


Abstract
We present evidence that stimulation of the human beta-3 adrenergic receptor (AR), expressed in Chinese hamster ovary/K1 cells, specifically activates the mitogen-activated protein kinases extracellular signal-regulated kinase (ERK)1 and 2, but not JNK or p38. The extent and kinetics of the ERK stimulation by the beta-3 AR are identical with those of the endogenic insulin receptor. However, insulin augments cellular proliferation, whereas beta-3 AR agonists inhibit proliferation due to the production of cyclic AMP. The pharmacological profile of the ERK activation by the beta-3 AR differs significantly from its activation of adenylyl cyclase. The order of potency and intrinsic activities of both natural ligands, norepinephrine and epinephrine, is inversed between both signaling pathways. In addition, BRL 37344 and propranolol, ligands that act as agonists in the stimulation of cyclase, act as antagonists for ERK activation. The activation of ERK1/2 is sensitive to pertussis toxin, suggesting that the beta-3 AR, in addition to its interaction with Gs, can couple to Gi/o. Furthermore, the activation of ERK by the beta-3 AR is sensitive to PD98059, wortmannin, and LY294002, indicating a crucial role for mitogen-activated protein kinase kinase and phosphatidylinositol-3 kinase (PI3K), respectively. Abeta-3 AR-mediated stimulation of PI3K is confirmed by the observation that the selective agonist CGP 12177A specifically activates protein kinase B. As was observed for the activation of ERK, the activation of protein kinase B is inhibited by preincubation with pertussis toxin and PI3K inhibitors, suggesting that both are a consequence of a Gi/o-mediated activation of PI3K.
The G protein-coupled beta-3 adrenergic receptor (AR) is primarily expressed in adipose tissues. In rodents, stimulation of thebeta-3 AR leads to a robust activation of Gs, resulting in stimulation of adenylyl cyclase and elevation in intracellular cyclic AMP (cAMP) concentration. The resulting activation of protein kinase A (PKA) induces phosphorylation and modulation of the activity of several target proteins, such as hormone-sensitive lipase that stimulates fat cell lipolysis (Strosberg, 1997b).
In human white adipose tissue, different results have been obtained concerning the functional role of the beta-3 AR. Although several investigators have observed a lipolytic activity ofbeta-3 AR agonists (Lonnqvist et al., 1993; Hoffstedt et al., 1995), others found no or only weak beta-3 AR activity (e.g., Rosenbaum et al., 1993; Tavernier et al., 1996). In human brown adipocytes, the beta-3 AR appears to be a predominant AR subtype, but it is poorly coupled to adenylyl cyclase (Zilberfarb et al., 1997; Jockers et al., 1998). This prompted us to investigate whether other signaling pathways are associated with this receptor.
G protein-coupled receptors generally signal through fast modulations of the intracellular concentration of second messengers like cAMP, diacylglycerol, inositol trisphosphate, and calcium. Certain G protein-coupled receptors can, however, also signal through tyrosine kinase signaling pathways (for a recent review, see Gutkind, 1998). This type of signaling, mostly activated by members of the superfamily of tyrosine kinase receptors, modulates long-term cellular responses associated with adaptations to, for example, growth or stress factors. The mitogen-activated protein kinase (MAPK)/ extracellular signal-regulated kinase (ERK)1/2 pathway is known to be involved in the control of proliferation and differentiation of many cell types, including adipocytes (Sale et al., 1995), and several studies have shown that G protein-coupled receptors can directly or indirectly influence these processes via modulation of the activity of the ERK1/2 pathway.
A role for the beta-3 AR in the regulation of adipocyte proliferation or differentiation has been suggested previously. Treatment with beta-3 agonists results in an increase in brown adipose tissue in rodents (Ghorbani et al., 1997) and dogs (Champigny et al., 1991). In cultured mouse brown adipocytes, activation of the beta-3 AR stimulates adipocyte differentiation (Bronnikov et al., 1992). In humans, the presence of brown adipose tissue is restricted to perinatal life but may reappear and develop in patients with pheochromocytoma, a tumor that secrets catecholamines (Himms-Hagen, 1990). In cultured human adipocytes, UCP1 expression is increased by beta-3 AR agonists, suggesting that dormant brown adipocytes are reactivated (Champigny and Ricquier, 1996). Taken together, these data indicate that the beta-3 AR can couple to pathways linked to cell growth and differentiation and led us to investigate the effect of beta-3 AR stimulation on the ERK1/2 signaling pathway.
In this report, we show that stimulation of the human beta-3 AR, expressed in Chinese hamster ovary (CHO)/K1 cells, specifically activates ERK1/2. This activation is mediated by the coupling of the receptor to a pertussis toxin (PTX)-sensitive G protein and is independent from the activation of adenylyl cyclase. Interestingly, we found significant differences in the pharmacological profile of the Gi/o-mediated increase in ERK activation compared with the Gs-mediated increase in cAMP. In addition, evidence is presented that this activation is mediated via the activation of phosphatidylinositol-3 kinase (PI3K).
Materials and Methods
Cells.
CHO/K1 cells stably expressing the humanbeta-3 AR (B max = 2.3 pmol/mg protein) were used as described previously (Gros et al., 1998). A second CHO/K1-beta-3 AR clone, expressing 470 fmol/mg protein, was used as a control.
SDS-Polyacrylamide Gel Electrophoresis (PAGE) and Immunoblot Analysis.
Cells were grown in multiwell (6) plates until semiconfluency, starved overnight, and stimulated with different ligands for 5 min at 37°C. The cells were washed once with ice-cold phosphate-buffered saline containing 1 mM orthovanadate and scraped in 80 μl of lysis buffer (125 mM Tris·HCl, pH 6.8, 4% SDS, 5% glycerol, 50 mM dithiothreitol, 1 mM orthovanadate, and 0.05% bromophenol blue). The samples were boiled for 5 min and loaded on an SDS-polyacrylamide gel containing 10% acrylamide and 0.13% bisacrylamide. The separated proteins were transferred to a nitrocellulose membrane (BDH), which was incubated with 1 μg/ml monoclonal antibody specifically recognizing ERK2 (Euromedex/UBI). ERK1 activity was revealed by an antibody recognizing only the phosphorylated form of both ERK1 and ERK2 (Promega, Madison, WI). P38 activity was revealed by an antibody recognizing the dually phosphorylated, active form of p38 (Promega Biotec). Protein kinase B (PKB) activity was revealed with an antibody recognizing only the (Ser) phosphorylated form of PKB (Bio-Lab, St. Paul, MN). After washing, the membranes were incubated with anti-mouse or anti-rabbit IgG peroxidase-coupled antibodies (Amersham Corp., Arlington Heights, IL), and signals were revealed using enhanced chemiluminescence (Amersham).
