


Abstract
Despite extensive study, the G protein coupling of dopamine D3 receptors is poorly understood. In this study, we used guanosine-5′-O-(3-[35S]thio)-triphosphate ([35S]-GTPγS) binding to investigate the activation of G proteins coupled to human (h) D3 receptors stably expressed in Chinese hamster ovary (CHO) cells. Although the receptor expression level was high (15 pmol/mg), dopamine only stimulated G protein activation by 1.6-fold. This was despite the presence of marked receptor reserve for dopamine, as revealed by Furchgott analysis after irreversible hD3 receptor inactivation with the alkylating agent, EEDQ (N-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline). Thus, half-maximal stimulation of [35S]-GTPγS binding required only 11.8% receptor occupation of hD3 sites. In contrast, although the hD2(short) receptor expression level in another CHO cell line was 11-fold lower, stimulation by dopamine was higher (2.5-fold). G protein activation was increased at hD3 and, less potently, at hD2 receptors by the preferential D3 agonists, PD 128,907 [(+)-(4aR,10bR)-3,4,4a,10b-tetrahydro-4-propyl-2H,5H- [1]benzopyrano[4,3-b]-1,4-oxazin-9-ol] and (+)-7-OH-DPAT (7-hydroxy-2-(di-n-propylamino)tetralin). Furthermore, the selective D3 antagonists, S 14297 ((+)-[7-(N,N-dipropylamino)-5,6,7,8-tetrahydro-naphtho(2,3b)dihydro-2,3-furane]) and GR 218,231 (2(R,S)-(dipropylamino)-6-(4-methoxyphenylsulfonylmethyl)-1,2,3,4- tetrahydronaphtalene), blocked dopamine-stimulated [35S]GTPγS binding more potently at hD3than at hD2 sites. Antibodies against Gαi/αo reduced dopamine-induced G protein activation at both CHO-hD3 and -hD2 membranes, whereas GαS antibodies had no effect at either site. In contrast, incubation with anti-Gαq/α11antibodies, which did not affect dopamine-induced G protein activation at hD2 receptors, attenuated hD3-induced G protein activation. These data suggest that hD3 receptors may couple to Gαq/α11 and would be consistent with the observation that pertussis toxin pretreatment, which inactivates only Gi/o proteins, only submaximally (80%) blocked dopamine-stimulated [35S]GTPγS binding in CHO-hD3 cells. Taken together, the present data indicate that 1) hD3 receptors functionally couple to G protein activation in CHO cells, 2) hD3 receptors activate G proteins less effectively than hD2 receptors, and 3) hD3 receptors may couple to different G protein subtypes than hD2 receptors, including nonpertussis sensitive Gq/11 proteins.
Dopaminergic neurotransmission is mediated by five receptor subtypes (D1 to D5) which can be grouped into two receptor families. D1-like receptors include the D1 and D5 subtypes, whereas D2-like receptors include the D2, D3, and D4 subtypes. D2 and D3 receptors, in particular, display marked sequence homology and pharmacological similarity in their in vitro ligand binding profiles (Levant, 1997; Missale et al., 1998). However, D3 receptors may be distinguished from D2 receptors by several factors. D3 receptors are concentrated in limbic rather than striatal brain regions (Liu et al., 1996; Hall et al., 1996). Furthermore, they mediate stimulation, rather than inhibition, of c-fos expression in striatal neurones (Pilon et al., 1994; Morris et al., 1997), and inhibition, rather than stimulation, of locomotor activity in rats (Svensson et al., 1994; Starr and Starr, 1995). In addition, whereas D2 receptors couple efficiently to second-messenger systems, markedly inhibiting adenylyl cyclase activity, such responses have proved elusive and complex for D3 receptors (e.g., Freedman et al., 1994;MacKenzie et al., 1994; Tang et al., 1994; Griffon et al., 1997). Indeed, D3 receptors couple selectively to inhibition of adenylyl cyclase type V, but not type I or VI, and only weakly to type II (Robinson and Caron, 1997; Watts and Neve, 1997). In vitro studies of agonist efficacy have employed other measures of receptor activation, including medium acification (Cox et al., 1995), and stimulation of mitogenesis (Pilon et al., 1994; Svensson et al., 1994; Sautel et al., 1995). However, these approaches measure responses “downstream” of the receptor in the intracellular activation cascade and the relevance of an increase in mitogenesis for postmitotic central nervous system neurones is unclear. A more promising approach may be to measure receptor-mediated G protein activation by stimulation of guanosine-5′-O-(3-[35S]thio)-triphosphate ([35S]GTPγS) binding: this corresponds to the first step of the intracellular activation cascade and directly reflects ligand binding events at the receptor itself (Pregenzer et al., 1997; Malmberg et al., 1998). Thus, the present study adopted this strategy to address several questions concerning, principally, the functional properties of human (h) D3 receptors. In addition, in some tests results at hD3receptors were compared with those at hD2receptors. First, differences in the second-messenger actions of D3 and D2 receptors may be related to differing capacities for stimulation of G proteins. We addressed this issue by investigating the ability of hD3 receptors to mediate dopamine-stimulated [35S]GTPγS binding. Second, the relationship between binding affinity and functional potency of dopaminergic agonists and antagonists was investigated using the most potent and selective D3 receptor ligands reported to date: the agonists (+)-7-OH-DPAT (7-hydroxy-2-(di-n-propylamino)tetralin) and PD 128,907 [(+)-(4aR,10bR)-3,4,4a,10b-tetrahydro-4-propyl-2H,5H- [1]benzopyrano[4,3-b]-1,4-oxazin-9-ol] (Pugsley et al., 1995) and the antagonists, S 14297 ((+)-[7-(N, N-dipropylamino)-5,6,7,8-tetrahydro-naphtho(2,3b)dihydro-2,3-furane]) and GR 218,231 (2(R,S)-(dipropylamino)-6-(4-methoxyphenylsulfonylmethyl)-1,2,3,4-tetrahydronaphtalene) (Millan et al., 1995b; Murray et al., 1996). The hD3/hD2 selectivities based on Ki ratios were compared with those based on EC50 and KB ratios (Burris et al., 1995; Levant, 1997). Third, the signal transduction differences between D3 and D2 receptors, such as the differential coupling to adenylyl cyclase isoforms, could be due to receptor interactions with different G protein populations. Indeed, at least 16 distinct G protein α subunits have been identified, divided into four families: Gi, GS, Gq/11, and G12/13 (Simon et al., 1991). Although a previous study suggested differences in coupling profiles of D2 and D3 receptors for modulation of outward K+ currents (Liu et al., 1996), no information is available from a functional test more proximal to the receptor and the G protein subtypes involved in D3 coupling are unclear (cf. Tang et al., 1994). The present study, therefore, examined G protein coupling specificity directly at the G protein activation level by challenging the receptor-mediated stimulation of [35S]GTPγS binding with specific antisera raised against different Gα subunits. In fact, antibodies raised against the COOH terminal part of Gα subunits have proved useful to determine the G protein specificity of several other 7-transmembrane domain receptors (Harris-Warrick et al., 1988; McFadzean et al., 1989; Lledo et al., 1992; Izenwasser and Côté, 1995).
