 and S207 in β-adrenergic receptors also causes impairment of agonist-mediated signal transduction (Strader et al., 1989), suggesting that the interaction between the catechol ring and TM5 is involved in the process of coupling ligand recognition to the emergence of the bioactive conformation in the receptor. Therefore, part of that lost binding energy may also reflect the reduced tendency of the receptor to interconvert into R* form.
In this study, we exploit the principle of energy conservation among simultaneous perturbations applied to a macromolecule binding process (Horovitz, 1987; Horovitz and Fersht, 1990; Hidalgo and MacKinnon, 1995; Faiman and Horovitz, 1996) to analyze the loss of agonist binding energy that follows the double deletion of S204 and S207 in the human β2-AR.
Using this approach, we estimate that a fraction of the mutation-induced change of binding energy that follows the removal of hydroxyl groups is attributable to a shift of the intramolecular equilibrium of the receptor toward the inactive state. Consistent with such measurements, we also find that the mutant receptor displays a significant reduction of ligand-independent activity compared with the wild type, indicating that serine deletion can cause some degree of constitutive inactivation of the receptor.
These data demonstrate that a contact region that is involved in agonist-induced activation of the receptor also controls the equilibrium between active and inactive receptor forms.
Materials and Methods
Receptor Mutagenesis.
The cDNA encoding the human β2-AR subcloned in pTZ (Pharmacia, Uppsala, Sweden) was mutated by polymerase chain reaction, using TaqDNA polymerase. Recombinant clones were isolated and sequenced. The mutated DNA fragment was digested with NcoI andBglII and cloned into the expression vector pBC12BI containing the cDNA encoding the wild-type β2-AR or its constitutively active mutant (cam) (Samama et al., 1993).
Cell Culture and Transfections.
COS-7 cells were grown in Dulbecco's modified Eagle's medium (high glucose) supplemented with 10% fetal calf serum, 100 U/ml penicillin G, and 100 μg/ml streptomycin sulfate, in a humidified atmosphere of 5% CO2 at 37°C. Cells were plated in 80- or 25-cm2 flasks and transfected transiently with plasmids (pBC12BI or pcDNA3) harboring either wild-type or mutant receptor cDNA using the DEAE-dextrane/chloroquine procedure (Cotecchia et al., 1990). Gradual levels of receptor expression were obtained by transfecting varying amounts of receptor cDNA with the total mass of input DNA (0.2 μg/cm2/0.1 ml) maintained constant through the addition of empty vector. Cells were harvested 48 h after transfection for the binding assay or plated 24 h after transfection in 24-well plates, and then grown for an additional 24 h before the determination of cAMP levels. For the generation of stably expressing clonal lines, Chinese hamster ovary (CHO) cells were grown in a 1:1 mixture of Dulbecco's modified Eagle's medium and Ham's F-12 medium. Cells were transfected using Lipofectin (Life Technologies, Paisley, Scotland) according to the manufacturer's instructions. Clones (30–40 for each transfected plasmid) resistant to Geneticin (400 μg/ml of active drug; Life Technologies) were isolated and tested for their ability to bind125I- pindolol (NEN Life Science Products, Boston, MA).
cAMP Determination in Intact Cells.
For determination of basal levels of cAMP, COS-7 cells were seeded in 24-well plates. After aspiration of the medium, the cells were incubated in a buffer containing: 135 mM NaCl, 2.7 mM KCl, 1.5 mM KH2PO4, 20 mM NaHEPES, 2 mM CaCl2, 1.2 mM MgSO4 , 1 mM EGTA, 11.1 mM d-glucose, 0.01 mM Rolipram, and 0.05% BSA, pH 7.4. Incubations lasted 20 min at 37°C and were arrested by the removal of the buffer and the addition of 0.5 ml of ice-cold 0.1 N HCl to each well. Plates were placed on ice, and an aliquot of the HCl extract was removed for the determination of cAMP concentration using radioimmunoassay, as described (Vachon et al., 1987).
Adenylate Cyclase Assays.
Membranes from frozen transfected cells were prepared as described previously (Vachon et al., 1987) and stored at −80°C (protein concentration 1–2 mg/ml) until used. The adenylyl cyclase reaction mix included 50 mM Tris/HEPES, 10 mM MgSO4, 0.5 mM ATP, 100 μM GTP, 5 mM phosphocreatine, 25 mM creatine phosphokinase, 150 mM NaCl (pH 7.5), and 10 μM Rolipram, in a final volume of 100 μl. Reactions were started by the addition of the membrane suspensions (2–5 μg of protein) and arrested after 10 min at 37°C by the addition of 0.1 ml of ice-cold 0.2 M HCl. The determination of cAMP formed was performed using radioimmunoassay.
Binding Assay in Membrane Preparations and Intact Cells.
The binding of 125I-pindolol was measured in 1 ml of 50 mM Tris-HCl and 0.1 mM EGTA (pH 7.4) for 90 min at room temperature using 0.1–10 μg of membrane proteins. The concentration of radiotracer was maintained constant at 10 pM in the presence of increasing concentration of unlabeled ligands. Reactions were terminated by rapid filtration onto GF/B glass fiber filtering microplates (Filtermate 196; Packard Instruments, Meriden, CT). Filters were washed three times in 1 ml of ice-cold 50 mM Tris-HCl pH 7.4 and allowed to dry for a few hours. The plates were counted in a Top Count (Packard Instruments) after the addition (25 μl) of Microscint 20 (Packard) to each well. To measure binding in intact cells, transfected COS-7 cells were seeded 24 h after transfection in opaque culture plates (Packard Instruments) and incubated the next day in a reaction buffer with a composition identical with that used for determination of cAMP in intact cells. The reaction mixture (1 ml) contained either 125I-pindolol (20 pM) or [3H]CGP 12177 (0.5 nM; NEN Life Science Products) and increasing concentration of cold ligands. The incubation lasted 90 min at 4°C and was terminated by rapid aspiration of the incubation buffer, followed by washing the monolayer twice in ice-cold PBS. After draining plates overnight onto filter paper, 250 μl of Microscint 20 was added to each well and the plates were counted in a Top Count.
Data Analysis and Calculations.
Equilibrium dissociation (K d) and association (K eq = 1/K d) constants were calculated by nonlinear fitting of the binding curves to a four-parameter logistic equation using the program ALLFIT (DeLean et al., 1978). When required, the binding affinity was also estimated with the computer program LIGAND (Munson and Rodbard, 1980). Free energy changes, except when indicated otherwise, are given in RT units (R, gas constant, T, absolute temperature) i.e.: ΔG = −ln(1/K d). Energy calculations (i.e., differences from wild type and sums of single mutations) were computed experiment by experiment before taking their averages, thus their variances reflect mostly true experimental variance and not accumulating errors that propagate when multiple calculations are applied to averaged affinity values. Similarly, statistics for ΔΔG and free energy coupling values (δG) were computed for the repeated measurements after converting data into free energy in each experiment. The variation of free energy attributable to mutation (ΔΔG) is the difference in binding energy between mutant and wild-type receptor, i.e., ΔΔG = ΔG(mut) − ΔG(wt). For a double-mutant cycle consisting of single mutations 1 and 2, and the double mutation (1,2), the free energy coupling can be computed as: δG 1,2 = ΔG(1,2) + ΔG(wt) − [ΔG(1) + ΔG(2)]. The theoretical background for these calculations is given below.
Theoretical Background.