In Vitro JNK Activity Assay.
The activity of JNK was determined as described recently (Etienne et al., 1998). In short, cellular extracts of Gst-Jun overexpressing Escherichia coli were incubated overnight with agarose beads. Cells were stimulated, extracted, and overnight incubated with the agarose-GST-Jun beads to precipitate JNK. The Jun/JNK beads were incubated with [γ-32P]ATP (NEN, Arlington, MA) for 30 min at 30°C, and the level of phosphorylation of Jun was determined after SDS-PAGE and autoradiography of the blotted gel.
Growth Curves.
Cells were seeded in multiwell (24) plates, and 24 h later, the medium was changed as indicated. The total amount of cells per well was determined using a Coulter Counter (Coulter Z1); 3 wells per time point were counted each 24 h. Experiments were done in triplicate.
In Vitro ERK Activity Assay.
For a quantitative determination of the activity of ERK, an in vitro kinase assay was used. Cells were grown in 60-mm dishes until semiconfluency and starved overnight. After the addition of ligands for 5 min at 37°C, the cells were washed with ice-cold phosphate-buffered saline containing 1 mM orthovanadate and scraped in 100 μl of lysis buffer [80 mM β-glycerophosphate, 20 mM EGTA, 15 mM MgCl2, 1 mM orthovanadate, 1 mM 4-(2-aminoethyl)benzenesulfonyl fluoride, 10 μg/ml leupeptin, 10 μg/ml aprotinin, and 10 μg/ml pepstatin]. Cellular extracts were sonicated and centrifuged, and the soluble protein content was determined using DC Protein Assay (Bio-Rad, Hercules, CA). Six to 10 μg of protein was incubated with 1 μmol of a synthetic peptide substrate of MBP (Neosystem Laboratoire) and 5 μCi of [γ-32P]ATP (NEN) for 10 min at 30°C. The reaction was stopped by adding 8% trichloroacetic acid and 0.3% bovine serum albumin, and the samples were centrifuged for 10 min at 13,000 rpm at 4°C. Six microliters of the supernatant was spotted onto Whatman P81 chromatography paper, dried, and washed three times in 175 mM phosphoric acid. The [γ-32P] incorporated into the peptide was measured by β-scintillation counting.K act values were calculated from at least three independent stimulations using the program GraphPad Prism (GraphPad Software Inc., San Diego, CA).
cAMP Assay.
The intracellular concentration of cAMP was determined as described previously (Gros et al., 1998).
Toxin Treatments.