Materials and Methods
Membrane Preparations of Chinese Hamster Ovary (CHO)-hD3 and CHO-hD2 Cells.
CHO cells expressing hD3 receptors were grown as described previously (Sokoloff et al., 1992). Cells were harvested from adherent culture and homogenized using a Kinematica Polytron (Kinematica GmBH, Littau, Switzerland) in a buffer containing 50 mM Tris (pH 7.4), 5 mM MgCl2. The suspension was then centrifuged at 20,000g for 15 min at 4°C and the pellet was resuspended in the appropriate binding buffer (see below) and stored at −80°C. CHO-hD2(short) cell membranes were purchased from Receptor Biology (Baltimore, MD). The “short” hD2 isoform, which lacks a 29-amino acid insert in the putative third intracellular loop, is processed faster to mature receptors at the cell surface than the “long” form and may couple more efficiently to certain G protein subtypes (Fishburn et al., 1995;Boundy et al., 1996).
[125I]Iodosulpride Binding to hD3 and hD2 Receptors.
Saturation binding at hD2 and hD3 receptors was carried out with 12 concentrations of [125I]iodosulpride (1000 Ci/mmol; Amersham, Les Ulis, France). For competition binding experiments, membranes (10 to 20 μg protein) of CHO-hD2 or CHO-hD3 cells were incubated with [125I]iodosulpride (0.1 nM for hD2 and 0.2 nM for hD3) at 30°C for 30 min in a buffer containing 50 mM Tris (pH 7.4), 120 mM NaCl, 5 mM KCl, 1 mM EDTA, and 5 mM MgCl2. Nonspecific binding was defined with raclopride (10 μM). Isotherms were analyzed by nonlinear regression, using the computer program PRISM (Graphpad Software Inc., San Diego, CA) to yield IC50 values. Inhibition constants (Ki values) were derived from IC50 values according to the Cheng-Prusoff equation. The goodness of fit was tested by runs test. For compounds that yielded P < .05 in the runs test and/or shallow inhibition isotherms (nH values markedly inferior to unity), 14-point competition binding experiments were carried out and one- and two-site fits were compared by F test.
Measurement of Agonist Efficacy and Antagonist Potency at hD3 and hD2 Receptors.
Receptor-linked G protein activation by dopamine at hD2 and hD3 receptors was determined by measuring the stimulation of [35S]GTPγS (1332 Ci/mmol; NEN, Les Ulis, France) binding induced by dopamine. CHO-hD2 membranes (30–40 μg protein) were incubated (60 min, 22°C) with agonists and/or antagonists in a buffer containing 20 mM HEPES (pH 7.4), 3 μM GDP, 10 mM MgCl2, 150 mM NaCl, and 0.1 nM [35S]GTPγS. CHO-hD3membranes (30–50 μg protein) were incubated (40 min, 22°C) with agonists and/or antagonists in a buffer containing 20 mM HEPES (pH 7.4), 3 μM GDP, 3 mM MgCl2, 100 mM NaCl, and 1.0 nM [35S]GTPγS. Nonspecific binding was defined with GTPγS (10 μM). Agonist efficacy is expressed relative to that of dopamine (100%), which was tested at a maximally effective concentration (10 μM) in each experiment. For all tests, membranes were preincubated with agonist and/or antagonist for 15 min before the addition of [35S]GTPγS.KB values for inhibition of dopamine (1 and 3 μM for hD3 and hD2respectively)-stimulated [35S]GTPγS binding were calculated according to Lazareno and Birdsall (1993):
KB = IC50/{[(2+(agonist/EC50)nH)nH−1] −1};
where IC50 is the inhibitory concentration50 of the antagonist, agonist is the dopamine concentration, EC50 is the effective concentration50 of dopamine alone, and nH is the Hill coefficient of the dopamine stimulation isotherm.
For dopamine concentration-response curves determined in the presence of fixed concentrations of the antagonist, GR 218,231, pA2 values were derived by Schild analysis. In isotopic dilution experiments, the basal and dopamine (10 μM)-stimulated binding of radiolabeled [35S]GTPγS was inhibited with unlabeled GTPγS. Saturation binding curves were derived to estimate the number of G proteins activated by dopamine, as described previously (Newman-Tancredi et al., 1997).
Experiments were terminated by rapid filtration through Whatman GF/B filters (pretreated with 0.1% polyethyleneimine in the case of [125I]iodosulpride binding) using a Brandel cell harvester. Radioactivity retained on the filters was determined by liquid scintillation counting. All data are expressed as mean ± S.E.M. of ≥3 independent determinations. Protein concentration was determined colorimetrically using a bicinchonic acid assay kit (Sigma Chemical Co., S. Quentin Fallavier, France).
hD3 Receptor Alkylation withN-Ethoxycarbonyl-2-Ethoxy-1,2-Dihydroquinoline (EEDQ).
CHO-hD3 cell membranes were treated by EEDQ at a final concentration between 0 and 300 μM (0, 10, 33, 100, and 300 μM). The membrane suspension (3 ml final volume) was vortexed and immediately centrifuged at 4°C for 15 min at 20,000g. The supernatant was discarded and the membrane pellet was resuspended in the appropriate buffer and [35S]GTPγS or [125I]iodosulpride binding was performed as described above. KA values were determined by Furchgott analysis, as described by Atkinson and Minneman (1992) andAdham et al. (1993), with CHO-hD3 membranes treated with 33 μM EEDQ. Plots were derived of 1/[A] versus 1/[A′]; where [A] and [A′] are equiactive concentrations for stimulation of [35S]GTPγS binding before and after receptor alkylation, respectively. KAwas calculated from
KA = (slope-1)/y-intercept.
Percentage receptor occupancy (O) was calculated by
O = 100 * L/(L +KA);
where L is the concentration of agonist.
Characterization of G proteins by Immunoblotting and ADP-Ribosylation.
Immunoblotting of Gα subunits was performed using antisera purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA) raised against Gαi/O (C10), GαS (C18), and Gαq/11(C19). Approximately 2 μg protein from CHO-hD2and CHO-hD3 membrane preparation was separated on 10% polyacrylamide gel and transferred onto nitrocellulose. Antisera were incubated at 1/1000 followed by enhanced chemiluminescence detection with horseradish peroxidase as secondary antibody (Amersham, Buckinghamshire, UK).
