


Abstract
Arachidonylethanolamide (AEA), the putative endogenous ligand of the cannabinoid receptor, has been shown to be a substrate for lipoxygenase enzymes in vitro. One goal of this study was to determine whether lipoxygenase-rich cells metabolize AEA. [14C]AEA was converted by human polymorphonuclear leukocytes (PMNs) to two major metabolites that comigrated with synthetic 12(S)- and 15(S)-hydroxy-arachidonylethanolamide (HAEA). Human platelets convert [14C]AEA to 12(S)-HAEA. 12(S)-HAEA binds to both CB1 and CB2 receptors with approximately the same affinity as AEA. 12(R)-HAEA, which is not produced by PMNs, has 2-fold lower affinity for the CB1 receptor and 10-fold lower affinity for the CB2 receptor than 12(S)-HAEA. 15-HAEA has a lower affinity than AEA for both receptors, with K i values of 738 and >1000 nm for CB1 and CB2 receptors, respectively. The addition of a hydroxyl group at C20 of AEA resulted in a ligand with the same affinity for the CB1 receptor but a 4-fold lower affinity for the CB2 receptor than AEA. 12(S)-HAEA and 15-HAEA are poor substrates for AEA amidohydrolase and do not bind to the AEA uptake carrier. In conclusion, the addition of a hydroxyl group at C12 of the arachidonate backbone of AEA does not affect binding to CB receptors but is likely to increase its half-life. The addition of hydroxyl groups at other positions affects ligand affinity for CB receptors; both the position of the hydroxyl group and the configuration of the remaining double bonds are determinants of affinity.
Two subtypes of cannabinoid receptor have been identified; CB1 is expressed primarily although not exclusively in brain (Matsuda et al., 1990), and CB2 is found primarily in cells of myeloid lineage (Munroet al., 1993; Galiegue et al., 1995). An endogenous ligand for the CB1 receptor has been isolated from porcine brain and identified as AEA (Devane et al., 1992). AEA mimics the behavioral effects of the active cannabinoids, including the production of hypothermia, analgesia, decreased locomotion, and catalepsy (Fride and Mechoulam, 1993). Activation of CB1 by AEA and other cannabinoid agonists results in inhibition of both adenylyl cyclase and voltage-operated calcium channels (Vogel et al., 1992, 1993; Mackie et al., 1993). AEA also binds to the CB2 receptor (Facci et al., 1995; Felder et al., 1995), but its efficacy at this cannabinoid receptor subtype is unclear. In one study, AEA was reported to inhibit adenylyl cyclase activity through the CB2 receptor (Felder et al., 1995); however, two other studies have reported that AEA has no CB2 agonist activity (Bayewitch et al., 1995; Facci et al., 1995).
Because AEA has an unmodified arachidonate backbone, the question arises of whether AEA is a substrate for the enzymes that metabolize AA and thereby plays a role as precursor for other biologically active molecules. Three enzymatic processes for the oxygenation of AA are known: (1) the cyclooxygenase pathway, resulting in the formation of prostaglandins and thromboxane; (2) the cytochrome P450 pathways, resulting in production of epoxyeicosatrienoic acids and HETEs, including 12(R)-HETE, compound D [12(R)-hydroxy-5Z,8Z,14Z-eicosatrienoic acid], and 20-HETE; and (3) the lipoxygenase pathway, resulting in hydroperoxyeicosatetraenoic acids, 5-HETE, 8-HETE, 11-HETE, 12-HETE, 15-HETE, and leukotrienes. Lipoxygenases that oxygenate AA at the 12 and 15 position can metabolize both free and esterified AA (Junget al., 1985), and others have recently shown that AEA is a substrate for porcine leukocyte 12-lipoxygenase (Ueda et al., 1995) and soybean 15-lipoxygenase and rat brain 12-lipoxygenase (Hampson et al., 1995). One purpose of this study was to investigate the metabolism of AEA by intact human cells: platelets, which contain 12-lipoxygenase, and PMNs, which contain both 5- and 15-lipoxygenases and cytochrome P450 ω-hydroxylase.
The second purpose of this study was to determine the relative affinities of the lipoxygenase metabolites of AEA and other hydroxy derivatives of AEA for the two known cannabinoid receptors. A previous study reported the affinities of 12(S)-HAEA and 15-HAEA for the CB1 receptor (Hampson et al., 1995); we have extended these studies to include determination of affinities for the CB2 receptor and the inclusion of other oxygenated AEA analogs. The results have given us several important and interesting insights into the ability of the CB1 and CB2 binding sites to discriminate AEA derivatives that have modifications along the arachidonate backbone.
Experimental Procedures
Materials.
All chemical syntheses were carried out with exclusion of moisture and air (dry nitrogen; septum-syringe technique) in glassware that was baked ≥4 hr at 150°. All solvents were of HPLC grade or higher. Methylene chloride was dried by distillation from calcium hydride immediately before use. [3H]CP55,940 (120 Ci/mmol) and [14C](U)-AA (920 mCi/mmol) were purchased from DuPont-New England Nuclear (Boston MA). [3H]AEA (210 Ci/mmol) was purchased from Amersham Life Sciences (Arlington Heights, IL). [14C](U)AEA labeled in the arachidonyl portion of the molecule was synthesized as described previously (Hillard et al., 1995).
Synthesis of AEA and derivatives.