The following agents have been used to explore the signaling pathway underlying the beta-3 AR-mediated activation of ERK: PTX (Sigma Chemical, St. Louis, MO), forskolin (Coger), H89 (Calbiochem Corp., La Jolla, CA), wortmannin (Sigma), LY 294002 (BIOMOL Research Laboratories, Plymouth Meeting, PA), PD 98059 (BIOMOL), 12-o-tetradecanoyl-phorbol 13-acetate (TPA) (Sigma), 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid-AM (Sigma), genistein (ICN Biomedicals, Costa Mesa, CA), and herbimycin (ICN Biomedicals). The conditions of incubation are specified in the figure legends.
Beta Adrenergic Ligands.
The following ligands were used: (−)-norepinephrine (Sigma), (−)-epinephrine (Sigma), (−)-isoproterenol (Sigma), CGP 12177A (generously provided by Ciba-Geigy), CL 316,243 (generously provided by Cyanamid), carazolol (Boehringer Mannheim, Indianapolis, IN), BRL 37344 (generously provided by SmithKline Beecham), (−)-propanolol (Sigma), and (−)-bupranolol (Schwarz Pharma AG).
Results
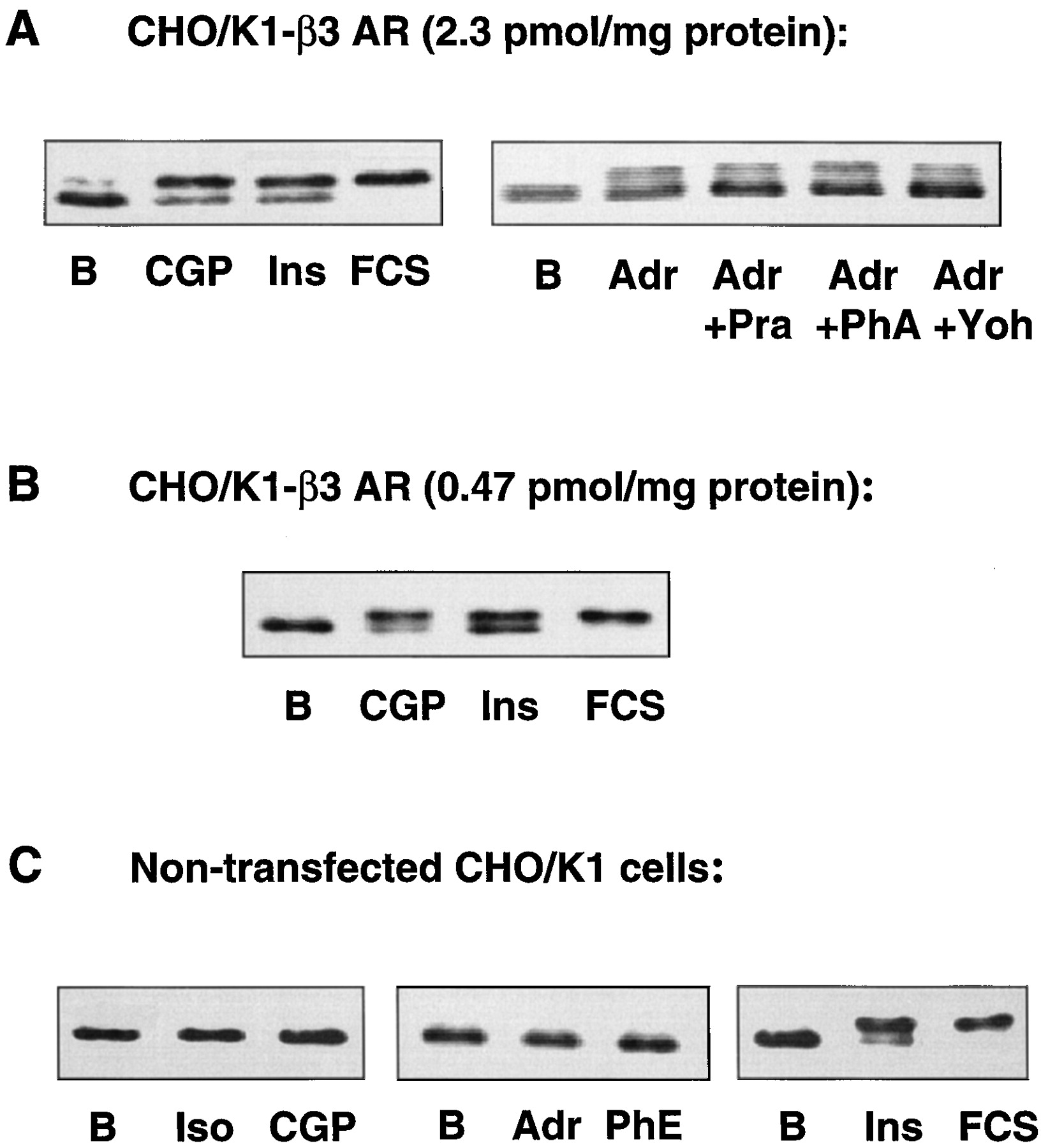
When CHO/K1 cells stably expressing the human beta-3 AR (B max = 2.3 pmol/mg protein) were stimulated with the beta-3 AR-selective agonist CGP 12177A, the activity of ERK2 was increased significantly (Fig.1A). The activity of ERK2 also increased after incubation of CHO/K1-beta-3 cells with epinephrine, and this activity was not inhibited by the alpha AR antagonists prazosin, phentolamine, or yohimbin (Fig. 1A). Stimulation of a CHO/K1 cell line exhibiting a lower beta-3 AR density (470 fmol/mg protein) similarly activated ERK2 (Fig. 1B). In nontransfected CHO/K1 cells, neither CGP 12177A nor the nonselective beta AR agonist isoproterenol induced a change in ERK2 activity. In addition, nontransfected CHO/K1 cells did not respond to epinephrine or to the alpha AR agonist phenylephrine (Fig. 1C). These results indicate that the activation of ERK2 by the beta-3 AR-specific agonist CGP 12177A is an effect specifically mediated by the beta-3 AR.
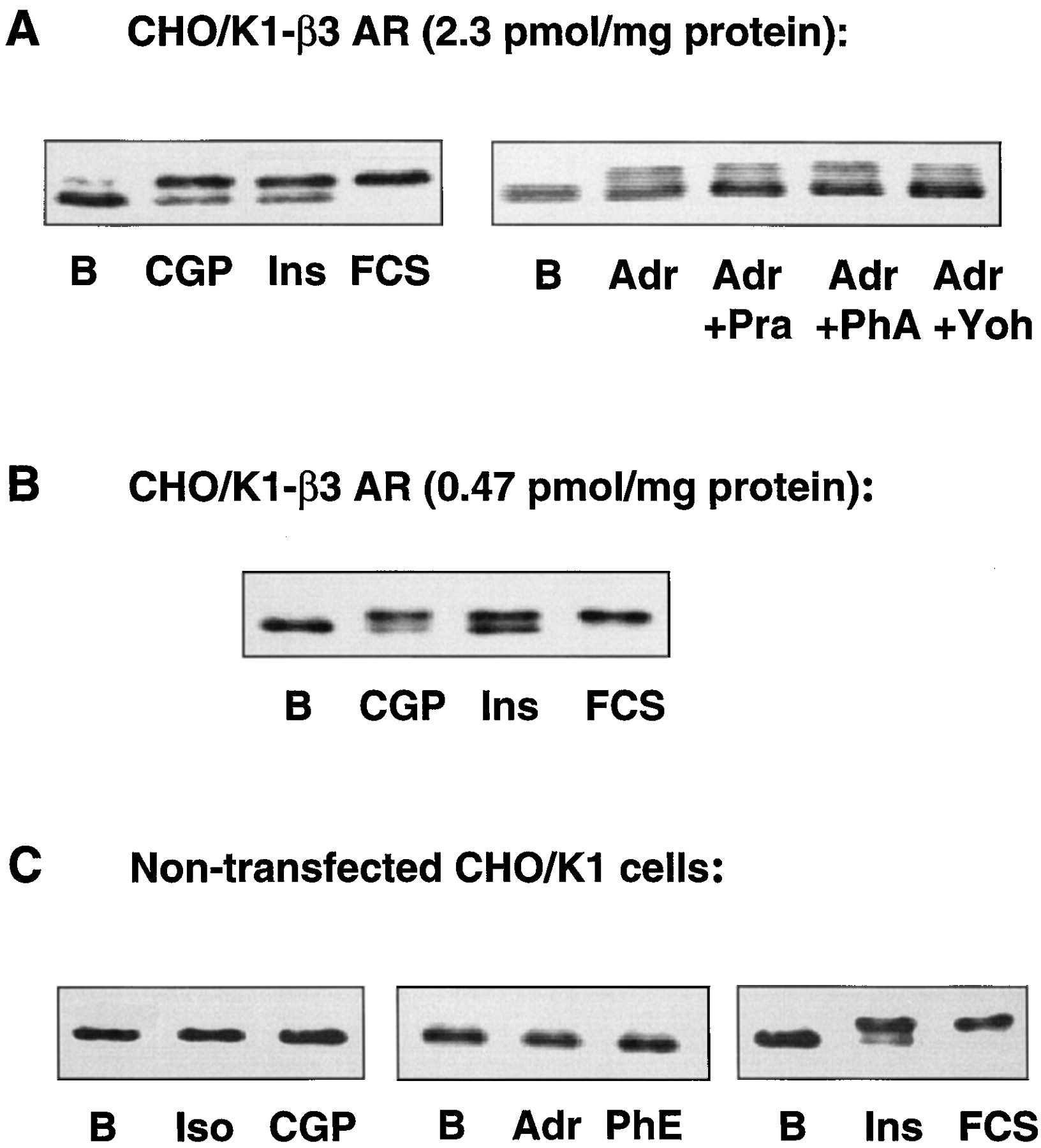
Band-shift assays indicating ERK2 phosphorylation in nontransfected CHO/K1 cells (A), CHO/K1 cells expressing humanbeta-3 AR at 2.3 pmol/mg protein (B), or CHO/K1 cells expressing human beta-3 AR at 0.47 pmol/mg protein (C). Cells were incubated with ligands for 5 min. Iso, 1 μM isoproterenol; CGP, 10 μM CGP12177A; Adr, 1 μM epinephrine; PhE, 1 μM phenylephrine; Ins, 1 μM insulin; FCS, 10% fetal calf serum; Pra, 2 μM prazosin; PhA, 2 μM phentolamine; and Yoh, 2 μM yohimbine. Whole cellular lysates were run on a 10% polyacrylamide gel and blotted. Blots were incubated with a polyclonal antibody recognizing both the phosphorylated and the nonphosphorylated forms of ERK2.
