


Abstract
To extend our knowledge of the pharmacological profile of human α4β2 neuronal nicotinic receptors, we investigated the action of hexamethonium on the major brain human nicotinic acetylcholine receptor (nAChR) stably expressed in human embryonic kidney 293 cells. This compound displays all of the characteristics of an open-channel blocker at the human α4β2 nAChR: a voltage-dependent inhibition (more pronounced at hyperpolarized potentials), absence of competition, and use dependence. Moreover, we observed that classicN-methyl-d-aspartate open-channel blockers amantadine, 3,5-dimethyl-1-adamantanamine (memantine), and dizocilpine [(+)-MK-801] and the calcium channel antagonist 8-(diethylamino)octyl-3,4,5-trimethoxybenzoate are powerful inhibitors of the human α4β2 nAChR. Dose-inhibition curves yield, at −100 mV, IC50 values in the micromolar range for all of compounds and Hill coefficients below unity. Whole-cell current-voltage relationships display a strong rectification profile at hyperpolarized potentials, and current blockades are fitted adequately by a mathematical model that describes the mechanism of an ion channel block. We conclude that these molecules are powerful human α4β2 open-channel blockers ranking in the following order of potency: amantadine > memantine = hexamethonium > 8-(diethylamino)octyl-3,4,5-trimethoxybenzoate ∼ (+)-MK-801.
Neuronal nAChRs belong to the superfamily of ionotropic LGCs (Bertrand and Changeux, 1995). Using the crayfish muscle preparation, it has been shown that muscle nAChRs are activated within a microsecond time scale on binding of the agonist (Franke et al., 1987). Very fast opening of the ionic pore results from the particular structure of these proteins that form both the ligand binding site and transmembrane channel. Given its physical dimensions, this aqueous pore is readily blocked by small molecules. Hexamethonium is a compound initially identified for its ability to block the ACh transmission in autonomic ganglia while leaving muscle nAChRs unaffected (Paton and Zaimis, 1951). The work of Blackman et al. (1963) and Ascheret al. (1979) suggested that hexamethonium inhibited the ACh-evoked currents in ganglionic neurons by sterically blocking the ionic pore of the nAChR. Extensive investigations have indicated that hexamethonium can be considered as a prototype OCB of the ganglionic nAChRs (Gurney and Rang, 1984) or of the reconstituted chick (Bertrandet al., 1990) and rat (Charnet et al., 1992) α4β2 nAChRs because this small molecule fulfilled the following characteristics: (1) its blocking effect is voltage dependent (Ascheret al., 1979; Gurney and Rang, 1984; Bertrand et al., 1990; Charnet et al., 1992), (2) it displays a use-dependent mode of action (Gurney and Rang, 1984), and (3) its blocking effect is more pronounced at higher agonist concentrations (Ascher et al., 1979). We therefore apply the term OCB to any compound that complies with these criteria.
Some of the physiological and pharmacological properties of the human α4β2 neuronal nicotinic receptor have been investigated using the patch-clamp technique (Buisson et al., 1996) and indicate that human nAChRs present a distinct profile compared with other vertebrate nAChRs. To further establish the pharmacological signature of the human α4β2 nAChR, we investigated the effect of hexamethonium and TMB-8, a calcium channel antagonist that has been identified as a noncompetitive nicotinic antagonist (Bencherif et al., 1995) (Fig. 1). In addition, we examined the properties of (+)-MK-801 (dizocilpine) at the human α4β2 nAChR and of two other classic NMDA OCBs: amantadine (1-amino-adamantane) and memantine (3,5-dimethyl-1-adamantanamine) (Fig. 1).

Chemical formulas of the compounds tested at the human α4β2 neuronal nAChR. Hexamethonium and (+)-MK-801 represent two prototypes of OCBs for central nAChRs and NMDA glutamate receptors, respectively. Amantadine and memantine inhibit NMDA receptors through an open-channel block mechanism. TMB-8 is a voltage-gated calcium channel antagonist displaying a noncompetitive mode of action at several nicotinic receptors (Bencherif et al., 1995). The net charge is 2 for hexamethonium and 1 for amantadine, (+)-MK-801, memantine, and TMB-8 at a physiological pH (7.4).