ADP-ribosylation by Bordetella pertussis toxin (PTX) was carried out as described by Cussac et al. (1996). Briefly, membranes (10 μg) from untreated CHO-hD3 cells and cells preincubated with PTX (100 ng/ml) or cholera toxin (1 μg/ml) for 6 h were incubated for 60 min in buffer containing 8 μM [32P]NAD (2 μCi), 70 mM Tris/HCl, pH 8.0, 1 mM ATP, 0.1 mM GTP, 1 mM EDTA, 25 mM dithiothreitol, 10 mM nicotinamide, 0.1 mM MgCl2, and 100 ng of PTX in a 40-μl assay volume. PTX was preactivated with 25 mM dithiothreitol for 30 min at 37°C. The reaction was stopped by addition of 40 μl of Laemmi buffer 2× and the sample was boiled 3 min at 95°C. Two micrograms of protein from the sample was then separated in 10% polyacrylamide gel and [32P]ADP-ribosylated Gi/O proteins were revealed by 8 h exposure of the dried gel to Hyperfilm (Amersham).
Antiserum Treatment of CHO-hD3 or -hD2Membranes.
CHO-hD3 and -hD2 membranes (30–50 μg protein) were preincubated at 4°C for 5 h with 3.3 μg of antisera against different Gα proteins. [35S]GTPγS binding was then performed in absence and in presence of dopamine (10 μM) as described above. The antisera used were the same as described above and were chosen for their capability to recognize the COOH terminal part of Gα subunits involved in receptor interactions. Another antiserum (C17) against c-Jun NH2-terminal kinase (JNK1), a target unrelated to G proteins, was also tested as a control to exclude nonspecific antibody effects.
Compounds.
(+)7-OH-DPAT was obtained from CNRS (Paris, France). PD 128,907 was purchased from RBI (Natick, MA); dopamine, haloperidol, EEDQ, and cholera and PTXs were purchased from Sigma. GR 218,231 and S 14297 were synthesized by J.-L. Peglion, Servier.
Results
Saturation Binding Experiments.
The receptor expression level of hD3 receptors, determined in [125I]iodosulpride saturation binding experiments, was almost 11-fold higher than that of hD2 receptors (Table1 and Fig.1).
Densities of recombinant receptors and agonist-activated G proteins in CHO cells stably expressing hD2 and hD3 receptors

Saturation binding of [125I]iodosulpride and [35S]GTPγS to CHO-hD3 and CHO-hD2 cell membranes. A, representative saturation binding isotherms of [125I]iodosulpride to CHO-hD2 and CHO-hD3 membranes. B, representative saturation binding isotherms of [35S]GTPγS to CHO-hD2 and CHO-hD3 membranes. Basal and dopamine (10 μM)-stimulated [35S]GTPγS binding were determined in the presence of increasing concentrations of GTPγS. These data were transformed as described in Materials and Methods to generate a saturation binding isotherm for net agonist-dependent [35S]GTPγS binding. Points shown are means of duplicate determinations from representative experiments repeated on at least four occasions. Bmax andKD/apparent KD(Kapp) data from these experiments are shown in Table 1.
The number of dopamine-activated G proteins, determined in [35S]GTPγS isotopic dilution experiments with unlabeled GTPγS, was higher in CHO-hD3membranes than in CHO-hD2 membranes (Table 1). These different expression levels of receptors (R) and G proteins (G) corresponded to a 3-fold higher R/G ratio in CHO-hD3 membranes than in CHO-hD2 membranes (4.6:1.5; Table 1). Treatment of CHO-hD3 membranes with EEDQ (33 μM) reduced hD3 receptor density by about half (Tables 1 and2). The effect of EEDQ was specific to the receptors: EEDQ did not significantly alter the number or affinity of [35S]GTPγS for dopamine-activated G proteins (Table 1).
Concentration-dependent actions of EEDQ on receptor density and function in CHO-hD3 membranes
hD3 Receptor Alkylation with EEDQ.
CHO-hD3 membranes were sensitive to EEDQ treatment. Addition of EEDQ to ice-cold membranes and immediate centrifugation (15 min, 4°C, 20,000g) was sufficient to reduce the number of hD3 binding sites in a concentration-dependent manner without a change in affinity of the radioligand (Table 2 and Fig.2). EEDQ also concentration dependently reduced the stimulation of [35S]GTPγS binding induced by dopamine (Table 2 and Fig. 3). At the maximal EEDQ concentration tested (300 μM), the density of hD3 receptors was reduced by over 80%, whereas dopamine-induced [35S]GTPγS binding was reduced by 64%. Subsequent experiments were carried out with an EEDQ concentration of 33 μM, which reduced dopamine-induced [35S]GTPγS binding by about 50% (Tables 1and 2). Under these conditions, the pEC50(−log EC50) of dopamine was slightly reduced to 7.87 ± 0.09 (compared with 8.00 for control membranes, Table 4), without alteration of basal [35S]GTPγS binding. TheKA value determined by Furchgott analysis was 53 ± 23 nM, which corresponded closely to the affinity of dopamine at hD3 receptors determined in competition binding experiments (54.9 nM, Table3). The resultingKA/EC50 ratio for dopamine was 5.3, indicating the presence of receptor reserve. This was confirmed in occupancy/response plots, which yielded hyperbolic curves, with the mean half-maximal response to dopamine being observed at 11.8 ± 2.3% occupation of hD3 binding sites (Fig. 3).