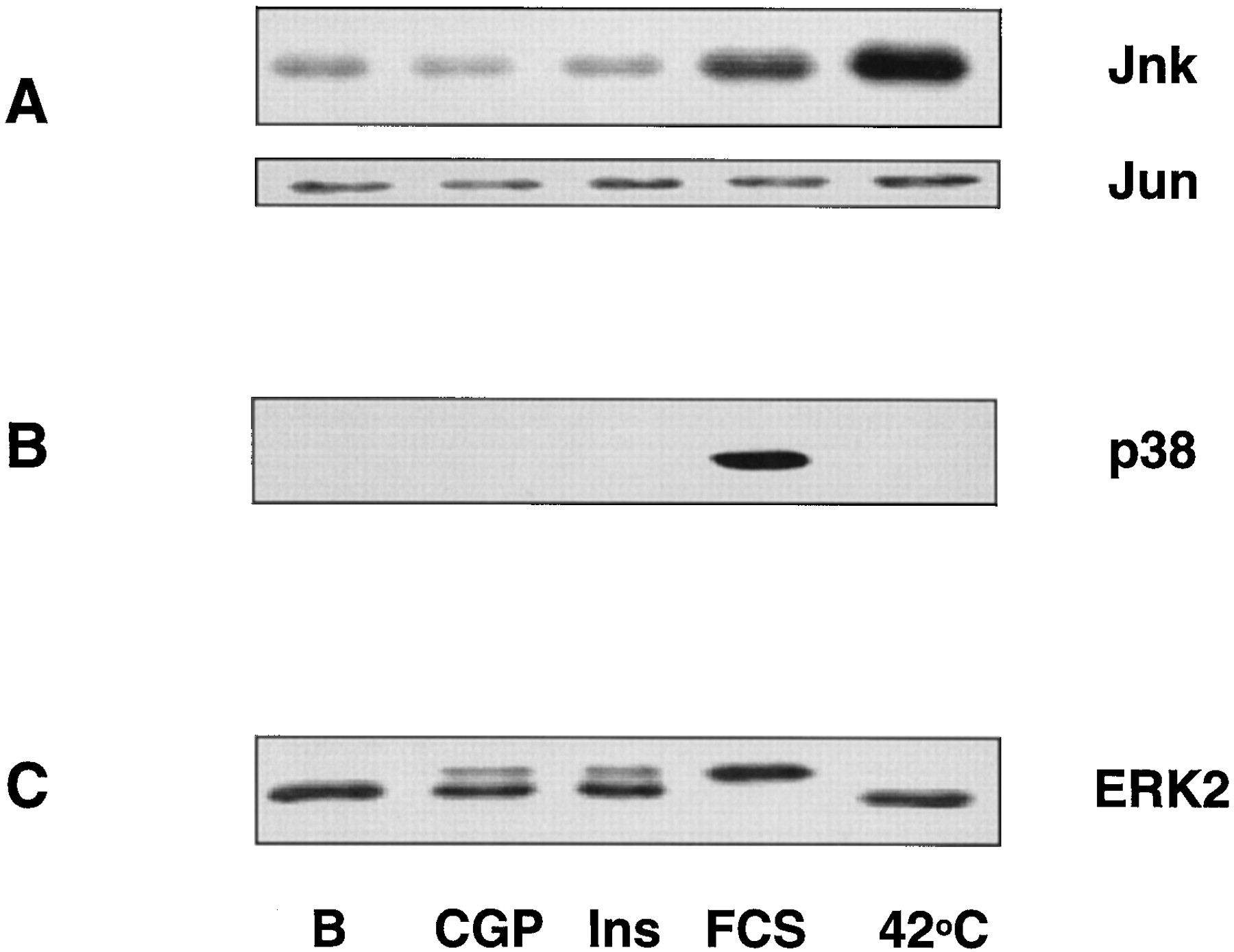
ERK belongs to the family of MAPKs, of which JNK and p38 are well characterized homologous members. To study whether ERK1/2 was the only MAPK pathway activated by the beta-3 AR, we tested the activity of JNK and p38 after stimulation of the beta-3 AR in CHO/K1 cells. Cells were stimulated 5, 15, 30, or 60 min with 10 μM CGP 12177A. As positive controls, we used a heat shock (20 min at 42°C) to activate JNK or 10% fetal calf serum to activate p38 and ERK. Figure 2 shows that stimulation of the beta-3 AR did not lead to a significant increase in the activity of either JNK or p38. In some experiments, however, a very faint activation of p38 appeared after overexposure of the films, at 5 min of incubation with CGP 12177A.