First we recall the meaning of apparent binding energy as derived from an experimentally measured equilibrium binding constant in an allosteric protein. Next, we will examine its implications in the analysis of multiple mutations cycles.
Apparent Binding Energy for an Allosteric Receptor.
Let us assume that a receptor (R) can exists in a large number (n) of interconvertible conformational states (Onaran and Costa, 1997). The transitions among all of them at equilibrium can be expressed with respect to an arbitrarily chosen reference state (s
0). The concentration of each individual state ([s
i]), (withi ≠ 0), is then given by a first-order equilibrium constant (j), such that j
i= [s
i]/[s
0]. Thus, [R] = [s
0](1 + ∑i=1
n j
i). If the receptor binds a ligand (H), the stability of each individual state in the bound receptor form will be perturbed by a factor (b), such that bi =[Hsi][s0]/[Hs0][si] . Hence, the concentration of bound receptor is [HR] = [Hs0](1 + ∑i=1
n bij
i), and the apparent second-order equilibrium affinity constant (Onaran and Costa, 1997) can be expressed as:
A mutation can change binding energy by affecting any one of the components or all. Therefore, an equivalent decrease of binding affinity caused by the deletion of a residue can result from three distinct, but indistinguishable, mechanisms: 1) the side chain of the mutated residue provides a docking site for the ligand and has little or no role on the conformational equilibria of the receptor(primary change of B); 2) conversely, the residue has no direct contact with the ligands but exerts a key role on intramolecular receptor motion before or after the binding of ligand (changes of Cb and/or Cf); or 3) a combinations of both mechanisms. It is also evident that there might be compensation between opposing effects when the two components are changed into inverse directions. Thus, even if mutagenesis of a residue produces minor or no change on binding affinity, we cannot rule out its role in the process of binding and activation of the receptor.
Free Energy Conservation for Alchemical Reaction Paths.
Whether additivity principles (i.e., the idea that several subconstituents of a system contribute linearly to its macroscopic behavior) can be used to dissect the components of free energy changes in proteins is a matter of heated theoretical debate (Boresch and Karplus, 1994; Mark and van Gusteren, 1994) and unresolved questions (Dill, 1997). However, the existence of free energy conservation among chemical (Weber, 1972, 1973) or alchemical (Horovitz and Fersht, 1990) perturbations applied to a macromolecule stands as a valid principle to analyze the presence or the lack of additivity in biomolecular interactions. The use of this strategy to study the effect of chemically or genetically engineered mutations was explained and discussed elsewhere (Horovitz, 1987; Horovitz and Fersht, 1990; Hidalgo and MacKinnon, 1995; Faiman and Horovitz, 1996). For convenience, we will summarize only briefly the key principles.
Consider a set of experimental measurements of the free energy of binding for a ligand-receptor interaction before and after either the single or the concurrent application of two nonidentical perturbations to the system. The effects on binding energy produced by the two perturbations obey the principle of free energy conservation and allow us to draw the following thermodynamic reaction cycle:
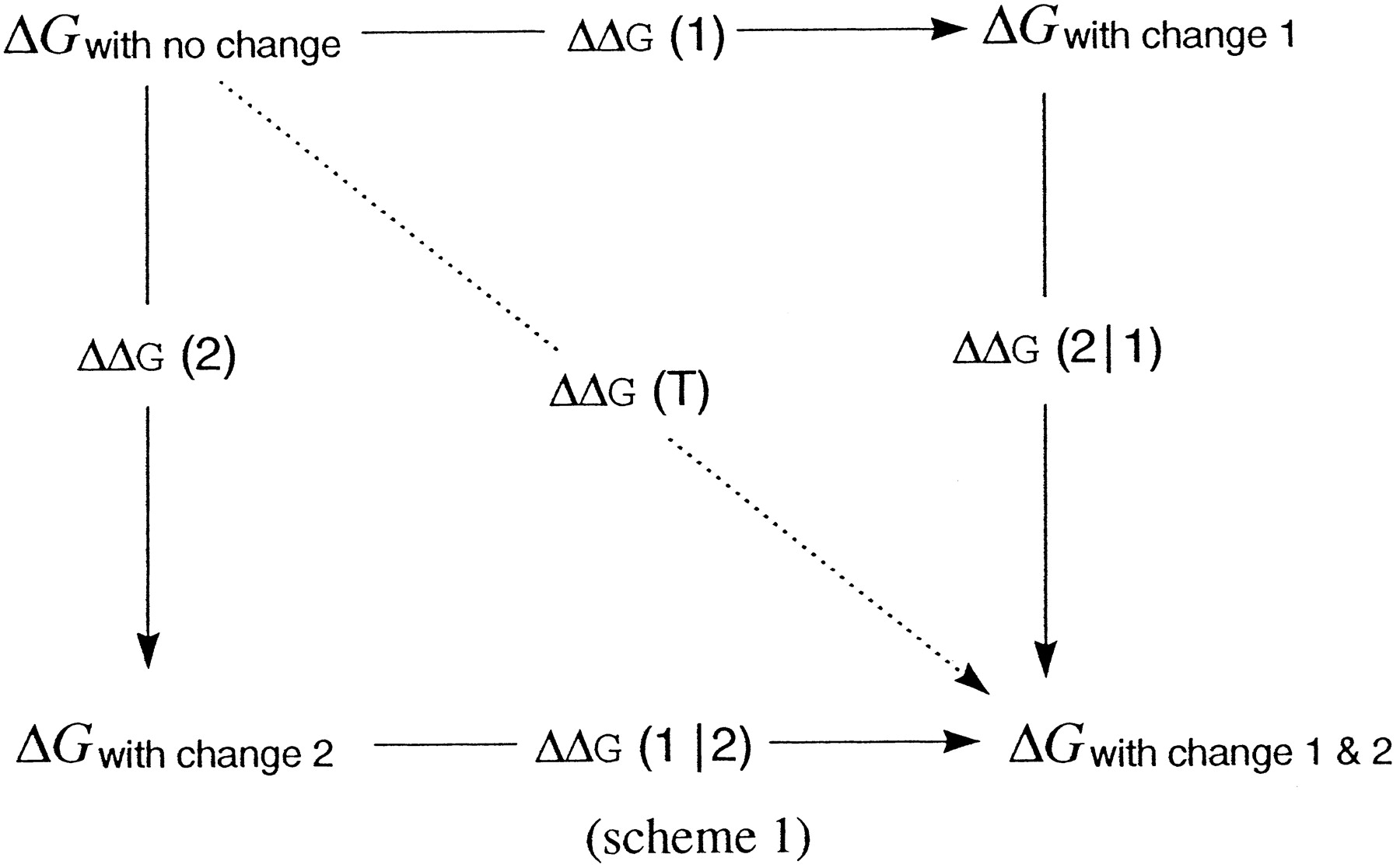
where ΔGs indicate free energy changes truly measured by experiment, and ΔΔGs depict alchemical free energy changes, because they are not measured directly, but inferred from the computed differences in ΔGs.
The overall free energy change for the transition from the unchanged state (in which no perturbation is yet present) to the final state (in which both have been applied) is path-independent. Thus:
Perturbation is meant in a general sense. It can be a covalent modification of either partners of a binding process, or the transfer to a different environment, or any sort of natural or unnatural mutations of the receptor. Similarly, general is the meaning of nonequivalence of the perturbations, because it is satisfied by either a diversity of location (e.g., an identical change applied to the ligand and the receptor or to distinct sites within the receptor) or a difference in the type of imparted change.