AEA was synthesized from arachidonyl chloride as described previously (Hillard et al., 1995) and was purified by thin layer chromatography (silica gel HL plates, 20 × 20 cm, 250 μm) using a solvent system of hexane/ethyl acetate/methanol (60:40:5). The band containing AEA (Rf = 0.24) was scraped and extracted with ethyl acetate.
Previously published methods were used for the synthesis of 15(S)-HETE (Baldwin et al., 1979), 12(R)-hydroxyeicosatrienoic acid (Compound D; Shin et al., 1989), 12(R)-HETE (Yadagiri et al., 1986), and 20-HETE (Manna et al., 1983). The ethanolamides of these oxygenated derivatives of AA (Fig.1) were synthesized by first converting the HETEs to the corresponding methyl esters by treatment with excess ethereal diazomethane, followed by the addition of 2 equivalents of 2-hydroxypyridine in ethanolamine for 60 min at 50°. The oxygenated derivatives of AEA were purified by RP-HPLC using a Nucleosil-C18, 5-m, 4.6 × 250 μm column and a 40-min linear gradient of 50–100% acetonitrile containing 0.1% acetic acid and water at 1 ml/min. Absorbance was monitored at 205 or 235 nm.

Chemical structures of the compounds used in the study.
Metabolite synthesis and separation.
[14C]AEA (30 μl of 28 μm in ethanol) or [14C]AA (30 μl of 100 μm in ethanol) was added to 3.0 ml of buffer (0.2m borate, pH 9.0, for the 15-lipoxygenase assay and 0.1m Tris·HCl, 5 mm EDTA, and 0.03% Tween 80, pH 7.5 for the 12-lipoxygenase assay). Five hundred units of soybean 15-lipoxygenase (Sigma Chemical, St. Louis, MO) or porcine 12-lipoxygenase (Cayman Chemical, Ann Arbor, MI) was dissolved in the respective buffer and added to the stirring solution. After incubation for 30 min at room temperature, 1 ml of 0.76 mmtriphenylphosphine in methylene chloride was added to the mixture to convert any hydroperoxides to hydroxyl products. After 5 min, the water layer was washed twice with 3 ml of methylene chloride, and the organic layer was removed. The organic layers were combined, and after removal of the solvent with a stream of nitrogen, the sample was analyzed by RP-HPLC as described above. Radioactivity of the column eluate was monitored by collecting 0.2-ml fractions, adding scintillation fluid, and counting.
Isolation and incubation of human platelets and PMNs.
Human platelets were isolated from blood of healthy donors who had not ingested nonsteroidal anti-inflammatory agents in the preceding 2 weeks. Platelets were separated from blood using the method of Callahanet al. (1985). The platelets were counted in phosphate-buffered saline and then suspended in HEPES buffer (10 mm HEPES, 150 mm NaCl, 5 mm KCl, 2 mm CaCl2, 1 mmMgCl2, 6 mm glucose, pH 7.4) at a concentration of 3.0 × 109 cells/ml. Platelets (0.5 ml) were preincubated at 37° with 10 μmof A23187 for 1 min. [14C]AEA or [14C]AA was added (final concentration, 29 μm), and the reaction was stopped after 15 min by the addition of ethanol (1.5 ml). The mixture was made 15% ethanol by the addition of deionized water, and the metabolites were extracted using solid-phase extraction as described previously (Pfister et al., 1988). The extracted metabolites were analyzed by RP-HPLC as described above. In the indomethacin experiments, platelets were pretreated with indomethacin for 5 min before the addition of A23187.
Human PMNs were isolated from heparinized blood of healthy donors who had not ingested nonsteroidal anti-inflammatory agents in the preceding 2 weeks. PMNs were separated from blood using a modified Hypaque-Ficoll technique (Boyum, 1968). The PMNs were counted in phosphate-buffered saline and then suspended in HEPES buffer (see above) at a concentration of 20–22 × 106 cells/ml. PMNs (0.5 ml) were preincubated at 37° with 10 μm of A23187 for 1 min. [14C]AEA or [14C]AA was added (final concentration, 29 μm), and the reaction was stopped after 15 min by the addition of 1.5 ml of ethanol. The mixture was diluted to 15% ethanol by the addition of deionized water, and the metabolites were extracted using solid phase extraction as described previously (Pfister et al., 1988). The extracted metabolites were analyzed by RP-HPLC as described above.
GC-MS.
TMS ethers of the lipoxygenase metabolites were synthesized by dissolving the sample in 0.1 ml of acetonitrile and adding 0.1 ml of bis(trimethylsilyl) trifluoroacetamide. After the sample was incubated for 60 min at 37°, the sample was dried with a stream of dry nitrogen. The samples were dissolved in 50 μl of acetonitrile and analyzed by GC-MS. The GC program was isothermic for 2.5 min at 100° and was increased to 300° at a rate of 20°/min; then, the temperature was held at 300° for 12.5 min. The column was a 15-m DB-5 (Supelco) with a 250-μm diameter. The peaks were detected using positive chemical ionization MS with methane as the ionizing gas.
Rat forebrain membrane preparation.