Effect of stimulation of the humanbeta-3 AR expressed in CHO/K1 cells on the activity of JNK (A), p38 (B), and ERK2 (C). CHO/K1-beta-3 AR cells were stimulated for 5 min with 10 μM CGP 12177A (CGP), 1 μM insulin (Ins), or 10% fetal calf serum (FCS) or were incubated at 42°C for 20 min. After extraction of the cellular lysates, JNK was precipitated using GST-Jun agarose beads, and the JNK activity was monitored by phosphorylation of Jun with [γ-32P]ATP, SDS-PAGE, and autoradiography of the Western blot (A). Bottom, equal amounts of Jun precipitated in the different samples. After precipitation with GST-Jun agarose beads, the remaining supernatants were run on SDS-PAGE and blotted. Blots were incubated with antibodies recognizing the phosphorylated form of p38 (B) or both the phosphorylated and the nonphosphorylated forms of ERK2 (C).
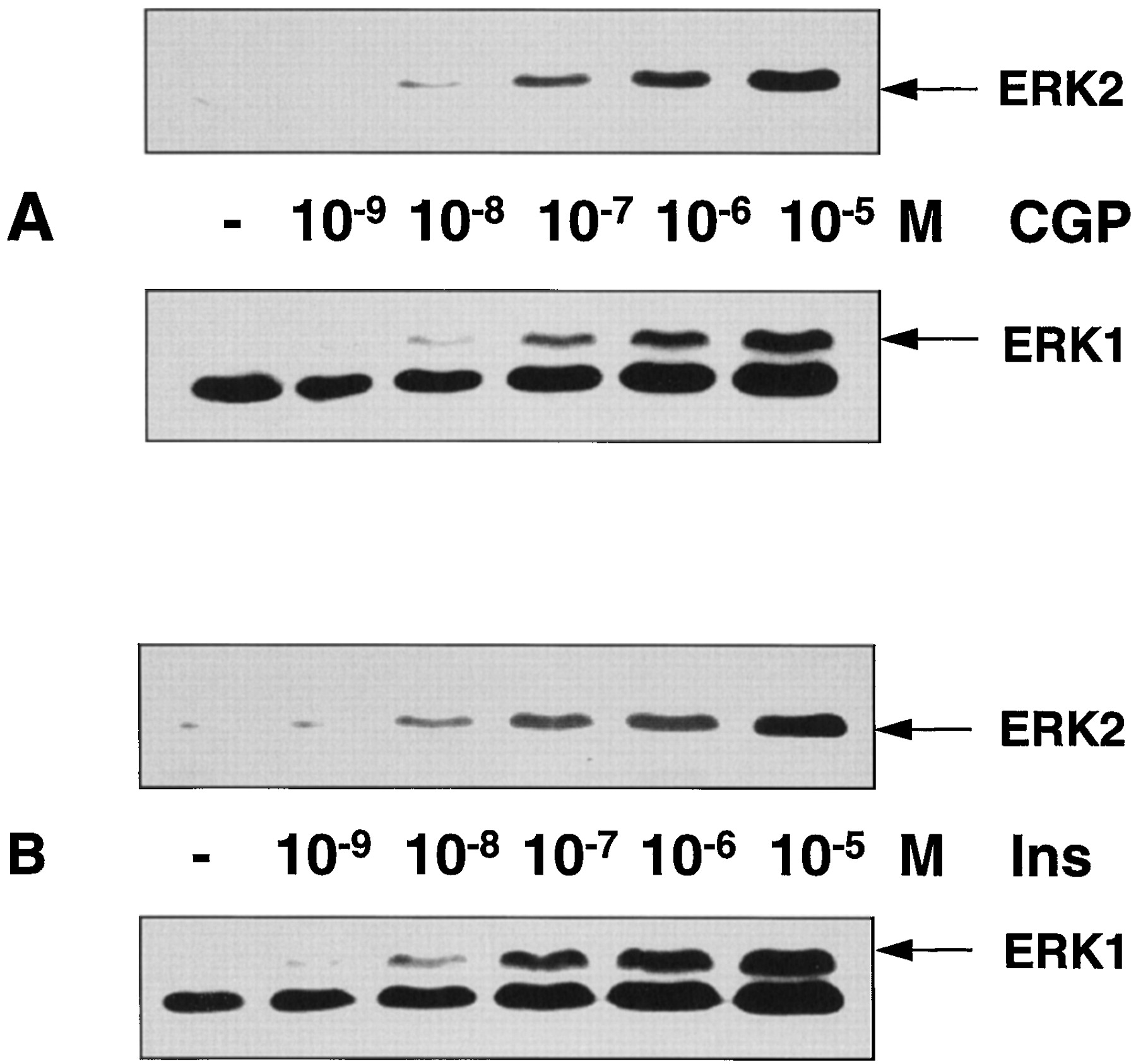
Because two isoforms of ERK exist, ERK1 (p44) and ERK2 (p42), we studied whether both forms were activated by stimulation of thebeta-3 AR. Using an antibody that recognizes only the activated form of both ERK1 and ERK2, we found that both isoforms of ERK were activated by the beta-3 AR, without any change in dose dependency (Fig. 3A). The intensity of this response was similar to that after stimulation of endogenous insulin receptors (Fig. 3B).

Dose-dependent activation of ERK1 and ERK2 by CGP 12177A (A) and insulin (B). Whole cellular lysates were run on a 10% polyacrylamide gel and blotted. Blots were incubated with a monoclonal antibody recognizing the phosphorylated forms of ERK1 and ERK2. Because the expression level of ERK1 is lower than that of ERK2, activated ERK1 is visualized after longer exposure of the same blots. Thus, the band located below the ERK1 signal corresponds to the saturated signal given by ERK2.
The kinetics of ERK activation by CGP 12177A and insulin were found to be similar, with a peak of activation after 5 min of stimulation, followed by a lower level of activation for at least 1 h (Fig. 4, A and B). Simultaneous incubation of insulin and CGP 12177A led to an additive stimulation of ERK (Fig. 4C).

Kinetics of the activation of ERK2 by 10 μM CGP 12177A (A), 1 μM insulin (B), or 10 μM CGP 12177A plus 1 μM insulin (C). CHO/K1-beta-3 AR cells were incubated with agonists for 1, 5, 15, 30, or 60 min. Whole cellular lysates were treated as described in the legend to Fig. 1.