Materials and Methods
Stably transfected cells (K177) were grown as described previously (Buisson et al., 1996; Gopalakrishnan et al., 1996) and used at passages 40–72. Cells were seeded onto 35-mm Petri dishes at low density and recorded 3–5 days later.
Electrophysiological recordings.
All experiments were performed at room temperature (20°) in salt solution (containing 120 mm NaCl, 5 mm KCl, 2 mmMgCl2, 2 mmCaCl2, 25 mm glucose, 10 mm HEPES, and 1 μm atropine (for blocking possible endogenous muscarinic receptors); pH 7.4 with NaOH. Patch pipettes (2–5 mΩ) were pulled from glass borosilicate (1.2-mm outer diameter) and filled with 5 mm NaCl, 10 mm CsCl, 120 mm CsF, 2 mmMgCl2, 10 mm HEPES, and 10 mm1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid, pH 7.4 with CsOH. Currents, which were recorded in isolated cells using an Axopatch 200A or 200B amplifier (Axon Instruments, Foster City, CA), were filtered at 1 kHz, digitized at 2–5 kHz, and stored on a personal computer equipped with an analog-to-digital converter (ATMIO-16D; National Instrument, Austin, TX) and the DATAC package (Bertrand and Bader, 1986). Data were analyzed on a Macintosh Performa 5200 using the MacDATAC program. Fast superfusion of the cells was performed with a custom-made multibarrel (Buisson et al., 1996; Bertrand et al., 1997) or a 300-μm glass theta-tube actuated by a piezoquartz device (Physik Instrument, Berlin, Germany). Both systems allow solution exchanges in the millisecond range (Frankeet al., 1987; Buisson et al., 1996). Chemicals were purchased from Sigma Chemical (St. Louis, MO), Fluka Chemical (Ronkonkoma, NY), and Research Biochemicals (Natick, MA).
Voltage-ramp protocols were performed as follows: the membrane was held at 40 mV, and a first ramp (from 40 to −140 mV in 2 sec) was applied in the standard saline medium (without agonist) for determination of the leak current. Three seconds later, 1 μm ACh was delivered for 3 sec, and the voltage-ramp was applied 400 msec after the onset of delivery. Data presented herein were obtained through subtraction from the leak current. For ramps starting at negative potential, the same protocol was used, but voltage command ranged from −140 to 40 mV.
Dose-inhibition curves.
Every 10 sec, a voltage-ramp protocol (see above) was performed first with 1 μm ACh and then with increasing concentrations of the inhibitor. The ACh-evoked currents were measured at −100 mV and normalized to the amplitude of the current elicited by ACh alone. Values were plotted against the concentrations of the inhibitor (on a logarithm scale) and fitted with the empirical Hill equation:
Fit of current ratios.
Analysis of the current blockade was done according to the model proposed by Zarel and Dani (1995); that is, current-voltage relationships were measured first under control conditions and then in presence of a given concentration of blocking agent, and the ratio of these currents was plotted as a function of the holding voltage. Data were fitted with the equation:
EC50 corresponds to the concentration of agonist evoking a current of half-maximal amplitude. IC50 corresponds to the concentration of blocking agent causing a 50% reduction in the current evoked by a pulse of agonist near the EC50 value (1 μmACh unless otherwise indicated).