The study reported here was approved by the Medical College of Wisconsin Animal Care Committee and was carried out in accordance with the Declaration of Helsinki and following the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Before their experimental use, male Sprague-Dawley rats (250–300 g) were maintained on a 12-hr light/dark schedule with free access to food and water. Forebrain membranes were prepared by homogenization in TME buffer (50 mm Tris·HCl, 1.0 mm EDTA, and 3.0 mmMgCl2, pH 7.4), followed by centrifugation at 11,300 × g for 20 min at 4°. The pellet was resuspended in buffer and stored at −80° until assay. Protein concentrations were determined in each membrane preparation using the dye binding method of Bradford (1976) using reagent and protein standard I obtained from BioRad (Richmond, CA).
Measurement of [3H]CP55,940 binding.
The affinity of the AEA analogs for the CB1 receptor was determined by competition with [3H]CP55,940 using membranes from rat forebrain according to Hillard et al. (1995). IC50 values were calculated from data determined at 8–12 concentrations of unlabeled ligand using nonlinear regression.Ki values were calculated from IC50 values according to the equation of Cheng and Prusoff (1973).
The affinity of the AEA analogs for the CB2 receptor was determined by competition with [3H]CP55,940. Binding was measured in membranes from CHO-K1 cells transfected with the human CB2 receptor obtained from Receptor Biology (Baltimore, MD). Membranes (diluted according to manufacturer’s instructions) were incubated with 1 nm [3H]CP55940 in 200 μl of buffer (10 mm HEPES, pH 7.4, 1 mm EDTA, and 1 mm MgCl2 containing 0.3 mg/ml fatty acid-free BSA). Competing ligands were added in 1 μl of dimethylsulfoxide; nonspecific binding was determined in the presence of 5 μm Win 55212–2. Incubations were carried out at 30° for 1 hr. Bound and free [3H]CP55940 were separated by filtration using a Brandel (Montreal, Quebec, Canada) Cell Harvester through glass fiber filters (#32; Schleicher & Schuell, Keene, NH) that had been presoaked in 0.5% polyethyleneimine. Filters were washed three times with 5 ml of cold wash buffer (10 mm HEPES, pH 7.4, containing 0.1% fatty acid-free BSA) and counted. IC50 values were calculated from data determined at 8–12 concentrations of unlabeled ligand using nonlinear regression. Ki values were calculated from IC50 values according to the equation ofCheng and Prusoff (1973).
Measurement of AEA amidohydrolase activity.
Rat liver microsomal membranes were prepared by homogenization in buffer (0.15m KCl and 0.25 mK2HPO4, pH 7.5). The homogenate was centrifuged at 1,000 × g for 5 min, and the supernatant was recentrifuged at 20,000 × g for 15 min. The resulting supernatant was centrifuged at 100,000 ×g for 60 min, and the resulting microsomal membranes were resuspended in TME buffer and stored at −80° until use. The effects of the HAEAs on AEA amidohydrolase activity were assessed by determining the ability of the analogs to reduce the hydrolysis of [14C]AEA. The liver membranes (0.05 mg/ml in a final volume of 1.5 ml TME buffer containing 1.0 mg/ml fatty acid-free BSA) were preincubated with competitors for 10 min followed by the addition of 9–16 nCi of [14C]AEA (final concentration, 23 μm). The reaction was allowed to continue for 30 min at 37° and were stopped with the addition of 2 ml of chloroform/methanol (1:2). After standing at room temperature for 30 min, 0.67 ml of chloroform and 0.6 ml of water were added. Aqueous and organic phases were separated by centrifugation at 1000 rpm for 10 min, and the radiolabeled species in the organic phase were separated by thin layer chromatography as outlined previously (Hillard et al., 1995). The amounts of substrate and product were determined using an Ambis radioanalytic detector (San Diego, CA).
Determination of AEA accumulation by cerebellar granule cells.
Cerebellar granule cells were prepared from neonatal rats and placed into primary culture as described previously (Hillardet al., 1997). The measurement of [3H]AEA accumulation by cerebellar granule cells was determined exactly as described previously (Hillard et al., 1997).
Results and Discussion
The 12- and 15-lipoxygenases use AEA as a substrate.