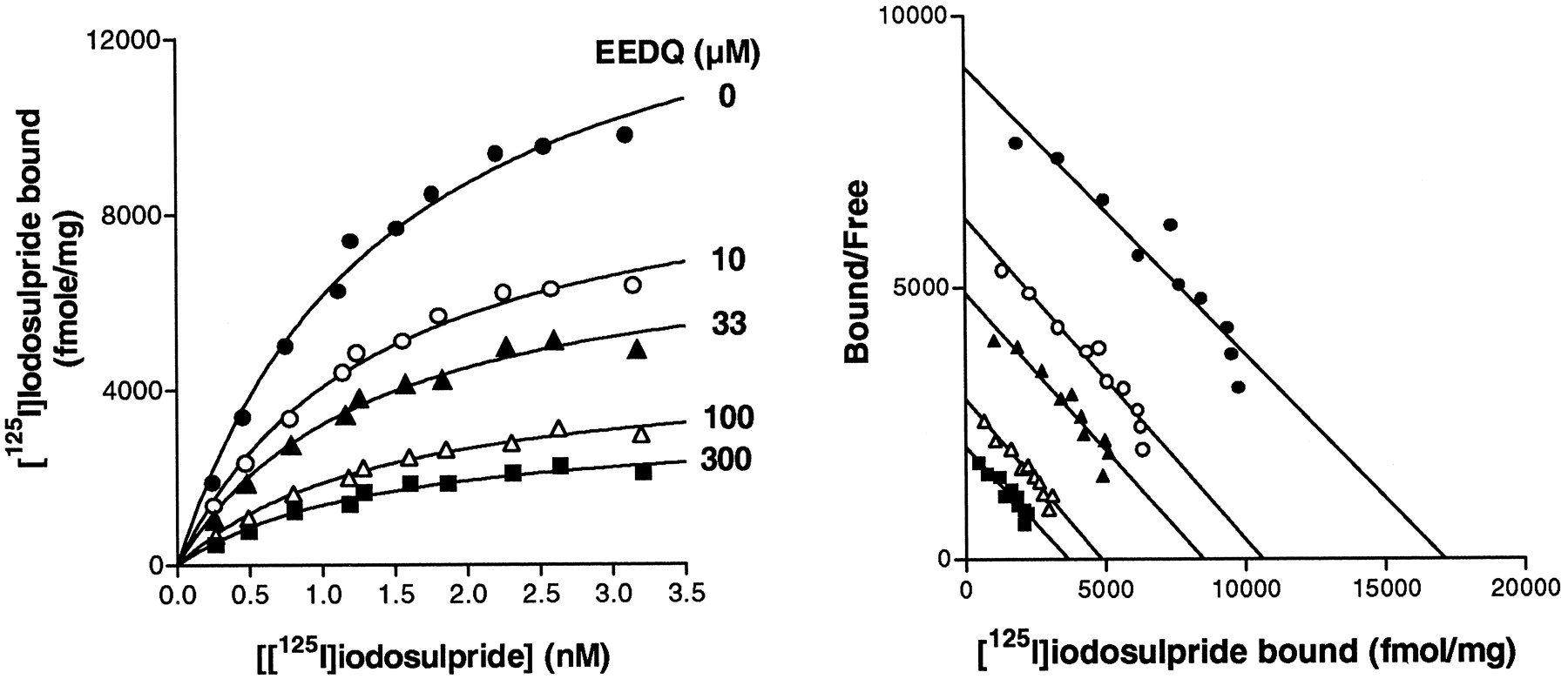
Concentration-dependent reduction of hD3receptor density by EEDQ. A, representative saturation binding isotherms of [125I]iodosulpride to CHO-hD3membranes pretreated with different concentrations of EEDQ. B, Scatchard representation of data from A. Points shown are means of duplicate determinations from representative experiments repeated on at least three occasions. Bmax andKD data from these experiments are shown in Table 2.

hD3 receptor inactivation with EEDQ reveals receptor reserve for dopamine-stimulated [35S]GTPγS binding to CHO-hD3 membranes. A, concentration-dependent reduction of dopamine-stimulated [35S]GTPγS binding by pretreatment with EEDQ (0 to 300 μM). Columns represent mean ± S.E.M. from at least three experiments carried out in triplicate. B, stimulation by dopamine of [35S]GTPγS binding to control or EEDQ (33 μM)-pretreated CHO-hD3 membranes. C, double-reciprocal plot of 1/[A] versus 1/[A′], where [A] and [A′] are equiactive concentrations for stimulation of [35S]GTPγS binding with and without EEDQ treatment, respectively. D, dopamine occupancy/response relationship, derived using the value ofKA from C. Hyperbolic isotherm indicates the presence of receptor reserve. For B, C, and D, points shown are means of triplicate determinations from a representative experiment repeated on at least three occasions. Mean KA value was 53 ± 23 nM. Mean half-maximal response to dopamine was observed at at 11.8 ± 2.3% occupation of hD3 binding sites.
Stimulation of [35S]GTPγS binding by dopaminergic ligands at hD2 and hD3 receptors
Competition binding of dopaminergic ligands at hD2 and hD3 receptors
[125I]Iodosulpride Competition Binding hD2 and hD3
At hD3 receptors, agonist competition binding isotherms were monophasic, although in some experiments with dopamine a small (∼10% of binding sites), high-affinity (pKH, −log KH, ∼ 9) component was apparent (data not shown). At hD2 receptors, agonist competition isotherms were biphasic and fitted better to a two-site model (p< .05, F test; Fig. 4), yielding estimates of affinity for the high- and low-affinity components (Table 3), presumably reflecting binding to G protein-coupled and -uncoupled states of the receptor, respectively. Selectivity ratios of affinity at hD2/hD3receptors were calculated by comparing theKi at hD3 receptors with theKH and the KL at hD2 receptors. Competition binding curves with antagonist ligands, haloperidol, S 14297, and GR 218,231, were monophasic for both hD2 and hD3 sites (Table 3).

Competition binding of dopaminergic agonists at hD3 and hD2 receptors. Representative [125I]iodosulpride competition binding isotherms for dopamine, (+)-7-OH-DPAT and PD 128,907 at hD2 and at hD3 receptors. At hD2 receptors the data fitted better to a two-site model (P < .05,F test). Points shown are means of triplicate determinations from representative experiments repeated on at least three occasions. pKi, pKH, and pKL data from these experiments are shown in Table 3.
[35S]GTPγS Binding Conditions at CHO-hD3 and CHO-hD2 Cell Membranes.
In preliminary experiments (not shown), conditions were defined that yielded optimal dopamine-induced stimulation of [35S]GTPγS binding. 1) Optimal stimulation was observed at NaCl concentrations of 100 and 150 mM for hD2 and hD3 membranes, respectively. 2) GDP concentration dependently reduced basal binding of [35S]GTPγS to both hD2and hD3 cell membranes. 3) MgCl2 increased dopamine-dependent [35S]GTPγS binding to a maximum at around 3 to 10 mM for both receptor subtypes. 4) Stimulation of [35S]GTPγS binding was linear with time over the period of the incubations. In view of the lower stimulation of [35S]GTPγS binding by agonists at hD3 receptors, a higher concentration of [35S]GTPγS was used (1.0 nM) than with hD2 (0.1 nM) to provide a stronger signal. Typical binding of [35S]GTPγS (0.1 nM) to CHO-hD2 membranes was 90 to 100 fmol/mg basal and 230 to 250 fmol/mg in the presence of dopamine (10 μM). Typical binding of [35S]GTPγS (1 nM) to CHO-hD3 membranes was 1000 to 1100 fmol/mg basal and 1500 to 1600 fmol/mg with dopamine (10 μM). In control experiments in which the concentration of [35S]GTPγS used for hD3receptors was 0.1 nM (as for hD2 receptors), the pEC50 for dopamine was 8.04 ± 0.03 (n = 3) nM, not significantly different from the pEC50 observed with a [35S]GTPγS concentration of 1 nM (8.00 ± 0.07, Table 4). Dopamine-induced stimulation was 45.8 ± 2.8% (n = 3).