Free Energy Coupling between Alchemical Changes in an Allosteric Receptor.
We examined how this strategy can be used to analyze apparent free energy changes at allosteric receptors. Let's consider two specific examples that are relevant to the work described in this study.
Deletions of Chemically Complementary Groups from the Ligand and the Receptor.
A set of complementary chemical groups are deleted from both the ligand and the receptor. Even if the two deletions produce similar or identical losses of binding energy, we cannot conclude that the two groups interact with each other in the ligand-receptor complex. In fact, the ligand group may interact elsewhere in the receptor, whereas the receptor group could be engaged in a network of intramolecular interactions that are crucial to the binding process. In terms of eq. 2, the decrease of binding energy attributable to the ligand deletion results from a primary effect onB, whereas that attributable to the receptor deletion comes from conformational contributions. Thus, the losses of binding energy can have the same magnitude even if the deleted groups do not interact with one another.
In principle, the construction of a thermodynamic cycle as in Scheme 1 and the calculation of the magnitude of free energy coupling (δG1,2) may provide a way of discrimination. The logic of interpretation is simple. If the groups interact directly, removing any of the two from either ligand or receptor should not produce more effect than removing both. Therefore, ΔΔG(1‖2) ≈ ΔΔG(2‖1) ≈ 0, and ΔΔG(1) ≈ ΔΔG(2) ≈ − δG1,2. This means strong interaction and, therefore, a total lack of additivity between the effects of the two deletions. Conversely, if the interaction is mediated indirectly through conformation: ΔΔG(1) ≈ ΔΔG(1‖2), ΔΔG(2) ≈ ΔΔG(2‖1), and δG1,2≈ 0. This means total lack of interaction and perfect additivity of the effects.
In practice, however, such “clear-cut” results are unrealistic and not likely encountered frequently in proteins such as receptors, in which binding and conformational change are linked inextricably. The question then is how large or small should the magnitude of coupling be, to suggest, respectively, interaction or lack of interaction. To answer, we will examine how intermolecular and intramolecular contributions to apparent binding energy can be affected differentially by perturbations and what the expected output in the analysis via thermodynamic cycles may be. Let's call “docking” and “conformational” the two types of contributions in eq. 2, and set 1 and 2 the deletion from receptor or ligand, respectively. The alchemical energy differences caused by the mutations can be written in terms of probable docking (B′) or conformational (C′b and C′f) components that add up to the overall binding energy as a consequence of each perturbation.
Indirect Interactions.
A receptor mutation that does not hit a docking site can decrease binding energy by turning into a destabilizing contribution the intramolecular equilibria that control bound and free receptor forms. If so, the effect of perturbation 1 is: ΔΔG(1) = C′b + C′f. When the mutation in the ligand instead suppresses a contact group, both docking and conformational contributions that are attributable to that group are affected. Thus, ΔΔG(2) = B′ + C′′b. If the receptor change does not disturb the conformational contribution of the ligand change (i.e., C′b and C′′b are independent), the total difference is: ΔΔG(T) = B′ + C′b + C′f + C′′b, and δG1,2 ≈ 0, (eq. 5). The effects are truly additive. However, if the conformational change caused by the receptor mutation mimics or cancels that from the loss of the ligand bond, ligand and receptor mutations may share a conformational contribution. Thus: ΔΔG(1) = C′b + C′f + C′′b. The resulting δG1,2 will not be zero, but −C′′b, and the analysis reveals that the effects are not perfectly additive, but show a small degree of interaction. How small should δG1,2 be to decide that the effects are predominantly additive? Because it is likely that C′′b ≪ ΔΔG(1) and C′′b < ΔΔG(2), then the magnitude of δG1,2 should be smaller than either changes attributable to each individual perturbation. Therefore, we may state in general that if free energy coupling is close to zero or significantly smaller than the individual changes produced by each single perturbation, we should discard the hypothesis that the groups deleted from the receptor and from the ligand form a direct bond.
Direct Interaction.
When mutations in both ligand and receptor suppress groups that bind to one another, the perturbations will share an identical “loss” of docking contribution and most likely also a similar conformational contribution that each group has on binding energy. If so, ΔΔG(1) ≈ ΔΔG(2) = B′ + C′b; ΔΔG(1‖2) ≈ ΔΔG(2‖1) ≈ 0 and δG1,2 ≈ −(B′ + C′b), which means strong interaction and total lack of additivity.
Under such conditions, δG1,2 is a reliable measure of the overall strength of interaction between the groups. However, there may be situations in which the binding of mutated ligand to mutated receptor is different from the binding of either of the two mutated molecules to the unmodified partner. For example, let's imagine a case in which mutated ligand binds to mutated receptor slightly better than to the wild type, because the simultaneous deletion of the pair of interacting groups from both ligand and receptor favors somewhat all the interactions at the other points of contact between the two molecules. Calling this extrainteraction C′′b, ΔΔG(1‖2) ≈ ΔΔG(2‖1) ≈ C′′b, and δG1,2 = C′′b − (B′′ + C′′b). Thus, depending on the sign of C′′b, δG1,2may be smaller or greater than B′ + C′b. Therefore, we can say in general that in case of direct interaction, the size of free energy coupling is comparable (even if not necessarily identical) to ΔΔG(1) or ΔΔG(2), and it will approach the actual value of −(B′ + C′b) as closer ΔΔG(1‖2) and ΔΔG(2‖1) tend to zero.
Direct Interaction with Additive Components.
The situation is identical with that of case 2, but the mutation of the receptor also affects the intramolecular equilibria of the “vacant” protein. In this case, ΔΔG(1) = B′+ C′b + C′f; ΔΔG(2) = B′ + C′b; ΔΔG(T) = B′ + C′b + C′f; and δG1,2 = −(B′ + C′b), which is exactly the same result of case 2. In fact, the contribution of the additive component will cancel out and does not disturb the detection of the strong interaction caused by the removal of complementary groups. A significant difference between ΔΔG(1) and ΔΔG(2) is the sole, but very important, clue to suspect a role of the intramolecular equilibria of the empty receptor. In general, a strong interaction evidenced by δG1,2 ≪ 0, with ΔΔG(1) > ΔΔG(2) suggests that the mutation in the receptor not only suppress the docking point for the ligand, but also changes binding energy by affecting intramolecular equilibria in the receptor itself.
Mutations in Two Separated Domains of the Receptor.
The principle of analysis is identical with that detailed above, but the implications of presence of interaction or lack of interaction are quite different. If two mutations applied to distant sites of the same receptor affect the energy of binding for a ligand in a nonadditive manner, that means that the two mutated sites are coupled through propagated intramolecular interactions in regulating the reactivity of the receptor binding site for that ligand (Horovitz, 1987; Horowitz and Fersht, 1990). Using this strategy thus is possible to map conformational “sensitive” residues in the ligand binding site. For example, suppose we know that a residue of the receptor is located in the ligand binding pocket and that mutation of a second far-apart residue of the molecule (which we can assume is not accessible to the ligand) affects ligand affinity, presumably via a conformational change. Let's call 1 and 2 the mutations of the first and second residue, respectively. If, by constructing a double-mutant thermodynamic cycle, the effect of the two mutations are additive (not coupled), then the change of binding energy induced by mutating the binding residue 1 does not involve any conformational contribution common to that induced by the mutation of the second residue. On the contrary, if there is interaction (lack of additivity), it means that the two mutations share a common conformational mechanism in changing binding affinity.