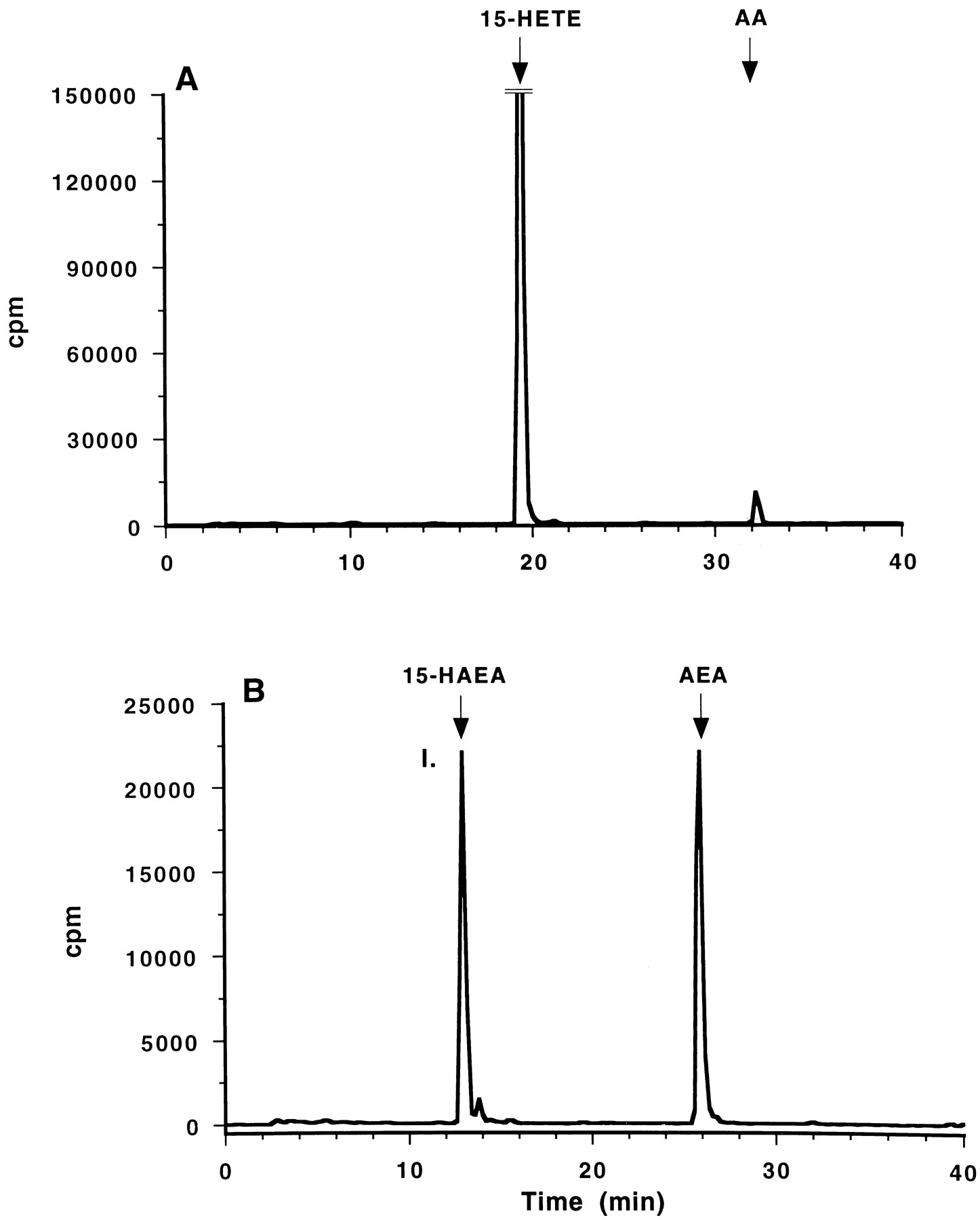
[14C]AEA or [14C]AA was incubated with either porcine leukocyte 12-lipoxygenase or soybean 15-lipoxygenase. As expected, the incubation of [14C]AA with 12- and 15-lipoxygenase resulted in the formation of 12-HETE and 15-HETE, respectively (Figs.2A and 3A). [14C]AEA also served as a substrate for both porcine leukocyte 12-lipoxygenase and soybean 15-lipoxygenase; both of the lipoxygenases metabolized [14C]AEA to more polar metabolites as shown in Figs. 2B and 3B. The major metabolite (I) from the incubation of [14C]AEA with 15-lipoxygenase comigrated with synthetic 15(S)-HAEA on RP-HPLC. The incubation of [14C]AEA with 12-lipoxygenase resulted in the production of a metabolite (II) that comigrated on HPLC with synthetic 12(S)-HAEA (Fig. 3B).

Chromatogram of radiolabeled products obtained from incubation of either [14C]AA (A) or [14C]AEA (B) with soybean 15-lipoxygenase. Elution times of known standards are indicated (top).
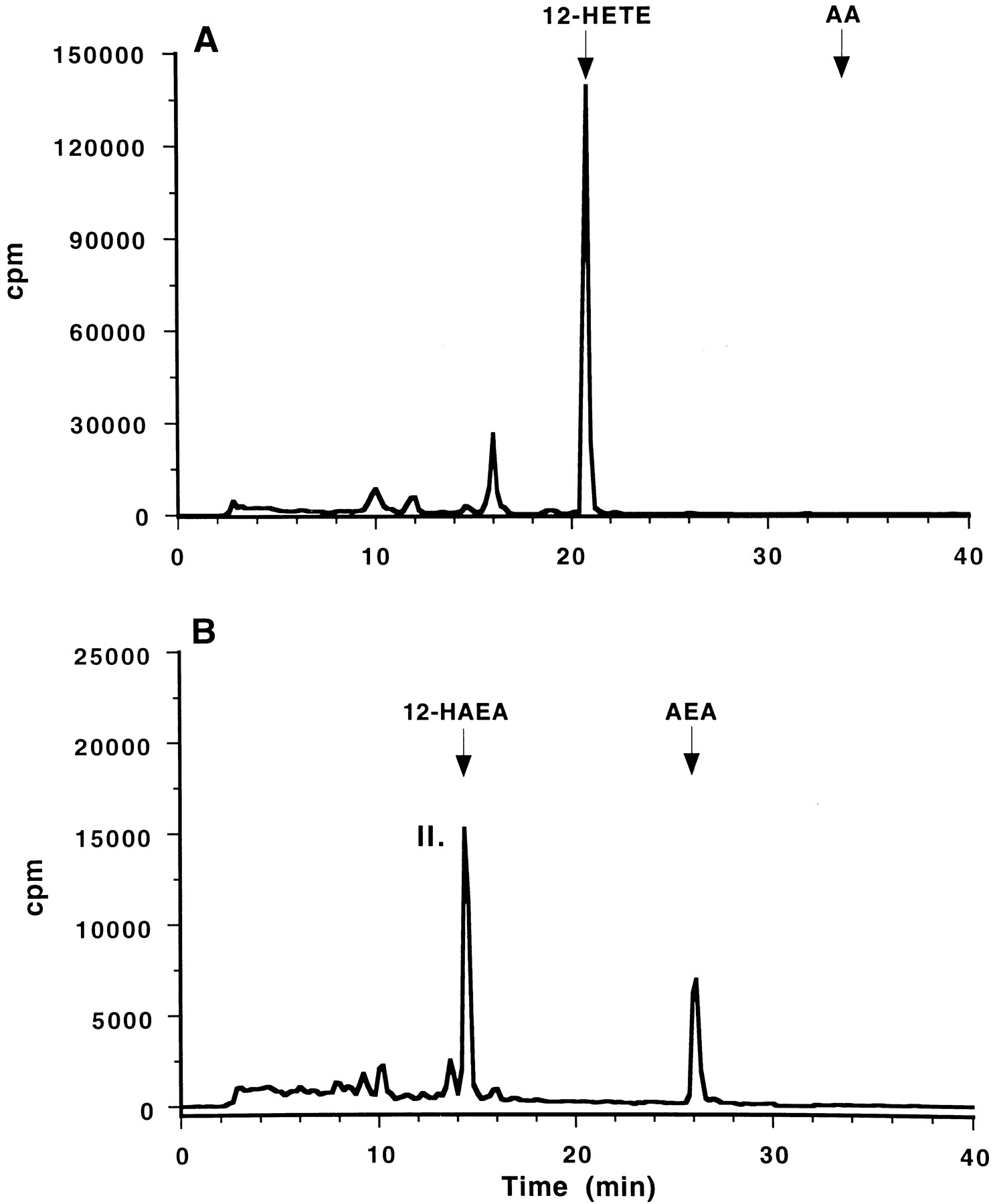
Chromatogram of radiolabeled products obtained from incubation of either [14C]AA (A) or [14C]AEA (B) with purified porcine 12-lipoxygenase. Elution times of known standards are indicated (top).
Metabolites I and II were derivatized to the TMS ethers and analyzed using GC-MS; the positive chemical ion (PCI) mass spectra of the TMS-derivatized metabolites are shown in Fig.4. Both spectra show peaks at 536, 508, 492, and 418 m/z. The 508-m/z peak corresponds to the molecular ion (M+) with the addition of a hydrogen (MH+); thus, both metabolites have the molecular weight of a derivatized HAEA (i.e., 507 m/z). The other ions represent 536 m/z(M++CH2CH3), 492 m/z (m—CH3), and 418m/z (MH+—TMSOH). In Fig.4A, the major fragments of metabolite I include 71m/z[CH2)4CH3] 173 m/z[TMSOCH(CH2)4CH3], 334 m/z[TMSO(CH2)2NHCO(CH2)3(CH⋕CHCH2)2(CH⋕CH)2)], and 437 m/z[TMSO(CH2)2NHCO(CH2)3(CH⋕CHCH2)2(CH⋕CH)2CHOTMS—H+]. The appearance of