[35S]GTPγS Binding at CHO-hD3 and CHO-hD2 Cell Membranes: Agonist Actions.
Dopamine, PD 128,907, and (+)-7-OH-DPAT increased [35S]GTPγS binding to CHO-hD3 and CHO-hD2membranes in a concentration-dependent manner, with EC50, Emax, and nH values shown in Table 4. S 14297 exhibited slight agonist actions at hD2 receptors (Emax = 20.6%) but no agonist activity was detected at hD3 receptors (Fig.5). PD 128,907 was almost twice as efficacious at hD2 as at hD3 receptors, whereas (+)-7-OH-DPAT was a partial agonist at both receptor subtypes. The ratios of EC50 values at hD2/hD3 were intermediate between theKH(hD2)/Ki(hD3) and theKL(hD2)/Ki(hD3) ratios in Table 3. In control experiments in which the concentrations of NaCl and MgCl2 were inverted between hD2 and hD3, we did not observe marked changes in EC50 andEmax values of (+)-7-OH-DPAT and PD 128,907 (data not shown), but a slight decrease in percentage of stimulation by agonists was noted.

Agonist stimulation of hD3 and hD2 receptor-mediated G protein activation. [35S]GTPγS binding is expressed as a percentage of maximal stimulation given by dopamine. A, fold stimulation of [35S]GTPγS binding by dopamine at hD2 and hD3 receptors. B, agonist concentration-response curves at hD2 receptors. C, agonist concentration-response curves at hD3 receptors (• dopamine; ♦ PD 128,907; ○ (+)-7-OH-DPAT; and ▴ S 14297). Points shown are means of triplicate determinations from representative experiments repeated on at least three occasions. Emax, pEC50, and nH data from these experiments are shown in Table 4.
[35S]GTPγS Binding at CHO-hD3 and CHO-hD2 Cell Membranes: Antagonist Actions.
Haloperidol, GR 218,231, and S 14297 did not alter [35S]GTPγS binding from basal levels at hD3 receptors or, except S 14297 (as described above) at hD2 receptors. The inhibition of dopamine-stimulated [35S]GTPγS binding (Table5 and Fig.6), yielded antagonist potencies (KB values), which conserved the same order of potency as the Ki values shown for these compounds in Table 3. The novel ligand GR 218,231 was shown to behave as a competitive antagonist, inducing a rightward parallel shift of the dopamine stimulation curve without loss of maximal efficacy (Fig.7), yielding a linear Schild plot (r = 0.96, slope = 1.06 ± 0.10) and a pA2 value of 9.34 similar to its affinity calculated by competition binding (pKi = 8.95, Table 3).
Antagonism of dopamine-stimulated [35S]GTPγS binding to CHO-hD2 and -hD3 membranes

Antagonism of hD3 and hD2receptor-mediated G protein activation. Antagonism of dopamine (3 μM)-stimulated [35S]GTPγS binding at hD2 receptors and of dopamine (1 μM)-stimulated [35S]GTPγS binding at hD3 receptors (○ haloperidol, • GR 218,231, and ♦ S 14297). Points shown are means of triplicate determinations from representative experiments repeated on at least three occasions. pIC50 and pKB data from these experiments are shown in Table 4.

Competitive antagonism of hD3receptor-mediated G protein activation by GR 218,231. A, concentration-response isotherms for stimulation of [35S]GTPγS binding by dopamine at hD3receptors in the presence of increasing concentrations of GR 218,231 (• 3 nM, ♦ 10 nM, ▴ 30 nM, and ▪ 100 nM). Points shown are means of triplicate determinations from representative experiments repeated on at least three occasions. B, Schild plot of the hD3 dopamine concentration-response experiments. Points shown are mean values from at least three independent experiments performed in triplicate.
Effect of Pertussis and Cholera Toxins on hD3 Receptor Coupling.
Membranes were prepared from CHO-hD3 cells treated with PTX (100 ng/ml) or cholera toxin (1 μg/ml) for 6 h. Pretreatment with PTX reduced, but did not totally suppress, dopamine-dependent [35S]GTPγS binding: it was attenuated by about 80% (81 ± 16 fmol/mg versus 430 ± 26 fmol/mg in control), without changes in basal [35S]GTPγS binding (Fig. 8). The incomplete suppression of dopamine-stimulated [35S]GTPγS binding was not due to an insufficiently long incubation of CHO-hD3 cells with PTX. Indeed, when membranes were prepared from CHO-hD3 cells after the 6-h incubation, no subsequent incorporation of [32P]ADP-ribose was observed, indicating that all the Gαi/o proteins present had already been ADP-ribosylated (Fig. 8).