Because the activation of ERK leads to a stimulation of cellular proliferation in many cells, we tested whether activation of ERK by either CGP 12177A or insulin leads to an increase in growth of CHO/K1-beta-3 AR cells. Figure5A shows that in the absence of any other growth factors, insulin stimulates the proliferation of CHO/K1-beta-3 AR cells. In contrast, CGP 12177A significantly decreases the number of cells in either the presence or absence of insulin. To determine whether the decrease in cell number was a consequence of the beta-3 AR-mediated activation of adenylyl cyclase, we stimulated this enzyme directly with 1 μM forskolin. Indeed, the number of cells decreased in time after incubation with forskolin; after 72 h of incubation, forskolin treatment lowered the number of cells in either the absence (48 ± 8%, n = 7) (Fig. 5B) or presence (21 ± 8%,n = 3) of insulin (data not shown). The decrease in cell number after treatment with either forskolin or CGP 12177A was found to be mediated by PKA in that incubation with 10 μM H89 reversed this effect (Fig. 5B). In the absence of H89, a 72-h treatment with CGP 12177A led to a decrease in cell number of 34 ± 2% (n = 6). In contrast, in the presence of H89, the number of cells incubated with CGP 12177A for 72 h represented 96 ± 9% (n = 4) of the cells treated with H89 alone.

Representative experiments showing the number of CHO/K1 cells expressing human beta-3 ARs in the absence of serum. A, cells were counted every 24 h while being incubated in the absence (■) or presence of 10 μM CGP 12177A (•), 1 μM insulin (▪), or both (○). B, relative number of cells cultured for 72 h in the absence or presence of 10 μM CGP 12177A (CGP) or 1 μM forskolin (Fsk). Light bars represent the number of cells treated with 10 μM H89 for 72 h.
The pharmacological characteristics of several beta AR ligands to activate or block the stimulation of ERK by thebeta-3 AR was evaluated using an in vitro kinase assay. The kinase activity of ERK was determined in response to agonist concentrations ranging from 10−10 to 10−5 m, which yielded the following rank order of EC50 values: isoproterenol < epinephrine, carazolol < CL 316,243 < norepinephrine, CGP 12177A (Fig. 6 and Table 1). BRL 37344, propranolol, and bupranolol were inactive and antagonized the effect of epinephrine on ERK activity. Interestingly, in the same cells, the rank order of potency for the activation of adenylyl cyclase was found to be different: isoproterenol < CGP 12177A < carazolol, norepinephrine < BRL 37344 < CL 316,243 < epinephrine < propranolol (Table 1). An especially striking difference was found when the EC50 values of epinephrine and norepinephrine were compared, whereas the EC50 cAMP is higher for epinephrine (121 ± 19 nM) than for norepinephrine (7 ± 1 nM); these potencies are reversed for the activation of ERK [EC50(epinephrine) = 17 ± 7 nM and EC50(norepinephrine) = 120 ± 4 nM]. In addition, the intrinsic activity of epinephrine is significantly higher for ERK activation (intrinsic activity = 1.5) than for adenylyl cyclase activation (intrinsic activity = 0.8). Also, BRL 37344 acted strikingly different in both signaling pathways; it was a potent, full agonist for the activation of adenylyl cyclase, but it was not able to significantly increase the activity of ERK. Propranolol, a weak agonist for adenylyl cyclase stimulation, was also found to be an antagonist for ERK activation. Furthermore, theK act of CGP 12177A was higher for the stimulation of ERK than for the stimulation of adenylyl cyclase. TheK act as well as the intrinsic activity of CL 316,243 and carazolol did not significantly differ between the two signaling pathways.

Dose-dependent activation of ERK activity by catecholamines isoproterenol (○), epinephrine (•), and norepinephrine (▪) (A) or by beta-3 AR-selective agonists carazolol (▪), CL 316,243 (○), and CGP 12177A (•) (B). Cells were stimulated for 5 min with different concentrations of agonists. ERK activity was determined in vitro by incubating whole cellular lysates with a peptide serving as ERK substrate and [γ-32P]ATP.
Effect of beta-AR ligands on human beta-3 AR-mediated activation of ERK and accumulation of cAMP
To test whether the different pharmacological profiles are a consequence of different beta-3 AR-G protein couplings, we investigated whether ERK is activated by the classic beta-3 AR-mediated increase in intracellular cAMP concentration. Direct activation of adenylyl cyclase with 1 μM forskolin increased the intracellular cAMP concentration 6- to 8-fold (data not shown). However, no significant increase in ERK activity was observed. Similarly, inhibition of the activity of PKA by preincubation with H89 did not inhibit the activation of ERK (Fig.7A). This result suggests that the activation of ERK is not mediated by Gs and prompted us to investigate whether Gi/o could be involved. Preincubation of the cells with PTX, which ADP ribosylates Gαi/o subunits and blocks their activity, led to a total inhibition of the CGP 12177A-mediated activation of ERK (Fig. 7B), indicating that the beta-3 AR couples to a Gi/o protein to activate ERK.

Activation of ERK by the beta-3 AR in the presence of forskolin and H89 (A) or PTX (B). Cells were stimulated with 10 μM CGP 12177A for 5 min or 1 μM forskolin for 15 min, with or without 20 μM H89 for 45 min (A) or after preincubation with 100 ng/ml PTX for 18 h or 200 ng/ml pertussis toxin for 6 h (B).
In many cells, PKC or calcium-activated kinases play a key role in the activation of ras or MEK kinase by Gi/o-coupled receptors. We tested whether calcium is involved in the beta-3 AR-mediated activation of ERK. Incubation of the cells in phosphate-buffered saline without calcium and in the presence of 2 mM EGTA did not lead to any changes in CGP 12177A-mediated activation of ERK. Furthermore, pretreatment of the cells with 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid-AM, a cell-permeable calcium chelator, did not inhibit the activation of ERK by CGP 12177A. In addition, down-regulation of PKC after overnight incubation with TPA completely inhibited the acute TPA-induced ERK activation but did not inhibit the activation of ERK by CGP 12177A (data not shown).
The involvement of protein tyrosine kinases in the activation of ERK by the beta-3 AR was tested by incubating the cells with genistein (a general tyrosine kinase inhibitor) or herbimycin A (a Src family tyrosine kinase inhibitor). Neither of these agents significantly changed the activation of ERK by CGP 12177A (data not shown).