these fragments support the conclusion that metabolite I is 15-HAEA (Fig. 4A). In Fig.4B, the major fragments of metabolite II include 213m/z(TMSOCHCH2CH⋕CH(CH2)4CH3), 294 m/z(TMSO(CH2)2NHCO(CH2)3(CH⋕CHCH2)(CH⋕CH)2), and 396 m/z(TMSO(CH2)2NHCO(CH2)3(CH⋕CHCH2) (CH⋕CH)2CHOTMS—H+). The appearance of these fragments from metabolite II indicate that it is 12-HAEA (Fig. 4B). These results are in accord with those reported previously (Hampson et al., 1995, Ueda et al., 1995).

Positive ion chemical ionization mass spectra of TMS ethers of 15-HAEA and 12-HAEA. [14C]AEA was incubated with either 15-lipoxygenase (Fig. 2) or 12-lipoxygenase (Fig. 3) as described in the text. Peaks that comigrated on HPLC with known 15-HAEA (A) or 12-HAEA (B) standards were isolated, converted to TMS ethers, and analyzed by GC-MS.
Human platelets convert AEA to 12(S)-HAEA.
Although it is clear that AEA is a substrate for purified lipoxygenasesin vitro, it is important to determine whether metabolism also occurs in an intact cellular system. Human platelets are a rich source of 12-lipoxygenase and rapidly convert exogenously added AA to 12-HETE (Fig. 5A). In the experiment shown, 46% of the total cpm comigrated with 12-HETE standard; 9% of the cpm was unmetabolized arachidonic acid, and 6% of the cpm comigrated with 12-HHT standard. Platelets metabolize AEA to 12(S)-HAEA (Fig. 5B). The conversion is significant; 19% of the total cpm comigrate with 12-HAEA, and 49% of the cpm is unmetabolized AEA. Pretreatment of platelets with 10 μmindomethacin had no effect on the metabolism of [14C]AEA to 12(S)-HAEA but inhibited the conversion of [14C]AA to 12-HHT (data not shown). These results demonstrate that platelets are capable of incorporating AEA and metabolizing AEA to 12(S)-HAEA. AA seems to be a better substrate for human platelet 12 lipoxygenase than AEA just as it seems to be a better substrate for porcine leukocyte 12-lipoxygenase (Fig. 3).