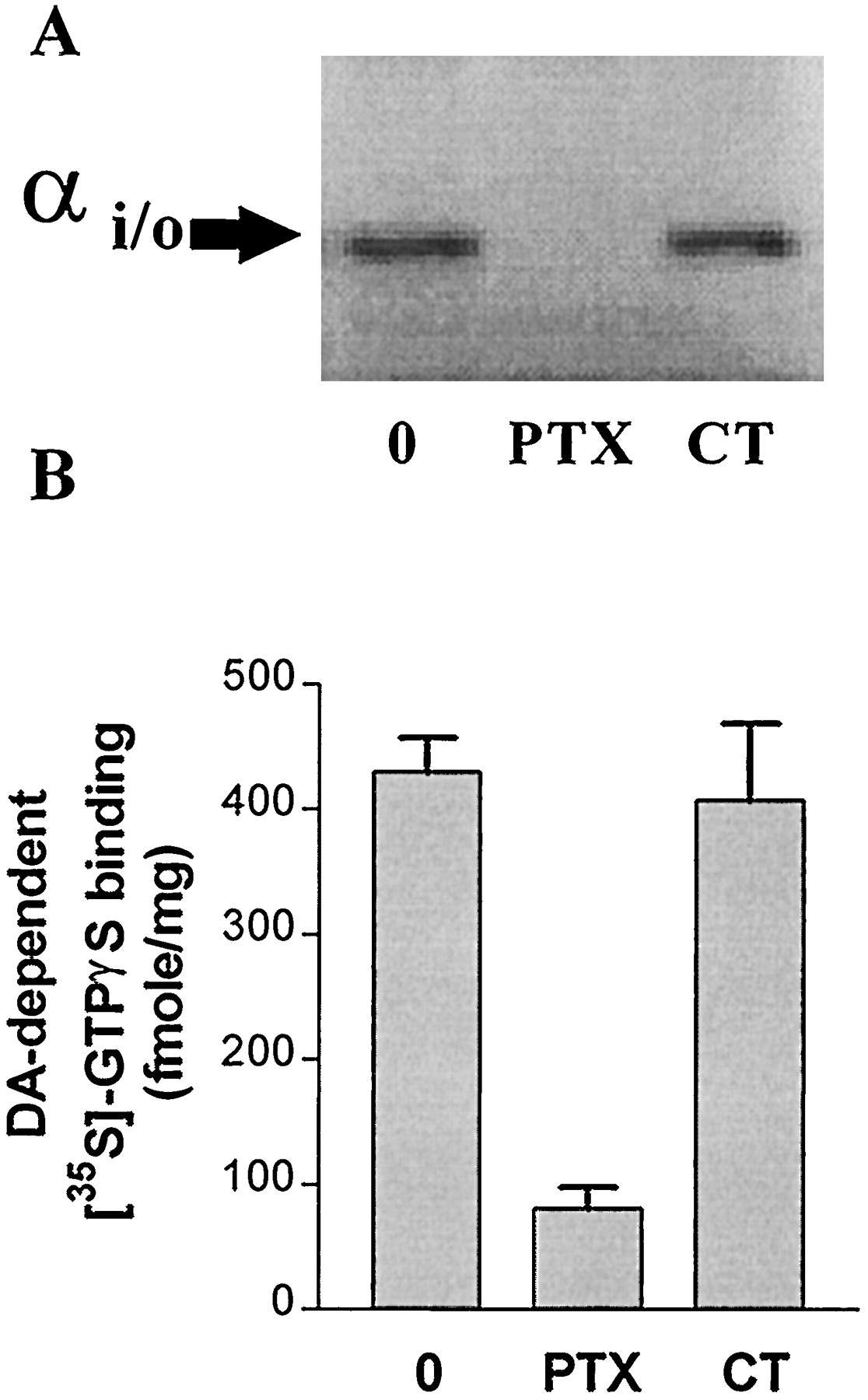
Partial attenuation of dopamine-stimulated [35S]GTPγS binding by PTX, but not cholera toxin. A, [32P]ADP-ribose incorporation catalyzed by PTX. ADP-ribosylation of CHO-hD3 cell membranes preincubated with or without PTX or cholera toxin (CT) for 6 h was carried out as described in Materials and Methods.[32P]ADP-ribosylated Gi/O proteins were revealed by a 8-h exposure to Hyperfilm. The data shows that PTX pretreatment abolished subsequent [32P]ADP-ribose incorporation. The same result was obtained in a second, independent experiment. B, Effect of PTX and CT on dopamine-stimulated [35S]GTPγS binding. CHO-hD3 cell were incubated for 6 h with PTX or CT and stimulation of [35S]GTPγS binding was determined with dopamine (10 μM). Bars represent mean ± S.E.M. values from at least three independent experiments performed in triplicate and are expressed in fentomoles per milligram of dopamine-induced [35S]GTPγS binding.
Dopamine stimulated [35S]GTPγS binding to membranes of CHO-hD3 cells treated with cholera toxin with an pEC50 of 8.04 ± 0.08 (n = 4), similar to that observed in control membranes (pEC50 = 8.00, Table 4), but basal [35S]GTPγS binding in cholera toxin-treated cell membranes was increased (1230 ± 80 fmol/mg versus 1030 ± 36 fmol/mg for control membranes). However, the amount of dopaminedependent [35S]GTPγS binding was unchanged (407 ± 61 fmol/mg versus 430 ± 26 fmol/mg in control membranes) (Fig. 8).
Effect of Antibodies on hD3 and hD2Receptor Coupling.
The presence of Gαi/o, GαS, and Gαq/11 in both CHO-hD3 and CHO-hD2 cell membranes was demonstrated by immunodetection with specific antibodies (Fig. 9). Preincubation of hD3 and hD2 cell membranes with anti-Gαi/αo subunit antiserum significantly (P < .05, Student’s pairedt test) attenuated dopamine-dependent [35S]GTPγS binding, at both hD3 and hD2 receptors (Fig.9). Anti-Gαq/α11antiserum significantly attenuated dopamine-dependent [35S]GTPγS binding to CHO-hD3 but not CHO-hD2membranes (P < .05, Student’s paired ttest). Antisera directed against GαS and an unrelated target, JNK1, did not affect [35S]GTPγS binding at either receptor (data not shown).
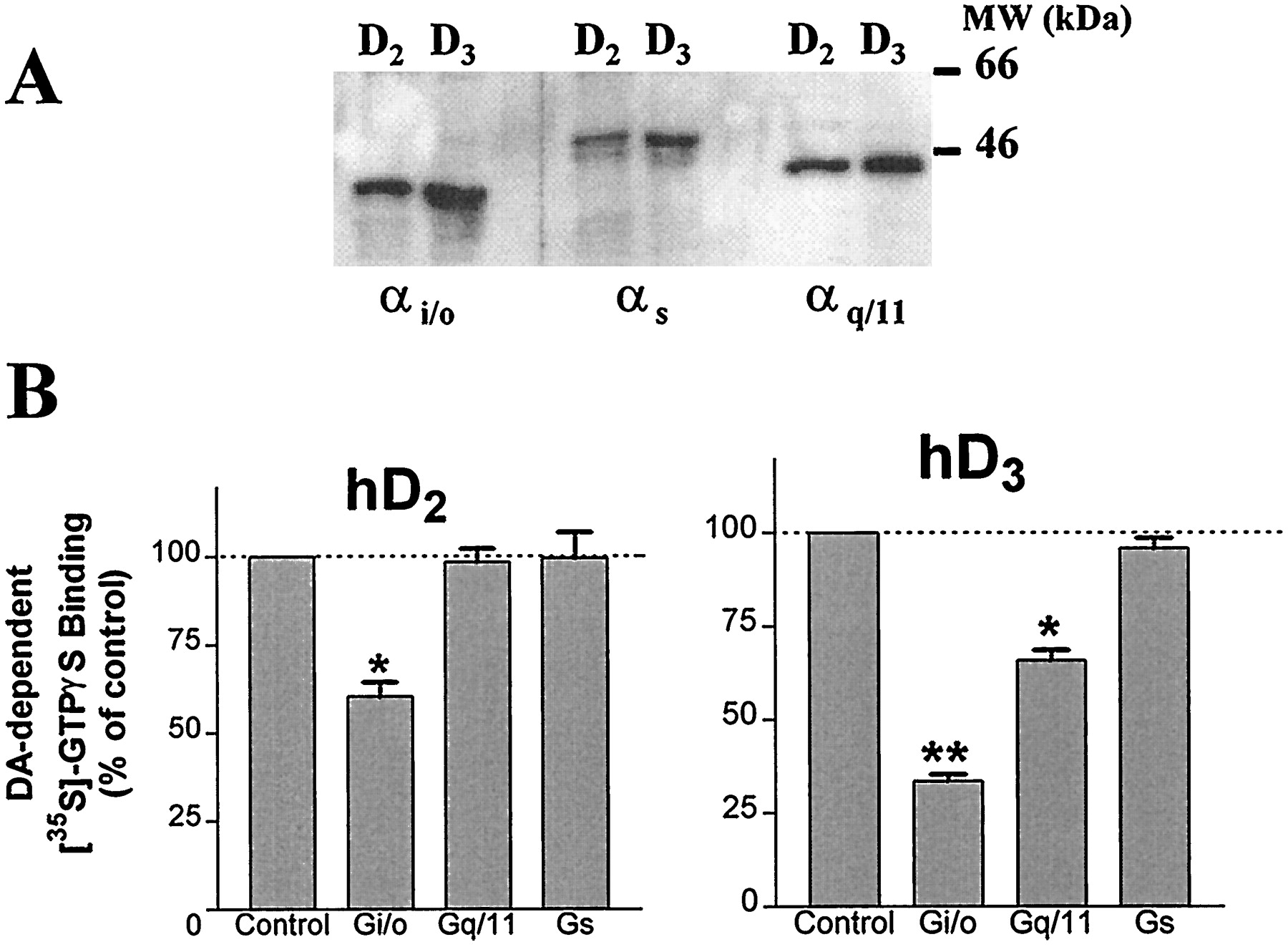
Inhibition by anti-G protein antibodies of dopamine-stimulated [35S]GTPγS binding to CHO-hD3 and CHO-hD2 cell membranes. A, immunodetection of Gαi/o, GαS, and Gαq/11 subunits in both CHO-hD3 and CHO-hD2 cell membranes was performed as described inMaterials and Methods. B, CHO-hD2 and CHO-hD3 cell membranes were incubated for 5 h at 4°C in the presence of antibodies used in A. Stimulation of [35S]GTPγS binding was determined with dopamine (10 μM). Bars represent mean ± S.E.M. values from at least three independent experiments performed in triplicate and are expressed as percentage of dopamine-dependent [35S]GTPγS binding observed in control (untreated) samples. *P < .05; **P < .01 versus control (2-tailed, pairedt test).
Discussion
The primary purpose of the present study was to investigate the G protein coupling of dopamine hD3 receptors. The results demonstrate that hD3 (and hD2) receptors mediate stimulation of [35S]GTPγS binding when expressed in mammalian CHO cells, indicating that they are capable of activating intracellular G proteins. A robust degree of stimulation was observed (Fig. 5), enabling a detailed investigation of the coupling of these receptor subtypes and the identification of some marked differences between hD3 and hD2 sites.