To investigate whether the activation of ERK by the beta-3 AR is mediated by activation of mitogen-activated protein kinase kinase (MEK), PD 98059, an inhibitor of MEK, was incubated for 30 min before the activation of the beta-3 AR. Pretreatment with PD 98059 led to a complete inhibition of the activation of ERK, induced by either CGP 12177A or insulin (Fig. 8A).

Activation of ERK by CGP 12177A or insulin in the presence of MEK inhibitor PD 98059 (A) or PI3K inhibitors wortmannin (B) and LY 294002 (C). Cells were preincubated with 50 μM PD 98059 for 30 min, 500 nM wortmannin for 20 min, or 50 μM LY 294002 for 20 min before stimulation with 10 μM CGP 12177A or 1 μM insulin for 5 min.
It has been suggested that G protein-coupled receptors activate ERK in a PI3K-dependent manner. To investigate the role of PI3K in thebeta-3 AR-mediated activation of ERK, we used two specific inhibitors, wortmannin and LY 294002. A total inhibition of the ERK activation by CGP 12177A as well as insulin was observed in the presence of wortmannin (Fig. 8B). LY 294002 completely inhibited thebeta-3 AR-mediated activation of ERK and partially inhibited the insulin-mediated activation of ERK (Fig. 8C).
Because the activation of ERK by the beta-3 AR is sensitive to PI3K inhibitors, activation of the beta-3 AR should also lead to activation of the PI3K target PKB. Using an antibody raised against the activated, Ser-473-phosphorylated form of PKB, we found that activation of the beta-3 AR indeed led to an activation of PKB (±8-fold over basal) (Fig. 9A). However, this activation was relatively small; it composed ±35% of the activation of PKB induced by serum and ±20% of the activation of PKB induced by insulin. Activation of CHO/K1 cells expressing a lower receptor density (470 fmol/mg protein) led to a similar activation of PKB (Fig. 9A). The activation of PKB by CGP 12177A was completely inhibited in the presence of PTX, wortmannin, or LY 294002 (Fig. 9B).

Activation of PKB by CGP 12177A, fetal calf serum (FCS), and insulin in CHO/K1 cells expressing humanbeta-3 ARs at 2.3 pmol/mg protein or at 0.47 pmol/mg protein (A). B, effects of PTX, wortmannin, and LY 294002 on the CGP-induced activation of PKB. Cells were stimulated for 5 min with 10 μM CGP 12177A, 10% fetal calf serum (FCS), or 1 μM insulin. Preincubations with inhibitors were as described in legends to Figs. 4and 5. Whole cellular lysates were run on a 10% SDS-PAGE and blotted. Blots were incubated with an antibody recognizing only the active, Ser-phosphorylated form of PKB.
Discussion
In this report, we show that the human beta-3 AR, expressed in CHO/K1 cells, can functionally couple to Gs and Gi/o to stimulate either cAMP production or ERK1/2 activity, respectively.
Both beta-3 AR-ligands and insulin activate ERK1/2 to the same extent and with the same kinetics. However, although insulin stimulates cell proliferation, beta-3 AR ligands inhibit proliferation. This inhibitory response was due to activation of adenylyl cyclase because it could be mimicked by forskolin. In addition, inhibition of PKA by H89 fully reversed the inhibitory effect of CGP 12177A. The mechanism by which cAMP inhibits cellular growth and/or survival in CHO/K1-beta-3 cells remains to be elucidated. Interestingly, it seems to be independent of ERK activity because beta-3 AR stimulation leads to a net increase in ERK1/2 activity and forskolin does not influence the activity of ERK in this cell type.
Using an in vitro ERK1/2-mediated kinase assay, we determined the intrinsic activity and EC50 of a number of most currently used beta-3 AR ligands. We compared these values with those obtained for the beta-3-induced increase in cellular cAMP levels (Table 1). Interestingly, the relative potency and efficacy of norepinephrine and epinephrine, the natural ligands of thebeta-3 AR, are inversed between activation of ERK and adenylyl cyclase. This ligand-selective dual signaling could provide a mechanism by which a cell can respond to different physiological stimuli via the same receptor subtype. Surprisingly, BRL 37344, which is a potent agonist in stimulation of adenylyl cyclase, blockedbeta-3 AR-mediated activation of ERK, suggesting that this ligand induces a particular receptor conformation specifically inducing one but not another signaling pathway. Propranolol, a partial agonist in stimulation of adenylyl cyclase (intrinsic activity = 0.4), also acted as antagonist in ERK activation. Isoproterenol, carazolol, and CL 316,243 were found to act similarly in both signaling pathways. These results suggest that at least three different receptor states exist: one favoring the signaling toward adenylyl cyclase (e.g., induced by norepinephrine or BRL 37344), one favoring the signaling toward ERK (e.g., induced by epinephrine), and one not discriminating between the two signaling pathways (e.g., induced by isoproterenol). It has been suggested previously that multiple active receptor conformations exist (Kenakin, 1995; Tucek, 1997). Each ligand stimulates the formation of a certain receptor conformation, and different receptor conformations may favor the coupling to different G proteins. Indeed, we have found that the activation of ERK is sensitive to PTX, implicating a role for G proteins of the Gi/o class, whereas the Gαs-mediated activation of adenylyl cyclase is not changed by PTX treatment (Pietri-Rouxel et al., 1997). The difference in pharmacology between the activation of adenylyl cyclase and the activation of ERK1/2 could thus be explained by the interaction of the beta-3 AR with two different G proteins. In addition to the ligand-induced receptor conformations, it has been suggested that the G protein may influence the conformation of the receptor and thereby change agonist efficacies (Samama et al., 1993; Kenakin, 1995).