If we place mutation 1 in the ligand binding pocket and mutation 2 in the G protein-binding area, this strategy could map which residues of the 7TM molecule are important for the transmission of conformational influences between the two binding domains.
Results
Lack of Additivity between Single and Double Substitutions of S204 and S207.
Strader et al. (1989) proposed that serines 204 and 207 in TM5 of β2-AR may act as hydrogen bond donors for the attraction of the two hydroxyl groups of the catechol moiety of adrenergic agonists. In their study (Strader et al., 1989), as well as others (Kikkawa et al., 1997, 1998), site-directed mutagenesis studies, single-substitutions of either S204 or S207 were examined. Each single replacement caused roughly similar decreases of agonist binding energy, which supports the idea that two hydrogen bonds may be formed betweenmeta and para hydroxyl groups of the ligand and the serine side chains of the receptor (Strader et al., 1989). It was not known, however, whether the effects of the two deletions were additive, as it may have been expected if the diminution of binding energy would primarily reflect the loss of stabilizing effect attributable to the individual bonds removed by each mutation.
To answer this question, we prepared alanine mutants of the human receptor where either each of the two serines (S204A β2-AR and S207A β2-AR) or both (S204A, S207A β2-AR) were substituted. Wild-type and mutant receptors were compared after transfection in COS-7 cells, by measuring the binding affinity of the agonist (−)isoproterenol and the antagonist (−)pindolol in competition isotherms for the sites labeled by 125I-pindolol. In agreement with previous findings, serine replacement in each site of TM5 produced similar diminution of agonist affinity (Table1). In contrast, no significant changes of pindolol binding affinity in comparison with the wild type were observed in either single- or double-mutated receptors (data not shown), indicating that the observed responses are agonist-specific in all cases. The 20- and 12-fold decrease in agonist binding affinity observed for S204A and S207A, respectively, correspond to a mutation-induced destabilization of +7.9 and +6.5 kJ/mol in the free energy change caused by isoproterenol binding. However, the double mutation S204A, S207A produced a decrease of affinity corresponding to 10.2 kJ/mol of binding energy. Thus, the removal of both hydroxyl side chains exerts a smaller effect than the sum of those attributable to each single deletion.
Comparison of agonist binding affinity for single and double mutations of serines 204 and 207 in TM5 of β2AR
Although the mutation of a residue into alanine most closely mimics the pure deletion of the side chain (Faiman and Horovitz, 1996), increasing the abundance of alanines in a transmembrane helix may also affect its orientation and overall conformation. To verify whether the nonadditive pattern may depend on the chemical nature of the chosen residues, we also prepared corresponding β2-AR mutants in which the same serines were replaced by cysteine. Serine and cysteine have side chains of equivalent length, but SH groups are poorer hydrogen bond donors or acceptors than hydroxyl ones. As shown in Table1, the results observed with such mutants perfectly overlap those obtained with alanine mutants, indicating that in either cases is the removal of hydroxyl groups responsible for the measured losses of binding energy, regardless of the residue used for replacement.
The lack of additivity shown here is not surprising, for hydrogen bonds cannot be removed without also affecting additional factors, such as basicity, dipole moment, repulsion and conformation (Perrin and Nielson, 1997). Thus, energetic contributions of individual hydrogen bonds deduced through deletion in proteins can only be taken as upper-limit estimates. We suspected, however, that the conformational contribution in this case should be worth of additional investigation, because it may be related to the agonist-induced transition of the receptor into active form. We elected the double mutant for further study, because only the elimination of both OH groups leads to an unambiguous conclusion that no hydrogen bond interactions are possible between the catechol ring and that site of TM5.
Thermodynamic Analysis for the Deletion of Both Hydroxyl Groups from Ligand and Receptor.
We first chose to determine the overall component of agonist binding energy that can be attributed to hydroxyl group interactions by computing the free energy couplings for concomitant mutations applied to each and both interacting partners of a ligand binding process (see “Theoretical Background” inMethods and Methods).
To this end, we prepared a number of transfected CHO cell lines stably expressing different levels of either wild-type (SS) or S204A, S207A mutated receptors (AA). In membranes obtained from such clones, we compared the binding affinities of a number of agonists, such as isoproterenol in both racemic and pure enantiomeric form, (±)-epinephrine, and its dehydroxylated analog (±)2-(methylamino)-1-phenyl-1-ethanol (mape). All measurements were performed in the presence and absence of GTP, to assess to which extent a possible interference of the G protein interaction could complicate the interpretation of the analysis.
As summarized in Table 2, the double serine deletion decreased the binding affinity of isoproterenol to an extent similar to that observed in COS-7 cells, for measurements made in the presence of GTP (70- ± 13-fold), or slightly greater in its absence (153- ± 18-fold). A similar pattern was observed for the affinities of epinephrine, although the shift was somewhat larger (186- ± 20-fold plus and 371- ± 45-fold minus GTP, respectively). In agreement with previous findings (Strader et al., 1989), the enantiomeric nature of the ligand had no influence on the shift of affinity induced by the mutation, as equivalent diminutions of binding affinity were observed for (+) and (−) enantiomers of isoproterenol or the racemic form (data not shown). The binding affinity of the noncatecholic agonist mape was affected very little by the AA mutation (2.2- ± 0.3-fold, ±GTP), which agrees with observations made using the isopropylamino analog of mape in hamster β2-AR (Strader et al., 1989).
Dissociation constant of adrenergic ligands for the binding to wild type and mutant adrenergic receptors
The free energy changes computed from the binding affinities of epinephrine and mape to wild-type and mutated receptors stand at the corners of a thermodynamic reaction cycle, as drawn in Table3. The differences (ΔΔG) among experimentally measured free energy differences represent the same “alchemical” reaction, i.e., removal of two hydroxyl groups, which is applied to the receptor (ΔΔG(1)), the ligand (ΔΔG(2)), or both (ΔΔG(1‖2) and ΔΔG(2‖1)). In fact, mape and epinephrine only differ for the presence of two hydroxyl groups (Table 3), just like the double alanine receptor differs from the wild type. The difference between parallel paths (free energy coupling) is a direct measure of the degree of interaction between the two perturbations. If δG 1,2 is null or very close to zero there is perfect additivity between the two effects, thus it is unlikely that the pairs of hydroxyl groups of ligand and receptor are involved in an interaction during the binding process. The greater the difference between δG 1,2 and zero, the more likely is the existence of a direct interaction.
Thermodynamic cycle for hydroxyl groups removal from ligand and receptor
We computed a free energy coupling between −4 (with GTP) and −5 (no GTP) RT units from the experimental data summarized in Table3, which demonstrates the existence of interaction between the hydroxyl groups of the agonist and the receptor. The negative sign and the size of the calculated δG 1,2 indicates strong “positive cooperativity” between the two perturbations, in the sense that the effect of abolishing hydroxyl groups from the receptor “facilitates” that of their removal from the ligand (and vice versa). Thus, there is almost no additivity between the two perturbations. In fact, if two sets of groups establish hydrogen bonds in the ligand-receptor interaction, the effect of removing the donors from one of the reacting partners cannot increase much further by additionally deleting the acceptors from the other.
However, the deletion of OH groups produced a greater loss of binding energy when applied to the receptor than to the ligand (ΔΔG(1) > ΔΔG(2), Table 3). This suggests that in spite of the overall coupling, there is also an additive component in the effect of the mutation of the receptor on binding affinity (see “Theoretical Background” in Methods and Methods). Thus, the change of affinity induced by serine mutagenesis in the receptor is not explained entirely by the loss of docking interactions with the ligand, but may also include a conformational mediated mechanism.