Chromatogram of products obtained after incubation of human platelets with [14C]AA and [14C]AEA. A23187-stimulated human platelets were incubated with [14C]AA (A) or [14C]AEA (B) for 15 min, followed by extraction and separation using RP-HPLC. The column eluate was monitored for radioactivity. Arrows, positions at which known standards migrate using this elution procedure. Shown is a representative chromatogram; the entire experiment was repeated twice with similar results.
Human PMNs convert AEA to 12(S)-HAEA and 15(S)-HAEA.
Human PMNs (a mixture of neutrophils, eosinophils, and basophils) are a rich source of lipoxygenases, and the major AA metabolites formed by these cells are 15-HETE and the leukotrienes, which are products of 5-lipoxygenase (Borgeat and Samuelsson, 1976). Isolated human PMNs were preincubated with the calcium ionophore A23187 to promote precursor uptake and metabolism followed by incubation with [14C]AEA or [14C]AA. Incubation of PMNs with [14C]AA resulted in production of a major metabolite that comigrated with 15(S)-HETE on HPLC (Fig.6A). 12(S)-HETE also was identified, and the cellular source of this metabolite is unclear. It has been shown recently that canine PMNs metabolize AA to 12-HETE and 20-HETE, suggesting that neutrophils contain 12-lipoxygenase and cytochrome P450 ω-hydroxylase (Rosolowsky et al., 1996). It also is possible that platelet contaminants in the PMN preparation are responsible for the production of 12(S)-HETE.

Chromatogram of products obtained after incubation of human PMNs with [14C]AA and [14C]AEA. A23187-stimulated human PMNs were incubated with [14C]AA (A) or [14C]AEA (B) for 15 min, followed by extraction and separation using RP-HPLC. The column eluate was monitored for radioactivity. Arrows, positions at which known standards migrate using this elution procedure. Shown is a representative chromatogram; the entire experiment was repeated three times with similar results.
The metabolites resulting from incubation of PMNs with [14C]AEA were resolved using HPLC, and a representative chromatogram is shown in Fig. 6B. Two major radioactive metabolites were seen (A and B); both products absorbed UV light at 235 nm. Metabolite A comigrated with synthetic 15(S)-HAEA and accounted for 1.7% of the total cpm. Metabolite B comigrated with synthetic 12(S)-HAEA and accounted for 1.3% of the total cpm. These results demonstrate that AEA is taken up by PMNs and is a substrate for PMN 12- and 15-lipoxygenases. Although the biological significance of this pathway is not known, these findings support the concept that AEA can serve as a precursor for other biologically active agents. Free AA also was observed, suggesting that PMNs contain AEA amidohydrolase that hydrolyzes AEA to AA and ethanolamine (Deutsch and Chin, 1993).
Affinity of oxygenated derivatives of AEA for the CB1 receptor.
A second aspect of these studies was to determine the affinity and efficacy of the HAEAs as ligands of cannabinoid receptors. There are two known subtypes of cannabinoid receptor: CB1, which is found predominantly in the brain (Matsuda et al., 1990) and has also been demonstrated in circulating cells of the immune system (Galiegue et al., 1995) and in reproductive organs (Daset al., 1995), and CB2, which is located predominantly on cells of myeloid lineage (Galiegue et al., 1995). The affinities of chemically synthesized, and purified HAEAs for the CB1 receptor binding site were determined in rat brain membranes. Both 12(S)-HAEA and 15(S)-HAEA compete for binding to the CB1 receptor; 12(S)-HAEA has slightly lower affinity than the parent compound AEA, and 15(S)-HAEA has 6-fold lower affinity for the receptor than AEA (Table1). These results are in basic agreement with the results reported by Hampson et al. (1995) except they reported that 12-HAEA had 2-fold higher affinity for the receptor than AEA.
Binding affinities for the hydroxylated derivatives of AEA for the two subtypes of cannabinoid receptor
Lipoxygenase metabolism of AA is stereospecific (Yamamoto, 1992), and it is known that the (S) and (R) isomers of 12-HETE have different biological activities (Fretland and Djuric, 1989). We were interested in whether the stereochemistry of the hydroxyl group was important in the binding of 12(S)-HAEA to the CB1 receptor. 12(R)-HAEA, the stereoisomer of 12(S)-HAEA that is not produced in any appreciable quantity by 12-lipoxygenase, has 2-fold lower affinity for the CB1 binding site than 12(S)-HAEA (Table 1). Because the isomers have a small difference in affinity for the CB1 receptor binding site, it is unlikely that the hydroxyl group contributes to the formation of bonds between the ligand and receptor. These results suggest that the addition of a hydroxyl to C12 in AEA introduces a slight steric hindrance to binding that is more severe when the hydroxyl is in the (R) configuration.