First, despite the 11-fold higher hD3 receptor expression level (15 pmol/mg), the dopamine-elicited increase in [35S]GTPγS binding (up to 1.6-fold) was less than that at hD2 receptors. Partial inactivation of hD3 receptors using the alkylating agent EEDQ showed that high hD3 receptor expression levels are necessary for stimulation of G protein activation, because EEDQ treatment reduced the stimulation of [35S]GTPγS binding induced by dopamine (Table2). Nevertheless, Furchgott analysis yielded a hyperbolic occupancy/response plot (Fig. 3), indicating the presence of marked receptor reserve for half-maximal stimulation of [35S]GTPγS binding by dopamine and is consistent with the 5-foldKA/EC50 ratio for activation of hD3 receptors (seeResults). Thus the limited stimulation of [35S]GTPγS binding to CHO-hD3 membranes appears to be a property of hD3 receptors themselves and not due to insufficient intrinsic efficacy of dopamine. It should be noted that the modest stimulation at hD3 receptors is not a consequence of augmented basal [35S]GTPγS binding, because basal [35S]GTPγS binding was unaffected by receptor inactivation (not shown). Furthermore, the low-fold stimulation in CHO-hD3 membranes is unlikely to be due to a global lack of activatable G proteins: the amount (Bmax) of dopamine-activated G proteins in CHO-hD3 cell membranes is about 3-fold higher than that in CHO-hD2 cell membranes (Table 1 and Fig. 1). Taken together, the present data suggest that stimulation of hD3 receptors less effectively induces the conformational changes necessary for G protein activation than at hD2 receptors (Chio et al., 1994), perhaps due to a slower rate of G protein coupling/uncoupling at hD3 receptors or, alternatively, to interaction with different G protein subtypes at hD3 versus hD2 receptors, a possibility discussed below.
Second, agonist efficacy varied between hD2 and hD3 receptors. S 14297, previously characterized as a D3 receptor antagonist in vivo (Millan et al., 1995a,b) exhibited residual intrinsic activity at hD2 receptors (Table 4) but no detectable agonist activity at hD3 receptors. In the present high-expressing CHO-hD3 cell membranes, it might have been expected that partial agonist actions at hD3 receptors would be “amplified” to yield maximal activation of [35S]GTPγS binding, like dopamine. Nevertheless, both (+)-7-OH-DPAT and PD 128,907 behaved as partial agonists at hD3 receptors (Table 4 and Fig. 5). It therefore appears that despite the high levels of hD3 receptors, the high R/G ratio in CHO-hD3 membranes, and, as discussed above, receptor reserve for activation by dopamine, the present [35S]GTPγS binding methodology more readily differentiates partial agonist efficacies at hD3receptors than certain downstream models of hD3(or hD2) receptor activation, such as mitogenesis (Sautel et al., 1995), where dopamine, (+)-7-OH-DPAT and PD 128,907 all behaved as full agonists. Furthermore, in the present study, the degree of selectivity of (+)-7-OH-DPAT and PD 128,907 for activation of hD3 versus hD2 receptors was greater than previously reported (Levant, 1997). (+)-7-OH-DPAT displayed an hD3/hD2EC50 ratio of 77 (Table 4) compared with 14 and 7 for inhibition of adenylyl cyclase and mitogenesis experiments respectively (Chio et al., 1994; Sautel et al., 1995). PD 128,907 was 382-fold as selective in this study compared with only 6-fold as selective in mitogenesis experiments (Pugsley et al., 1995). The source of these differences is unclear but probably relates to the fact that mitogenesis and extracellular acidification measure responses that are distal to agonist-induced receptor/G protein conformational changes. In contrast, [35S]GTPγS binding measures G protein activation, which provides a more proximal indication of agonist/antagonist actions at the receptor itself.