Effect of the Hydroxyl Groups' Deletion on Signal Transduction.
It is not possible to compute the free energy coupling for the effect of the removal of hydroxyl groups on the conversion of the receptor into active form, because the signaling activity of the receptor cannot be described as a free energy change. However, we compared on a qualitative basis how dehydroxylation of either ligand and receptor affects signal transduction.
Concentration-response curves for epinephrine and mape-mediated stimulation of adenylyl cyclase activity were obtained in wild-type and mutant receptors using cell lines displaying various levels of receptor expressions. There was no measurable adrenergic response nor specific pindolol binding in untransfected cells or control CHO lines expressing the neo-resistance plasmid (data not shown).
The maximal effect for epinephrine-mediated stimulation of enzymatic activity was reduced 40 to 50% by removal of hydroxyl groups from the receptor (compare epinephrine in Fig.1, A and B). A similar reduction was caused at wild-type receptors after removal of hydroxyl groups from the ligand (epinephrine versus mape, Fig. 1A). As a consequence, mape is partial agonist in wild-type receptors, but an agonist as full as epinephrine and isoproterenol in mutated receptors (Fig. 1B). The mutation also increased the EC50 for full agonist-mediated stimulations, to an extent comparable with the shift of affinity measured in binding studies, but had little effect on that of mape (Fig. 1D). There was little influence of receptor density on the EC50 of agonists, and the relation betweenE max and apparent receptor concentrations determined in various clones did not deviate significantly from linearity, in both wild-type and mutant receptors (Fig. 1C). This means that there is little amplification between receptor occupation and cyclase response in this system, thus the differences in apparent intrinsic activity of ligands may be assumed proportional to the differences in their efficacies.

Adenylyl cyclase responses mediated by wild-type and (S204A, S207A) β2-AR. CHO lines expressing wild-type (SS) or (S204A, S207A) β2-AR (AA) were prepared as described in Materials and Methods. Top panels, concentration-response curves for epinephrine or mape-stimulated enzymatic activity (10 min) in membranes prepared from CHO cells expressing wild-type (A) or mutant (B) receptor. The points are averages of triplicate determinations and were divided for theB max values measured in the membrane of the two cell lines (12.5 and 11.2 pmol/mg in wild-type and mutant-expressing cells, respectively). Bottom panels, concentration-response curves as those shown in the top panels were generated in membranes prepared from CHO cells expressing different concentrations of wild-type and mutant receptors, as indicated. Maximal (E max, C) and half-maximal (EC50, D) stimulations were estimated using ALLFIT (DeLean et al., 1978) and are plotted as a function of receptor concentration measured in the corresponding membranes.
In conclusion, hydroxyl groups removal produces the same decrease of agonist efficacy either when the deletion affects the ligand or the receptor, which means that the effects of the two perturbations are cooperative (nonadditive) not only on agonist affinity, but also on agonist-induced activation of the receptor.
This was further confirmed by examination of the effects of mutating ligand and receptor on guanine nucleotide-induced shifts of agonist affinity. The mutated receptor displayed a significantly reduced effect of GTP on agonist binding isotherms. Likewise, the effect of GTP on mape binding was negligible either in wild-type and in mutant receptor (Fig. 2). Thus, removal of hydroxyl groups from the ligand (mape versus epinephrine, Fig. 2) reduces GTP effect just like it does the removal of hydroxyl groups from the receptor (isoproterenol and epinephrine in wild type versus mutant receptors, Fig. 2). We do not know why dehydroxylation of either ligand or receptor seems to suppress GTP effect on binding entirely, whereas in both cases at least 50% of cyclase response can still be detected (Fig. 1). As shown in frog erythrocytes, GTP shift and agonist intrinsic activity are correlated (Lefkowitz et al., 1976; DeLean et al., 1980). However, the sensitivity of the detection of GTP shifts depends on the conditions of the binding assay and on the cell membrane under study. It is nonetheless clear that the reduction of such parameter induced by mutations of ligand or receptor are very similar.

Effect of GTP on the binding isotherms of adrenergic agonists in wild-type and [S204A, S207A]β2-AR. The binding of the indicated agonists was studied as competition for the sites labeled by125I-pindolol in membranes of CHO cells expressing wild-type or mutant receptors, either in the presence or absence of GTP (100 μM). The points are means of data generated in three independent experiments, each performed as duplicate determinations.
Interaction between Hydroxyl Group Deletion and Constitutive Activation.
The data presented suggest that the interactions between catechol hydroxyl groups of the ligand and TM5 serines of the receptor contribute to both agonist affinity and agonist-induced conversion of the receptor into R*. In addition, the double mutant analysis of this interaction suggests that the loss of binding free energy attributable to the removal of serine residues from the receptor may involve a conformational shift. To verify this hypothesis we used a second double-mutant cycle. We measured the possible interaction between two receptor mutations that affect binding energy through different mechanisms. One is the double-alanine substitution (AA) investigated above. The second is a mutation enhancing the constitutive activity of the receptor. As described previously, mutagenesis of residues located in the C-terminal portion of the third intracellular loop of the receptor enhance ligand-independent activity but also increase agonist affinity in a manner related to agonist efficacy (Samama et al., 1993). This increase in affinity is mediated allosterically because the targeted residues cannot directly contact the ligand, and it can be explained if we assume that such mutation shifts the equilibrium of the receptor toward the active form R* (Samama et al., 1993).
Therefore, the two mutations cam and AA represent mechanistically distinct perturbations of binding affinity borne into functionally different sites of the same molecule. The free energy coupling, i.e., the extent of linkage, between these two perturbations has interesting implications. If the serines targeted by the AA mutation only provided pure ‘docking’ forces for the ligand and did not contribute at all to the conformational equilibrium that turns the receptor into active form, then the effects of the two mutations AA and cam should be independent of one another (perfectly additive), with free energy coupling equal to zero. A nonzero value instead implies interaction and would suggest that a proportion of the effect of the AA mutation can be attributed to an allosteric mechanism.
Receptor cDNAs carrying the AA mutation, the cam mutation, or both (camAA), were transfected into CHO cells, and agonist equilibrium affinities were measured in permanently expressing clonal lines (Table2). The computed free energy changes for ligand binding to wild-type (SS) and the three mutated receptors (AA, cam, and camAA) form the corners of a close thermodynamic cycle (Table4). Their differences mark four “alchemical” reaction paths, the linkage among which measures the extent of long-range intramolecular interaction between perturbations originating at distinct sites of the same macromolecule.
Thermodynamic cycle for the effects on ligand affinity of mutations applied to the agonist (SS → AA) or to the G protein side (SS → Cam) of the receptor
Using the agonist isoproterenol, the magnitude of the computed free energy coupling, although small, was significantly greater than zero, according to measurements made both in the presence and absence of GTP (Table 4, X = isoproterenol). Similar values (0.93 ± 0.21 and 2.1 ± 0.17 without and with GTP, respectively) were determined for the agonist (±)- epinephrine (not shown in Table 4). In contrast, the free energy coupling measured for the binding affinities of the antagonist pindolol was not significantly different from zero (Table 4, X = pindolol).