Results
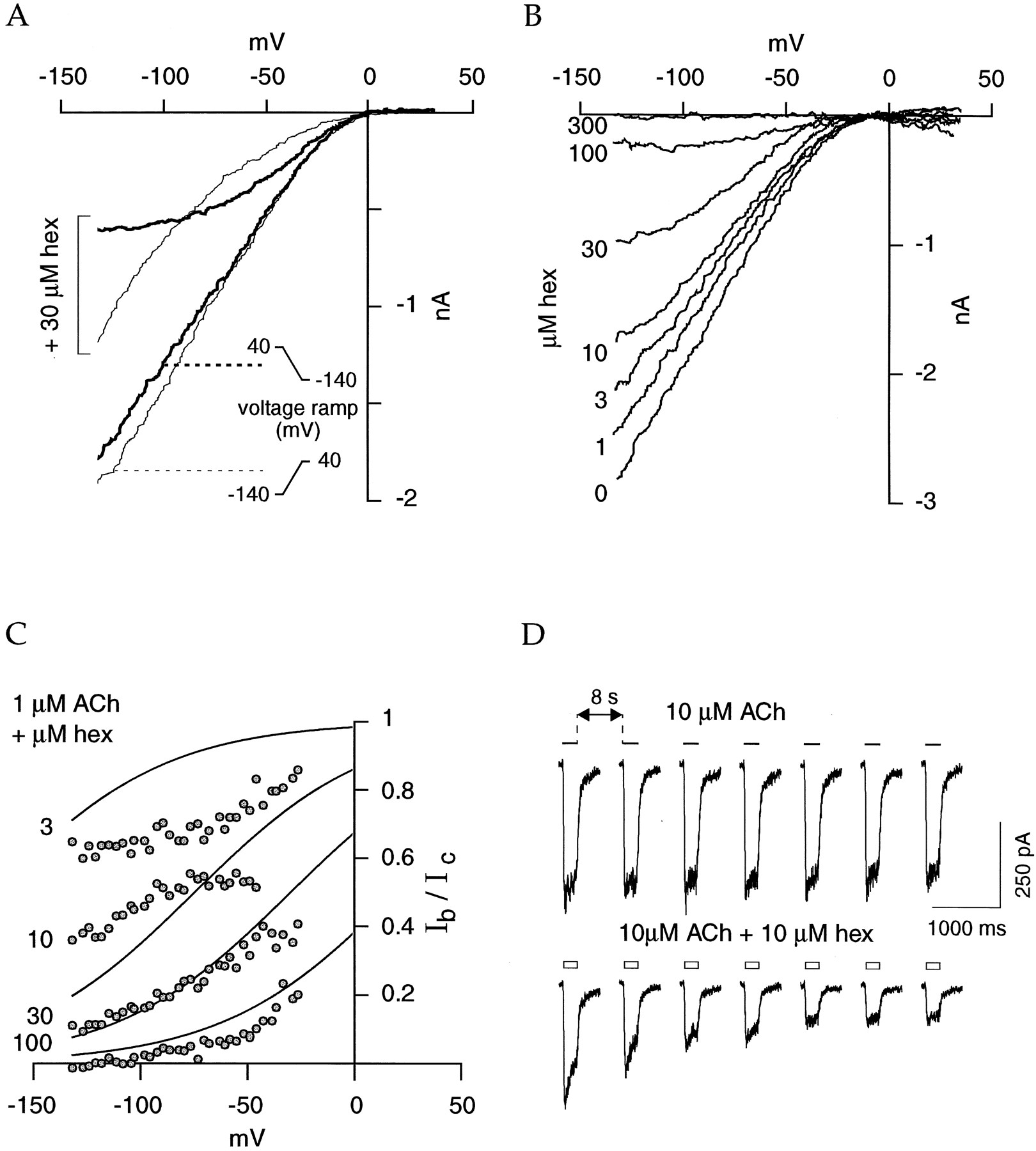
We first examined the action of the well known ganglionic inhibitor hexamethonium (Fig. 1) on the human α4β2 nAChR (Fig.2). Previous work has indicated that hexamethonium is a potent inhibitor of the chick (Bertrand et al., 1990) and rat (Charnet et al., 1992) α4β2 nAChRs reconstituted in the oocyte system, with a blocking effect strongly dependent of the membrane potential. As illustrated in Fig.2A, coapplication of 3 μm hexamethonium efficiently inhibits ACh-evoked currents. The application of hexamethonium to a steady concentration of ACh causes a fast decrease in the ACh-evoked current that reverses readily when the drug is removed (Fig. 2B). Blockade and recovery are both voltage and time dependent, as shown by recordings obtained at −100 and −60 mV, respectively. Although slowly reversible, full recovery from hexamethonium blockade typically is observed after a wash for a few minutes (Fig. 2C). A plot of the percentage of the current inhibition (determined with the voltage-ramp protocol; see Materials and Methods) as a function of the hexamethonium concentration yields an IC50 value of 6 μm and a Hill coefficient of 0.8 (at −100 mV; Fig. 2D). The mean values are summarized in Table1. Hexamethonium does not significantly modify the EC50 value of ACh for the human α4β2 nAChR, as illustrated in Fig. 2E; the ACh dose-response curve performed in the presence of 10 μm hexamethonium gives an EC50 value of 1.5 μm with a Hill coefficient of 1.2 (six cells), which is close to the values determined in the absence of hexamethonium. In the presence of 1 μm ACh, voltage-ramps yield overlaying current-voltage relationships independent of the ramp polarity (Fig.3A). In contrast, when the same protocols are repeated in the presence of 30 μmhexamethonium, a marked difference in the blockade is observed, with ramps starting at a positive voltage displaying a stronger voltage-dependent blockade (Fig. 3A). Therefore, all the current-voltage relationships presented below were recorded in this configuration. Voltage-ramps performed at 1 μmACh with increasing concentrations of hexamethonium (1–300 μm) reveal a marked voltage-dependent mechanism of block: the fraction of the current inhibited is larger at hyperpolarized membrane potential values (Fig. 3B). This effect can be interpreted in the frame of an open-channel blocking mechanism (Ascheret al., 1979; Bertrand et al., 1990; Charnetet al., 1992). To examine further this hypothesis, we used the single-site model of an ion channel block (Woodhull, 1973). Widely used, this model was adapted later for many LGCs and can be used to fit the current ratio Ib/Ic, where Ib is the current recorded during the blockade, and Ic is the value measured in control. As shown for NMDA, this ratio is described adequately by eq. 2 (Zarel and Dani, 1995). Similar measures