The CHO/K1 cell line used in this work expresses a relatively high number of beta-3 receptors (2.3 pmol/mg protein). We chose this cell line to compare the EC50 values for ERK activation with those obtained for activation of cAMP that we determined previously in these cells because EC50values may depend on receptor density (Burt et al., 1996). Nevertheless, CHO/K1 cells expressing a lower beta-3 AR density (470 fmol/mg protein), corresponding to the density that was observed in immortalized human brown adipocytes (596 fmol/mg protein) (Zilberfarb et al., 1997), ERK1/2 as well as PKB are similarly activated. This suggests that the additional coupling of thebeta-3 AR to a PTX-sensitive G protein is not an artifactual coupling due to the high level of receptor expression. Moreover, activation of promiscuous signaling pathways usually requires higher agonist concentrations than those necessary to activate primary pathways. We found that several beta AR agonists possess EC50 values for the stimulation of ERK1/2 that are similar, or even lower, than those obtained for the stimulation of adenylyl cyclase. In addition, endogenously expressed beta-3 ARs have been reported to couple to PTX-sensitive G proteins, such as in rat adipocytes (Chaudhry et al., 1994) and in heart (Gauthier et al., 1996). Thus, the coupling of beta-3 ARs to a Gi/o protein observed in this work is probably not a cell line-specific effect and is likely to be of physiological significance.
The related beta-2 AR, endogenously expressed in human embryonic kidney 293 cells, was recently shown to stimulate ERK1/2 via the beta-gamma subunit of a PTX-sensitive G protein (Daaka et al., 1997). Interestingly, the coupling to Gαi required the receptor to be phosphorylated by PKA because ERK activation was sensitive to the PKA inhibitor H89. Unlike the beta-2 AR, the beta-3 AR does not require the activity of PKA because ERK activation was not inhibited by H89. This could be related to the lack of consensus sequences for PKA-mediated phosphorylation in the beta-3 AR and, in particular, to the replacement of Ser262 in the beta-2 AR by an alanine in the beta-3 AR. Ser262, when phosphorylated by PKA, has been shown to be involved in the coupling of thebeta-2 AR with Gi (Okamoto et al., 1991).
Recent studies have shed light on the molecular mechanisms by which G protein-coupled receptors can activate ERK (for a review, see Gutkind, 1998). For many G protein-coupled receptors that activate MAPK, calcium-requiring steps play a crucial role. In the case of Gi/o-coupled receptors, the activatedbeta-gamma subunit may activate phospholipase Cβ, leading to diacylglycerol-mediated increases in PKC activity, as well as phosphatidyl inositol-1,4,5-trisphosphate-mediated liberation of intracellular calcium. PKC can activate raf-1 by stimulation of formation of active ras–raf-1 complexes (Marais et al., 1998). Calcium, via activation of calcium-sensitive tyrosine kinases like Pyk-2, can activate Src family proteins that are upstream of the formation of an active Shc-Grb2-Sos complex (Dikic et al., 1996; Della Rocca et al., 1997). Interestingly, although the beta-3 AR in CHO/K1 cells functionally couples to a Gi/o protein, we found that neither PKC nor calcium is involved in the activation of ERK1/2 by thebeta-3 AR.
In contrast, we found that the activation of ERK1/2 by thebeta-3 AR is completely abolished in the presence of PI3K inhibitors wortmannin and LY 294002. PI3Kγ has been suggested to be “the missing link” between Gi/o-coupled receptors and Src or Src family members that stimulate formation of an active Shc-Grb2-Sos complex leading to ERK stimulation (Hawes et al., 1996; Lopez-Ilasaca et al., 1997). The Gβ-γ subunit can directly activate PI3Kγ (Stoyanov et al., 1995), and the second messengers phosphatidylinositol bisphosphate and phosphatidylinositol trisphosphate, generated by PI3K, may directly activate Src-like proteins via binding to their SH2 domain (Rameh et al., 1995). In this study, we show not only the sensitivity to the PI3K inhibitors but also the activation of PKB, one of the main targets of PI3K (Franke et al., 1997), by the beta-3 AR. The maximal level of PKB activation induced by the beta-3 AR is, however, much lower than that induced by the insulin receptor. This difference may reflect a different potency of the G protein-coupled PI3Kγ and the insulin receptor-coupled PI3Kα/β to activate PKB. PKB can be also activated in a way insensitive to PI3K inhibitors, such as by elevated cAMP (Sable et al., 1997). Moreover, isoproterenol has been shown to activate PKB independently of PI3K and cAMP in rat adipocytes (Moule et al., 1997). However, the activation of PKB by the humanbeta-3 AR expressed in CHO/K1 is sensitive to PTX, wortmannin, and LY 294002. This suggests that the PKB activation by CGP 12177A is a direct consequence of Gi/o-mediated PI3K activation.
In this work, we demonstrate that the beta-3 AR can be coupled to a Gi/o protein to stimulate ERK and PKB. The coupling of the beta-3 AR to signaling pathways usually associated with growth factors and insulin may be of physiological importance. Both insulin and beta adrenergic hormones play crucial roles in the regulation of energy metabolism. Alterations in the beta AR signaling have been observed in rodent models of obesity, and in humans, a polymorphism (Trp64→Arg) in the beta-3 AR gene has been associated with obesity and type 2 diabetes (Strosberg, 1997a). Beta-3 AR agonists have been proposed as potential drugs for the treatment of obesity and diabetes in humans (Arch and Wilson, 1996). The identification of a novel beta-3 AR-mediated signaling pathway, which displays pharmacological properties distinct from that of the activation of adenylyl cyclase, will have important implications for the screening ofbeta-3 AR agonists that are effective in humans.
Acknowledgments
We thank Marie-Françoise Drumare and France Pietri-Rouxel for providing us with some of the data shown in Table 1 on thebeta-3 AR-mediated production of cAMP.
Footnotes
- Received July 28, 1998.
- Accepted October 15, 1998.
-
Send reprint requests to: Dr. T. Issad, Institut Cochin de Génétique Moléculaire, Centre National de la Recherche Scientifique, Unité Propre de Recherche 415, Laboratoire d’Immuno-Pharmacologie Moléculaire, 22, rue Méchain, 75014 Paris, France. E-mail:Issad{at}cochin.inserm.fr
-
This work was supported by the Centre National de la Recherche Scientifique, the Institut National de Santé et de Recherche Médicale, and the Fondation pour la Recherche Médicale.
Abbreviations
- AR
- adrenergic receptor
- ERK
- extracellular signal-regulated kinase
- PI3K
- phosphatidylinositol-3 kinase
- PAGE
- polyacrylamide gel electrophoresis: PK, protein kinase
- PTX
- pertussis toxin
- MAPK
- mitogen-activated protein kinase
- MEK
- mitogen-activated protein kinase kinase
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}