Thus, the effects produced by the two perturbations are perfectly additive on the affinity of the antagonist, but show an interactive component on that of the agonist. This means that there is linkage between the two mutations, which is only apparent although if the ligand has the ability to convert the receptor into active form. As reflected in the negative cooperative nature of such interaction (positive sign of the free energy coupling), AA and cam exert inverse effects on agonist affinity. Yet, the small value of free energy coupling indicates that part of such opposite effect is cooperative, and thus mediated through a common intramolecular mechanism. Because the cam mutation enhances agonist affinity as a result of the shift of the equilibrium toward the active receptor form, the “negative” linkage found here implies that the AA mutation also changes the same equilibrium, into the opposite direction.
A finer way to perform this analysis is to compute the free energy coupling for all three perturbations together, as shown in Table5. Free energy changes for epinephrine and mape binding to the wild-type and the three mutant receptors can be assembled into a three-dimensional reaction scheme representing three concomitant “alchemical” changes: 1) dehydroxylation of the receptor, 2) dehydroxylation of the ligand, and 3) constitutive activation. Free energy coupling for this triple-mutation cycle yields the global linkage among the three effects, independently of the interactions observed in all possible pairwise comparisons. This higher order coupling is a direct measure of the effect that each of the three perturbations has on the interaction between the other two. It tells, for example, whether and to which extent constitutive activation is linked to the interaction among hydroxyl groups of ligand and receptor, or vice versa. We measure a non-null value of −1 and −2 RTunits (±GTP) of free energy for this linkage (Table 5), which means that the conformational mechanism that enhance binding energy in response to constitutive activation share a common component with the conformational consequences of OH interactions between agonist and receptor.
Three concurrent perturbations: Hydroxyl group removal and the two receptor mutations
Taken collectively, the thermodynamic analyses of double or triple mutant cycles presented suggest that the deletion of TM5 serines from β2-AR not only severs an important docking site for the agonist, as discovered previously, but also shifts the conformational equilibrium of the receptor toward the inactive form. If this is true, the ligand-independent activity of the mutated receptor should be decreased.
Serine Deletion Diminishes the Constitutive Activity of the Receptor.
We first endeavored to test such prediction using stably transfected CHO lines exhibiting a suitable range of receptor expression. But the relation between basal adenylyl cyclase activity and receptor density failed to show any consistent degree of constitutive activity for wild-type receptors (Fig.3A), which makes consequently impossible to determine a potential inhibitory effect of the AA mutation on such parameter. Enhancement of constitutive activity was readily detectable instead in clones transfected with the cam mutation, despite the much lower level of receptor expression (Fig. 3B). Through the comparison of cells expressing cam or camAA mutant receptors we examined the effect of superimposing the OH deletion on the constitutive activated mutation. There is a definite trend for a decrease of constitutive activity in the receptor carrying both modifications compared with cam alone (Fig. 3B). However, given the very modest range of expression of the camAA mutant, such result provides only presumptive evidence that OH groups deletion may lower the constitutive activity induced by mutagenesis.

Basal and agonist-stimulated adenylyl cyclase activity as a function of receptor concentration. Adenylyl cyclase activity was measured in membranes from CHO cells expressing wild-type and mutant β2 AR. The enzymatic activity was determined using three time-points in duplicate (2, 4, and 8 min) either in the absence (BAS) or presence of 100 μM (−)isoproterenol (ISO) and was calculated from the slope of the linear regressions. The data are plotted as a function of the receptor density measured in the same membranes by 125I-pindolol binding curves, and are means ± S.E. of three independent experiments. A, comparison between cells expressing wild-type (SS) and [S204A, S207A] mutant (AA) receptor. Note that no receptor-dependent increase of basal activity can be detected. B, comparison between cells expressing cam and receptors carrying both mutations (cam-AA).
We thus turned to a transient transfection system in COS-7 cells, where the constitutive activity of the receptor can be detected effectively as enhancement of basal intracellular cAMP concentrations that follows peak-expression of receptor cDNA (Parma et al., 1993; Samama et al., 1993; Shenker et al., 1993).
When COS cells are transfected with progressively increasing abundance of coding plasmids, the relation between pmol of expressed receptors and μg of coding cDNA is strictly linear (Fig.4). Although the range of receptor expression varied extensively among different experiments, relative differences between slopes were fairly constant and seem to reflect an intrinsic property of each mutant. The AA mutant gave the highest levels of expression, the camAA mutant the least. By pooling a number of such experiments the relation between intracellular cAMP levels and receptor density in COS cells shows no significant deviations from linearity for wild-type or mutant receptors (Fig.5). Therefore, the ratio between net mol of cAMP induced by transfection and mol of expressed receptor can be taken as a reliable measure of constitutive activity.

Receptor density as a function of the concentration of coding cDNA. COS-7 cells were transfected with cDNA coding for wild-type or mutant receptors as indicated. Different ratios of empty and coding pBC vectors (as indicated on the x-axis) were used to maintain the mass of transfected DNA constant. Receptor density was measured in membranes from the binding isotherms of125I-pindolol.

Enhancement of cAMP levels by transient receptor expression. COS-7 cells were transfected in duplicate 25-cm2 flasks, using increasing concentrations of coding plasmids as described in Fig. 4. At 24 h post-transfection, cells from one of the duplicate flasks were harvested and seeded into 24-well plates. At 48 h post-transfection, cells in the second duplicate flask were harvested for membrane preparation and determination of receptor concentration, whereas the multiwell plated cells were used for the assessment of intracellular cAMP concentration as described inMaterials and Methods. The actual range of receptor expression varied between experiments, despite the use of the same dilution range of coding plasmids. The plots are thus an overlay of three independent experiments in each of which wild-type and all the mutants were compared within the same transfection. Each point represents triplicate cAMP determinations and aB max computed by nonlinear fitting of a 12-dilution binding isotherm of radiolabeled Pindolol. To assess the significance of the difference between mutant and wild-type receptors, the data were fitted by linear regressions (solid and dotted lines). The calculated slopes (mole cAMP/mol receptor) with lower-upper 95% confidence limits (in parentheses) are as follows: SS, 0.53 (0.46–0.6); AA, 0.24 (0.20–0.28); cam, 5.5 (4.3–6.7); and camAA, 2.9 (2.2–3.6). The differences in slopes were tested by F statistics. For the comparison SS versus AA, F (1,33) = 20.3, P < .0001; For the comparison cam versus camAA, F (1,25) = 5.5, P = .027. The slope of the AA mutant curve is significantly different from zero, F (1,18) = 102, (P < .0001), indicating that the constitutive activity of this receptor is reduced compared with the wild-type, but is not abolished.
As summarized in Figs. 5 and 6, the constitutive activity of the AA mutant thus measured was significantly smaller than that of the wild type (37 ± 2%, n = 10), indicating that OH deletion indeed reduces the intrinsic signaling ability of the receptor regardless of the presence of an agonist. The same deletion also diminished the ligand-independent activity of the constitutively active receptor as indicated by the comparison between cam and camAA mutants (Fig. 6). Although the decrease in this case seems to be smaller than that induced by the AA mutation on wild type, it is not clear whether the difference reflects reality or experimental noise, as it was difficult to achieve an ample range of expression for the camAA mutant even in COS cells.