performed for several hexamethonium concentrations allowed determination of the mean values of δ and Kd (Fig. 3C, Table2). Other features of the OCBs are the use-dependent effects (i.e., at a fixed OCB concentration, the fraction of current blockade increases with repetitive agonist stimulations) (Neher and Steinbach, 1978; Gurney and Rang, 1984). As illustrated in Fig. 3D, coapplication of 10 μm hexamethonium induces a progressive inhibition of the currents elicited by repetitive pulses of ACh (10 μm, 200 msec). Because hexamethonium enters the nAChR ionic pore, it induces a reduction in the open time of these channels that could be quantified in burst analysis (Colquhoun and Hawkes, 1995). However, given the fast run-down of the α4β2 nAChRs in outside-out patches (Buisson et al., 1996), we could not record under steady state conditions that allow computation of mean open-time histograms to investigate the effect of hexamethonium at the single-channel level. Despite this lack of single-channel measurement, all other evidence indicates hexamethonium behaves as a potent OCB of the human α4β2 nAChR.

Hexamethonium, a typical OCB of the human α4β2 nAChR. A, Inhibition by hexamethonium (hex) of currents evoked by three ACh concentrations. The cell was held at −100 mV.Bars, drug applications. B, Time course of hexamethonium blockade and recovery. The application of 30 μmhexamethonium for 2 sec (open bar) during a steady exposure to a low ACh concentration (1 μm, 30 sec) causes a fast reduction in the ACh-evoked current. Amplitude of the blockade is voltage dependent [see top (−100 mV) andbottom (−60 mV) traces]. C, Hexamethonium blockade and recovery of ACh-evoked currents. Currents were recorded before and after a 1-sec hexamethonium blockade (30 μm, open bar). ACh pulses (horizontal lines) were applied once every minute. Full recovery (right trace) was obtained after a ∼4-min wash. D, Dose-response inhibition curve of hexamethonium at −100 mV. ACh-evoked currents (1 μm ACh) measured in presence of several concentration of hexamethonium are plotted as a function of the antagonist concentration (nine cells; see Materials and Methods). Line through data points, best fit of the empirical Hill equation. E, Hexamethonium does not modify the EC50 value of ACh for the human α4β2 nAChR. Dose-response curve to ACh obtained in control (squares) or presence of 10 μm hexamethonium (circles). Top continuous line, best fit of empirical Hill equation (EC50 = 3 μm, n H = 1.2; seeBuisson et al., 1996). Calibrated to the mean current recorded with 1 μm ACh alone, data recorded in presence of 10 μm hexamethonium are fitted (bottom continuous line) by the empirical Hill equation, yielding an EC50 of 1.5 μm and Hill coefficient of 1.2 (six cells) (Hill equation computed for the standard ACh dose-response curve multiplied by a scaling factor of 0.34).