Third, the antagonist rank order of potency of haloperidol, S 14297, and the novel selective antagonist, GR 218,231 (Murray et al., 1996; Figs. 6 and 7) at hD3 and hD2 receptors corresponded to the order of affinity determined in competition binding experiments, although a reduced preference for hD3 sites was observed in functional tests (Table 5). It is noteworthy that, whereas the pKB values of the antagonists resembled their respective pKi values, the antagonists did not exhibit negative efficacy at either hD3 or hD2 receptors at concentrations up to 10−5m. At higher concentrations, [35S]GTPγS binding was somewhat reduced below basal levels in some experiments but this was taken to be a nonspecific effect, because it occurred at concentrations >1000-fold greater than their binding affinity, the effects did not show a discernible correlation with the order of potency, and a similar trend was observed in untransfected CHO cell membranes (D. Cussac, unpublished observations). A recent study using hD3receptors expressed in CHO cells (Malmberg et al., 1998) reported that basal [35S]GTPγS binding could be increased by dopaminergic agonists and decreased by antagonists. However, in that study dopamine-induced stimulation was very low (only ∼1.2-fold), negative efficacy was only observed at very high drug concentrations (≥10−6m), and control untransfected CHO cells were not examined. Nevertheless, further investigation of the issue of negative efficacy is desirable, because the conditions used for [35S]GTPγS binding both in the present study and that of Malmberg et al. (1998)(high concentrations of GDP and NaCl) favor suppression of constitutive hD3 and hD2 receptor activation (Gardner et al., 1996). Indeed, some studies reported that haloperidol shows negative efficacy in models of D3 and D2 receptor activation (mitogenesis, Griffon et al., 1996; prolactin secretion,Nilsson et al., 1996).
Fourth, G protein activation by dopamine at hD3receptors is PTX sensitive (Fig. 8), implicating Gi/o G proteins. This is analogous to the known PTX sensitivity of D2 receptors (Neve et al., 1989; Lajiness et al., 1993; Seabrook et al., 1994; Swarzenski et al., 1996; Hall and Strange, 1997). However, marked differences were observed between hD3 and hD2 receptors in antibody tests. In the present study, dopamine-stimulated [35S]GTPγS binding at hD3 and hD2 receptors was inhibited by an antiserum that recognizes the three αi subunits (αi1/i2/i3) and, more weakly, αO subunits (Cussac et al., 1996). This antiserum inhibited dopamine-stimulated [35S]GTPγS binding to CHO-hD3 membranes more strongly than to CHO-hD2 membranes (67% versus 40% inhibition, Fig. 9). The greater effect at hD3 receptors may be due to a coupling by hD3 to both Gi and GO proteins, whereas hD2 receptors may couple only to members of the Gi protein family. This would be consistent with a study that found that an attenuation of D2receptor-mediated inhibition of adenylyl cyclase activity was achieved by pretreatment with anti-Gαi1/i2 but not by anti-GαO antibodies (Izenwasser and Côté, 1995). Alternatively, hD3receptor functional coupling to G proteins may be more labile than that at hD2 receptors. Thus, the hD3 receptor/G protein interaction may be more susceptible to the steric hindrance of antibody binding to Gα subunits, in accordance with the apparently less “efficient” G protein coupling of hD3 receptors discussed above.
Fifth, an interaction of hD3 receptors with a G protein other than Gi or GOis suggested by the observation that, when CHO-hD3 cells were treated with PTX, there remained a residual capacity of dopamine to stimulate [35S]GTPγS binding. This was not due to an insufficient incubation period with PTX, because control experiments (Fig. 8) indicated that 6 h were sufficient to completely ADP-ribosylate Gi/o proteins in CHO-hD3 cells, in agreement with previous studies in pituitary cells (Cussac et al., 1996). Thus, a component of dopamine-dependent [35S]GTPγS binding at hD3 receptors may be mediated by a G protein that is not PTX sensitive. This possibility is supported by the inhibition of dopamine-dependent [35S]GTPγS binding at hD3 receptors by anti-Gαq/11 antibodies (Fig. 9). Such an effect was not observed at hD2 receptors, although αq/11 (as well as αi/Oand αS) subunits are expressed in both cell lines (Fig. 9). Furthermore, the inhibition by anti-Gαq/11 antibodies is not simply due to nonspecific Ig interactions, because the same concentration of antisera against an unrelated target (JNK1) did not affect dopamine-dependent [35S]GTPγS binding (not shown). Thus, these data suggest that hD3 receptors in the present CHO-hD3 cell line may interact with Gαq/11 and, potentially, modulate phosphatidyl inositol turnover. Although a previous study of hD3 receptors expressed in CHO cells did not find such an effect (Freedman et al., 1994), that may have been due to a 50-fold lower hD3 expression level (0.3 versus 15 pmol/mg in the cells used in this study).
In conclusion, the [35S]GTPγS binding strategy employed in this study enabled the characterization of G protein coupling at hD3 receptors. The data suggest that the coupling of hD3 receptors to G proteins is less efficacious than that at hD2receptors, yielding a less pronounced stimulation of [35S]GTPγS binding despite the high expression levels of receptors and G proteins, and the presence of receptor reserve for dopamine. In addition, unlike hD2 receptors, hD3receptors may couple to G proteins other than Gi, such as GO and/or Gq/11proteins. The precise G protein subtypes involved in hD3 receptor coupling and their relevance to physiological actions require further investigation.
Acknowledgment
We thank Lucy Sezguin for quality technical assistance.
Footnotes
-
Send reprint requests to: Adrian Newman-Tancredi, Ph.D., Department of Psychopharmacology, Institut de Recherches Servier, 125, Chemin de Ronde, 78290 Croissy-sur-Seine Paris, France. E-mail:newman_tancredi{at}hotmail.com
-
↵1 These two authors made equivalent contributions to this work.
- Abbreviations:
- EEDQ
- N-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline
- GR 218
- 231, 2(R,S)-(dipropylamino)-6-(4-methoxyphenylsulfonylmethyl)-1,2,3,4- tetrahydronaphtalene
- [35S]GTPγS
- guanosine-5′-O-(3-[35S]thio)-triphosphate
- (+)7-OH-DPAT
- 7-hydroxy-2-(di-n-propylamino)tetralin
- PD 128
- 907, (+)-(4aR,10bR)-3,4,4a,10b-tetrahydro-4-propyl-2H,5H-[1]benzopyrano[4,3-b]-1,4-oxazin-9-ol
- S 14297
- (+)-[7-(N, N-dipropylamino)-5,6,7,8-tetrahydro-naphtho(2,3b)dihydro-2,3-furane]
- Received July 8, 1998.
- Accepted December 16, 1998.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}