The metabolism of AEA by 12-lipoxygenase results in both the addition of hydroxyl at the 12 position and an alteration in the position and orientation of the double bond (Fig. 1). In 12(S)-HAEA, atrans double bond occurs between C10 and C11, whereas in the parent compound AEA, a cis (Z) double bond occurs between C11 and C12. One explanation for our finding that 12(S)-HAEA has 2-fold lower affinity for the CB1 receptor than AEA is that the CB1 receptor binding site can accommodate a double bond between either C11 and C12 or C10 and C11 but the transorientation of the double bond in 12(S)-HAEA is sterically less favorable for binding. To explore the role of the orientation and saturation of the C10 and C11 bond in ligand binding to the CB1 receptor, we synthesized the ethanolamide of 12(R)-hydroxyeicosatrienoic acid or 10,11-dihydro-12(R)-HETE, also called Compound D (Fig. 1). 10,11-Dihydro-12(R)-HAEA has a 10-fold lower affinity for the CB1 receptor than AEA (Table 1). Because the data comparing AEA and 12(S)-HAEA suggest that the addition of a hydroxyl to C12 does not significantly affect binding, this result suggests the double bond in the region of C10–C12 is important for binding and its loss outweighs any benefit gained from loss of steric hindrance. Another AEA analog in which the C10–C11 region of the molecule has been modified is the conjugated triene derivative of AEA, 5Z,7E,9E,14Z-eicosatetraenoyl-N-ethanolamide (Wise et al., 1996). This analog was found to have 6-fold lower affinity for the CB1 receptor than AEA, which may be due to the loss of optimal π-bond interactions at C10–C11. Similarly, 11-HAEA, in which C11 is not involved in a double bond, has 15-fold lower affinity for the CB1 receptor than AEA (Hampson et al., 1995). Taken together, these data regarding the relationship between ligand structure and affinity for the CB1 receptor support a hypothesis that a π-bond interaction occurs between the C10–C12 region of the ligand and the receptor; in particular, C11 must be involved in a double bond for optimal binding to occur.
15(S)-HAEA binds to the CB1 receptor with 4–6-fold lower affinity than AEA (Table 1). This suggests that either the addition of the hydroxyl at this site or the alteration of the 14Z bond of AEA to 13E interferes with binding. Although we have not carried out studies to distinguish between these possibilities, the ethanolamide of mead acid (5Z,8Z,11Z-eicosatrienoic acid) has been shown to have affinity for the CB1 receptor equal to AEA (Prilleret al., 1995). Because loss of the double bond at C14 does not affect binding affinity, it is likely that steric hindrance introduced either by the hydroxyl group or “kink” in the acyl chain is responsible for the loss in affinity.
We investigated the affinity of 20-HAEA for the CB1 receptor. In this analog, the character of the aliphatic pentameric “tail” of the molecule is altered. There is evidence that this region of the molecule is important for high affinity binding to the CB1 receptor. For example, ethanolamides of shorter chain fatty acids have low affinity for the CB1 receptor (Sugiura et al., 1995) and increase the hydrophobic character of this region, as occurs in 16,16-dimethyl arachidonyl ethanolamide, which results in increased affinity of the ligand for the receptor (Ryan et al., 1997; Seltzmanet al., 1997). The addition of a hydroxyl group in the 20 position of AEA did not affect the affinity of the ligand for the CB1 receptor (Table 1), which suggests that ligand binding can tolerate a decrease in the hydrophobicity of this region of the molecule.
Affinity of oxygenated derivatives of AEA for the CB2 receptor.
The major cannabinoid receptor present in the periphery and therefore the cannabinoid receptor that would be most likely to encounter PMN-derived HAEAs is the CB2 receptor (Munro et al., 1993, Galiegue et al., 1995). Several cannabinoid receptor ligands have been shown to have differing affinities for CB2 and CB1 receptors, including cannabinol (Munro et al., 1993). As seen in Table 1, there are several interesting differences between the pattern of binding of the HAEAs to the CB2 receptor and the CB1 receptor. First, AEA has equal affinity for the CB1 receptor of brain membranes and the human CB2 receptor expressed in CHO cells (Table 1). This finding does not agree with the original observation ofMunro et al. (1993), who reported that AEA had fairly low affinity for the CB2 receptor; however, recent reports from several other investigators have cited Ki values in the range of 33–85 nm for AEA (Bayewitch et al., 1995; Facci et al., 1995).
The affinity of 12(S)-HAEA for the CB2 receptor is similar to the affinity of AEA for the CB2 receptor (Table 1). However, the CB2 receptor is more sensitive to changes in the stereochemistry of the hydroxyl group at C12; 12(S)-HAEA has ≥10-fold higher affinity for the CB2 receptor than 12(R)-HAEA. In addition, the ethanolamide derivative of Compound D has considerably higher affinity for the CB2 receptor than the CB1 receptor. This finding suggests that the binding of AEA to the CB2 receptor is not dependent on π orbital interactions in the region of C10–C12. This conclusion is supported by data from another laboratory that the ethanolamide of palmitic acid, a 16-carbon saturated fatty acid, binds to the CB2 receptor with high affinity (Sugiura et al., 1995). In addition, 20-HAEA has 4-fold lower affinity for the CB2 receptor than the parent compound AEA, suggesting that the CB2 receptor is more sensitive than the CB1 receptor to a reduction in the hydrophobicity of the pentameric tail.
Resistance of oxygenated derivatives of AEA to catabolism by liver AEA amidohydrolase or reuptake by neurons.