Sensitivity of OCBs

Hexamethonium blockade is voltage and use dependent. A, Hysteresis of the current-voltage relationship under hexamethonium (hex). A voltage-ramp (see Materials and Methods) was performed during a 1 μm ACh application first in control and then in the presence of hexamethonium. Although no significant differences were observed in ramp polarities for the current-voltage relationships under the control condition, an important difference is observed when hexamethonium is present with a stronger blockade for the ramp starting at a positive voltage. Thin lines, ramp starting from a negative voltage. Thick lines, ramp starting from a positive value. B, Blockade of hexamethonium is concentration and voltage dependent as illustrated by currents evoked by 1 μm ACh during a voltage-ramp protocol from 40 to −140 mV (in 2 sec). A first ramp was performed during exposure to ACh alone and then during coapplication of increasing hexamethonium concentrations (left). C, Current ratios (Ib/Ic, as described in Materials and Methods) are plotted as a function of the holding voltage. Lines through data points, fit obtained with eq. 2 with a K d value of 62 μm and a δ value of 0.62. Left, values correspond to the concentration of hexamethonium (hex) coapplied with ACh. D, Hexamethonium (hex) blockade is use dependent. Short pulses of ACh (10 μm, 200 msec) were delivered every 8 sec for a control period (top); 10 μmhexamethonium then was coapplied with ACh (bottom).
Parameters of eq. 2
The voltage-gated channel antagonist TMB-8 (Fig. 1) has a noncompetitive mode of inhibition at muscle and ganglionic nAChRs, and it was proposed to act via an open-channel block mechanism (Bencherifet al., 1995). We therefore investigated its action on the human α4β2 nAChR. Coapplication of TMB-8 at micromolar concentrations induces a marked reduction in ACh-evoked currents comparable to that obtained with hexamethonium (data not shown). A dose-inhibition curve (Fig. 4A) at −100 mV yields an IC50 value of 15 μmand a Hill coefficient of 0.7 (mean values given in Table 1). Moreover, as illustrated in Fig. 4B, the effect of TMB-8 is voltage dependent, and the current ratio Ib/Iccan be described with the use of eq. 2. These results indicate that as proposed initially (Bencherif et al., 1995), TMB-8 should bind within the ionic pore of α4β2 nAChRs.

The calcium channel antagonist TMB-8 inhibits the human α4β2 nAChR. A, Dose-inhibition curve determined at −100 mV (squares, five cells; see Materials and Methods). Data are fitted with the empirical Hill equation (continuous line). B, Plot of the current ratios (Ib/Ic) as a function of cell voltage.Lines through data points, fit obtained with eq. 2 with a K d value of 160 μm and a δ value of 0.19. Left, values correspond to the concentration of TMB-8 coapplied with ACh.
Dose-inhibition protocols performed with increasing concentrations of (+)-MK-801 yielded an IC50 value of 15 μm and a Hill coefficient of 0.7 at −100 mV for the human α4β2 nAChR (Fig. 5A; mean values given in Table 1). The voltage-dependence of the (+)-MK-801 effect (see Fig. 5B) is illustrated by the correlation observed between the current ratio Ib/Ic and predictions made on the basis of eq. 2 (see Materials and Methods and Table 2). Together with the results of others (Ramoa et al., 1990; Amador and Dani, 1991; Briggs and Mckenna, 1996), our data confirm that (+)-MK-801 is a powerful OCB of central nAChRs.

(+)-MK-801 inhibits α4β2 nAChRs. A, Dose-inhibition curve (squares) measured at −100 mV (five cells; see Materials and Methods). Continuous line, data are fitted with the empirical Hill equation (see Materials and Methods). B, (+)-MK-801 blockade is voltage dependent. Plot of the current ratios (Ib/Ic) as a function of the holding voltage. Lines through data points, fit obtained with eq. 2 with aK d value of 7.5 μm and a δ value of 0.39. Left, values correspond to the concentration of MK-801 coapplied with ACh.