Comparison of the constitutive activity of wild-type and mutant β2-AR COS-7 cells were transfected with cDNA coding for mutants, wild-type receptors, and empty vector. Receptor density and cAMP levels were measured in parallel as explained in Fig. 5. Constitutive activity is computed as the ratio between net pmols of cAMP produced and net pmol of expressed receptor, after subtraction of the basal cAMP levels (range, 15–25 pmol/mg) and the basal concentration of endogenous β2-AR (range, 0.3–0.5 pmol/mg) measured in cells transfected with noncoding vector in parallel. These ratios are equivalent to the slopes of the regression lines measured in Fig. 5. The data are means ± S.D. of the number of independent experiments shown on top of each bar. In each experiment, cAMP levels were determined in quadruplicate wells, andB max was estimated from computer analyses of a 12-dilution binding isotherms of pindolol.
We noted that the ratio between cam and wild-type receptor expression obtained at a given concentration of transfected DNA is smaller when determined in membrane than in intact cells. This phenomenon, which probably reflects the intrinsic instability of receptors carrying activating mutations noted earlier (Gether et al., 1997), may influence the estimates of the extent of constitutive activity for the mutations. Thus, we also compared the constitutive activity of mutants using intact cells to assess the density of surface receptors. As shown in Fig. 7, the fold enhancement of constitutive activity over that of the wild type produced by the cam mutation is two-times smaller whenB max is determined in cells than in membranes (Fig. 6). However, it is clear also in these experiments that receptors carrying OH deletions have a significantly reduced constitutive activity compared with wild type or to cam mutants.
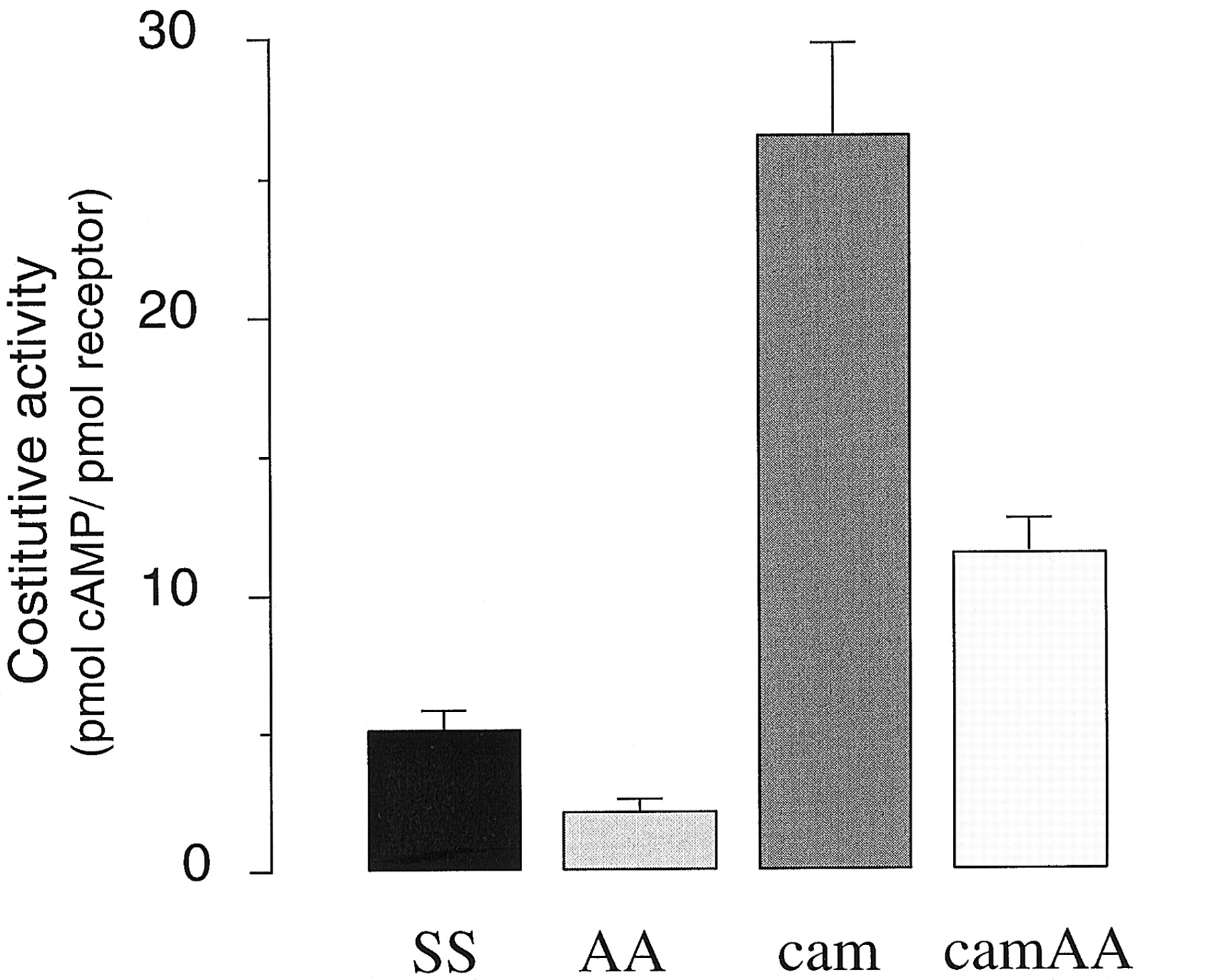
Constitutive activity of wild-type and mutant β2-AR determined in intact cell assays COS-7 cells were transfected and processed as described in Figs. 5 and 6. However, the concentration of expressed receptors was determined in intact cells (see Materials and Methods) using the hydrophilic adrenergic antagonist [3H]CGP 12177.
In conclusion, the deletion of S204 and S207 side chains exerts a negative allosteric effect on the basal (ligand-independent) biological activity of the receptor and confirms the predictions resulting from the analysis of free energy conservation among multiple mutations. Both types of information support the idea that the mutation can shift the conformational equilibrium of the receptor toward the inactive form. Moreover, these data indicate that the inhibitory effect of mutagenesis of serines 204 and 207 on the basal activity of the vacant receptor (Figs. 6 and 7) is comparable with that exerted on agonist-induced receptor activity (Fig. 1). This further supports the notion that the mutation can alter the intrinsic equilibrium of the receptor, not only its response to the ligand.
We wondered whether an alternative explanation could account for the reduction of constitutive activity caused by the AA mutation. For example, a mutation can decrease constitutive activity without altering the intramolecular equilibrium of the molecule if it changes the ability of the receptor to trigger on binding a conformational change in the G protein. In this case, the receptor can still be converted into R* form by the agonist and still bind to the G protein to the same extent as the wild type. However, the G protein-boundR* form would produce less G protein activation than the wild type. This can cause diminutions of constitutive activity, of GTP effects, and agonist-induced maximal stimulation just like we observe for the AA mutant.
This sort of modification (that we may define “dominant negative”) would be difficult to distinguish from that resulting from a shift of the equilibrium toward the inactive receptor form. Nonetheless, an indirect approach can be used to evaluate this possibility. We reasoned that if the reduction of constitutive activity results from a shift in the conformational equilibrium of the receptor, the overall affinity between receptor and G protein (i.e., MJ) is diminished. In contrast, for a dominant negative mutant, this remains unchanged. Therefore, only a dominant negative mutant may be expected to compete for and antagonize the effect of a constitutively active receptor when both are present in a suitable stoichiometric ratio in cotransfected cells. To examine this possibility, cDNAs coding for the cam and AA mutations, mixed to generate excess expression of AA mutant, were cotransfected in COS cells, and the net enhancements of basal cAMP levels were compared with those induced by each individually transfected mutant. When the exact fraction of basal activity due to each mutant was calculated after computer analysis of agonist binding isotherms in cells expressing the mixed populations of receptors, we found no evidence for the existence of competition between the two receptors (Fig.8). These and the preceding data taken together indicate that there is only intramolecular (Figs. 6 and 7), but not intermolecular (Fig. 8), antagonism between the two mutations. We thus suggest that the negative allosteric effect of hydroxyl group deletion on constitutive and ligand-dependent activity is mediated through a shift of the conformational equilibrium of the receptor rather than impairment of transduction at the receptor-G protein interface.