Exogenously administered AEA has a relatively short half-life in vivo(Smith et al., 1994). One possible consequence of the conversion of AEA to 12(S)-HAEA by platelets is the formation of a cannabinoid receptor agonist with longer bioavailability than AEA. Two mechanisms for the inactivation of AEA have been proposed. First, AEA is hydrolyzed to arachidonic acid and ethanolamine in the liver and brain by a membrane associated enzyme, AEA amidohydrolase (Deutsch and Chin, 1993). AEA amidohydrolase is responsible for inactivation of AEA by membrane preparations in vitro (Childers et al., 1994), and a recent study of the metabolism of AEA in mice supports the contention that AEA amidohydrolase plays a role in the inactivation of AEA in vivo (Willoughby et al., 1997). Second, AEA is accumulated by neuronal cells via an uptake carrier that is driven by the concentration gradient for AEA (Hillard et al., 1997). Inhibition of AEA uptake results in a potentiation of the hypotension produced by AEA in guinea pigs (Calignano et al., 1997).
Although unlabeled AEA inhibits the hydrolysis of [14C]AEA by AEA amidohydrolase in liver membranes, the addition of a hydroxyl group at the 12 or 15 position of AEA results in analogs that are poor substrates for AEA amidohydrolase of liver membranes (Table 2). Similar results were seen when the assay was carried out using forebrain membranes (Data not shown). These results are in agreement with those of Ueda et al. (1995). Of the three analogs tested, only 20-HAEA significantly inhibited the hydrolysis of [14C]AEA, although it was less effective than the parent compound.
Effects of hydroxylated derivatives of AEA on the inactivation of AEA by amidohydrolase and cellular reuptake
AEA, at a concentration of 100 μm, produces a 75% reduction in the accumulation of [3H]AEA by cerebellar granule cells. The addition of hydroxyl groups to AEA decreases its affinity for the uptake carrier of cerebellar granule cells (Table 2). Of the oxygenated analogs, only 12(S)-HAEA inhibits the uptake of [3H]AEA by a small amount (<30% at a concentration of 100 μm). These results suggest that the addition of hydroxyl groups to AEA results in decreased affinity for the uptake carrier. Therefore, although the addition of a hydroxyl at the 12 position of AEA does not affect affinity for the cannabinoid receptor to a great extent, 12(S)-HAEA is a poor substrate for AEA amidohydrolase and has lower affinity for the AEA uptake carrier than AEA. As a result, the conversion of AEA to 12(S)-HAEA by platelets results in an agonist for the CB1 and CB2 cannabinoid receptors that is equiefficacious to AEA but has an increased half-life. A possible physiological consequence of the oxygenation of AEA by 12-lipoxygenase and 15-lipoxygenase is a prolongation of the duration of action of AEA in the circulation. Future studies will explore this hypothesis directly.
Conclusions.
We demonstrated that AEA is taken up and metabolized to 12(S)-HAEA by human platelets and, to a lesser extent, to 15-HAEA by human PMNs. These results are the first demonstration in human cells that AEA can serve as a precursor for other metabolites with potential biological activity. We demonstrated that 12(S)-HAEA binds to both the CB1 and CB2 receptors with an affinity that is very similar to that of AEA itself. However, 12(S)-HAEA is not a substrate for AEA amidohydrolase and is a poor substrate for the AEA uptake carrier. These results suggest that one possible consequence of the conversion of AEA by lipoxygenases in platelets is to prolong the lifetime of a cannabinoid receptor agonist in the circulation and site of action.
Studies of the affinities of 12(S)-HAEA, 12(R)-HAEA, and 15-HAEA for the CB1 and CB2 receptors have provided interesting insights into the structural requirements for AEA-based ligands for binding to these receptors. In particular, we have found that the affinities of these and other HAEAs for the CB1 receptor are dependent on the presence of a double bond either between C11–C12 or C10–C11. In addition, ligand affinity for the CB1 receptor is indifferent to the presence of a hydroxyl at either C12 or C20; however, the addition of a hydroxyl to C15 significantly reduces affinity.
There is very little information about the structural requirements of AEA for the CB2 binding site. Earlier studies had shown that binding to the CB2 receptor is enhanced when AA is replaced with palmitic acid, suggesting that the presence of double bonds in the acyl chain impose a steric hindrance to binding. This supposition is supported by the binding data obtained for the HAEAs, particularly the finding that the stereochemistry of the hydroxyl group at C12 significantly affects binding. More studies with other structural analogs are needed to support definitive conclusions, but it is likely that the CB2 binding pocket is more sensitive to steric interference along the acyl chain than the CB1 binding pocket.
Acknowledgments
We thank Mr. Bruce Peltier and Ms. Marcie Greenberg for their excellent technical assistance.
Footnotes
- Received September 10, 1997.
- Accepted March 30, 1998.
-
Send reprint requests to: Cecilia J. Hillard, Ph.D., Department of Pharmacology, Medical College of Wisconsin, 8701 Watertown Plank Road, Milwaukee, WI 53226. E-mail:chillard{at}mcw.edu
-
This work was supported by United States Public Health Service Grants DA09155, HL51055 (W.B.C.), and GM31278 (J.R.F.).
Abbreviations
- AEA
- N-arachidonylethanolamine
- AA
- arachidonic acid
- BSA
- bovine serum albumin
- CB1
- cannabinoid receptor subtype 1
- CB2
- cannabinoid receptor subtype 2
- CHO
- Chinese hamster ovary
- GC
- gas chromatography
- MS
- mass spectroscopy
- HAEA
- hydroxyarachidonylethanolamide
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HETE
- hydroxyeicosatetraenoic acid
- PMN
- polymorphonuclear leukocyte
- RP
- reverse phase
- HPLC
- high pressure liquid chromatography
- TMS
- trimethylsilyl
- TME
- Tris/MgCl2/EDTA
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}