Lack of antagonism between cam and AA mutant receptors for the transfection-induced accumulation of cAMP in COS-7. Cells were transfected with either individual plasmids coding for the cam (0.19 μg/cm2) or AA (0.01 μg/cm2) mutation or cotransfected with the mixture of both. The total mass of input cDNA (0.2 μg/cm2) in the individual transfections was equalized by the addition of empty vector. The levels of cAMP and receptor expression were measured as described in Fig. 6. A, comparison of cAMP levels in cells transfected with empty vector (pBC), individual mutants (AA or cam) or cotransfected with both (cam + AA). Receptor densities measured for the various conditions are presented in bold on top of each bar. The actual concentrations of cam and AA mutants expressed in the membranes of cotransfected cells were resolved by computer analysis (see C, below). Note that the relative levels of the two mutants expressed in the cotransfection cannot be predicted from the individual transfection, even if performed in parallel. B, the extent of activity attributable to each of the mutants expressed in the cotransfected cells was computed by multiplying the ratios: net cAMP/net B max (measured from the single transfection of each mutant in A, second and third bars) for the pmol of each mutant receptor estimated in the membranes of cotransfected cells by computer analysis. The data are then converted into fractions of the total activity determined in cotransfected cells and plotted in histogram form. It is clear that the total activity observed in the cotransfection is equal to the exact sum of the activities expected for the pmol of each mutant present in the mixture. Thus, even if present in stoichiometric excess, the AA mutant cannot antagonize the basal increase induced by the cam mutant. C, isoproterenol binding isotherms for inhibition of 125I-pindolol binding in membranes prepared from cotransfected cells. Because the difference in agonist affinity between cam and AA mutant is very large (Table 2), the agonist binding curve is clearly biphasic and show two high- and low-affinity components. When data were fitted according to mass action law models for multiple binding sites (LIGAND, Munson and Rodbard, 1980), a two-site model (solid line) provided a very significant improvement in the fit (P < .0001). The total binding capacity of the membranes (55 pmol/mg) was resolved into a high-affinity (K eq 2.6 ± 1.2 × 108 M −1) component (i.e., the cam mutant) accounting for 13.9 ± 1.1 pmol/mg, and a low-affinity (K eq 8.9 ± 0.3 × 104 M −1) component (i.e., the AA mutant) accounting for 41.1 ± 2.1 pmol/mg. The data illustrate the results of a single representative experiment. Two additional experiments, which resulted in different ratios of coexpressed AA and cam mutant (8.6:1 and 3.7:1, respectively), gave identical results.
Discussion
We used an approach based on mutant cycles to analyze the energetic contribution of the postulated interaction between OH groups of the catecholic moiety of a bound adrenergic agonist and serines 204 and 207 of the fifth transmembrane domain of the β2-adrenergic receptor. We tested the closure of two nested thermodynamic reaction cycles. In one, the same deletions of OH groups is applied to distinct molecules, ligand, and receptor. In the other, two diverse perturbations, OH deletion and constitutive activation, are imposed onto separate functional domains of the same receptor. From this analysis we can draw two conclusions.
First, we reconfirm the notion that the two sets of OH groups in the ligand and the receptor interact directly, probably via the formation of a pair of hydrogen bonds, as originally proposed (Strader et al., 1989).
Second, we uncover an additional and unexpected role for S204 and S207 of TM5. They seem to control the equilibrium between the active (R*) and inactive (R) forms of the receptor. This suggestion is verified by the finding that the ligand-independent basal activity of the double-alanine mutant is significantly smaller than that of the wild-type receptor. This means that dehydroxylation in that particular agonist-docking spot of β2-AR also decreases its constitutive activity.
The results presented in this study bear a number of interesting implications. One concerns our understanding of the nature of the agonist-binding site. If serines 204 and 207 in TM5 provide an anchor site for the agonist and also regulate the setpoint for the equilibrium of the R* form, this suggests that several, if not all, of the subsites where the agonist binds to the receptor may also contribute key intramolecular interactions for the conversion of the molecule into active form. The agonist binding pocket is thus a multidimensional map of residues that take part in the molecular motion underlying the emergence of a bioactive conformation in the receptor. Although obvious on an intuitive basis, this notion has received little experimental evidence thus far. Our data show that a fraction of the agonist binding energy lost on removal of serine residues is associated with the shift of intramolecular equilibrium of the receptor toward inactive conformations. This not only supports the idea that experimentally observable binding energies of any ligand reflect both intermolecular and intramolecular forces, but also demonstrates that for an agonist, the intramolecular component must be necessarily related to the process that generates the biologically relevant conformation. It is likely that agonist-receptor complexes have been shaped through evolution to optimize the maximum number of contacts with conformationally sensitive points of the receptor rather than the formation of the most stable energy-minimized assembly. We may speculate that the key difference in the mode of binding between agonist and antagonist is more energetic than topographic. A fine-tuned energy balance between intermolecular stabilization and intramolecular perturbation within the configuration of the binding pocket may be the fundamental requirement for full agonism, far more than the selective interaction with a particular set of critical docking residues.
A second implication regards our views on the mechanism by which agonists induce receptor activation. It was thought that receptors were essentially “silent” molecules in the classical view and that agonist binding would endow them with the ability to generate biological signals. The discovery that certain residues of their sequence exert a very stringent role in suppressing ligand-independent activity (Kjelsberg et al., 1992; Scheer et al., 1993) has suggested an opposite paradigm. A receptor may have signaling activity by default that is clamped down by structural factors and released by agonist binding (Kjelsberg et al., 1992; Lefkowitz et al., 1993; Samama et al., 1993). The finding in this study that an agonist docking site can also control the level of receptor constitutive activity brings additional support to this notion. It is interesting to note that alanine mutation of serine residues inhibits in parallel receptor constitutive activity and agonist-induced maximal stimulation. Thus, the suppression of an agonist-docking site restricts biological activity by an essentially identical mechanism, regardless of whether the receptor is bound or not to the agonist. This suggests that the role of bound agonist is to amplify or catalyze a process that is intrinsically wired into the receptor, not to impart novel molecular properties. In other words, the bioactive receptor conformers that are “induced” by an agonist are improbable, but not impossible, in its absence.
Footnotes
-
Send reprint requests to: Dr. Tommaso Costa, Laboratorio di Farmacologia, Istituto Superiore di Sanità, Viale Regina Elena 299, 00161 Rome, Italy. E-mail:tomcosta{at}iss.it
-
We acknowledge support from the EU BIOMED 2 program “Inverse Agonism. Implications for Drug Design” (to T.C.) and from the Fonds National Suisse de la Recherche Scientifique, Grant 31-51043.97 (to S.C.).
- Abbreviations:
- 7TM
- 7 transmembrane domains
- β2-AR
- β2-adrenergic receptor
- AA
- S204A, S207A mutation
- CHO
- Chinese hamster ovary
- cam
- constitutive active mutant
- mape
- (±)2-(methylamino)-1-phenyl-1-ethanol
- Received May 24, 1999.
- Accepted October 1, 1999.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}