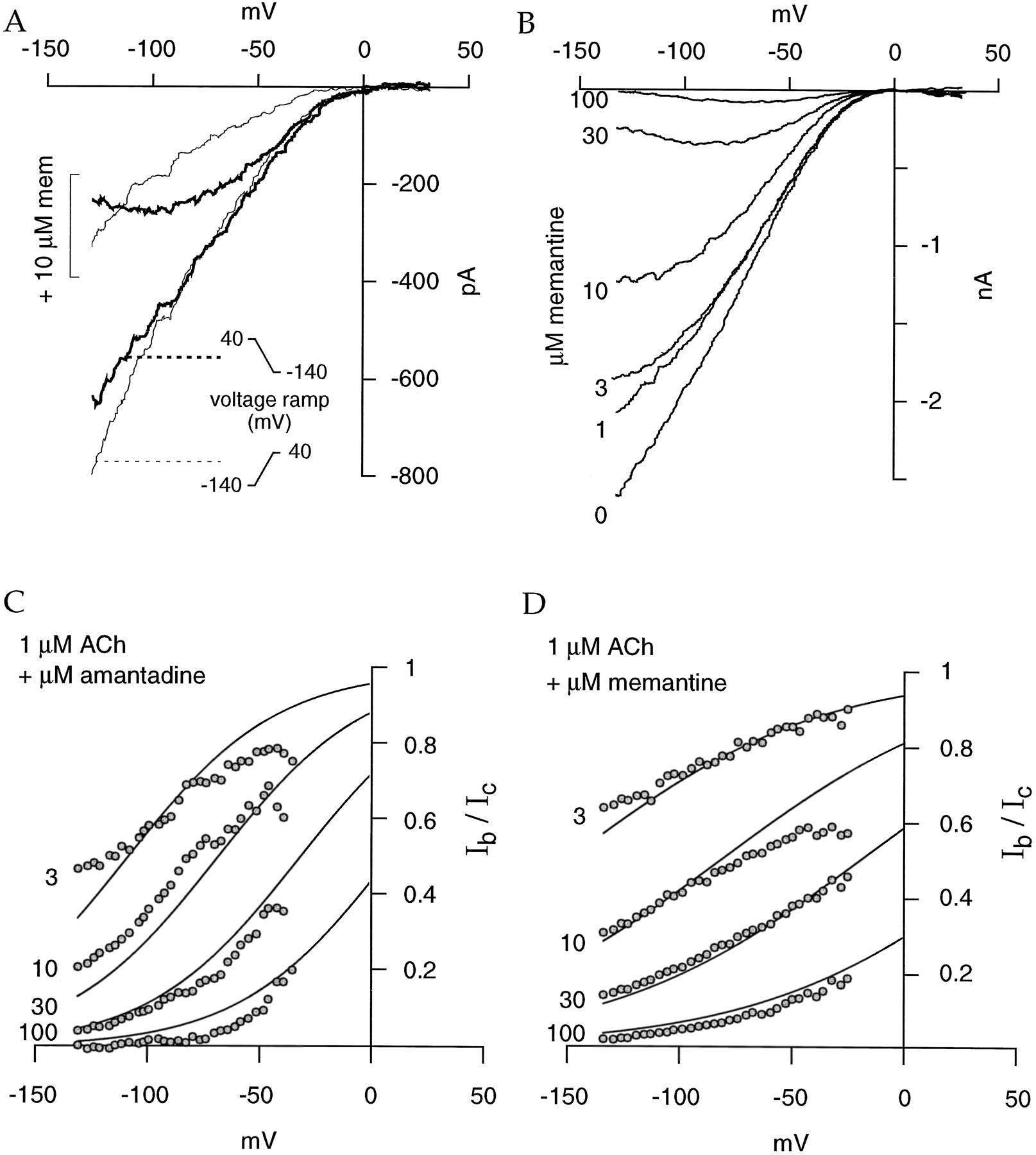
As presented in Fig. 6A, we observed that coapplication of 10 μm memantine with 10 μmACh induced a progressive and use-dependent inhibition of the α4β2-evoked currents. Partial to full recovery can be observed after an extensive washout and is dependent on the holding voltage, as illustrated in Fig. 6B. Similar results were obtained with amantadine (data not shown). Dose-inhibition curves determined at −100 mV reveal the very high potency of both compounds to inhibit the human α4β2 nAChRs. Amantadine displays the lowest IC50 value of all the compounds investigated in the current study (Fig. 6C, Table1), whereas memantine potency is comparable to that of hexamethonium (Fig. 6D, Table 1). Noncompetitive antagonists, such as hexamethonium, decrease the ACh maximal amplitude but do not modify its EC50 value for the nAChR (see above and Fig. 2D). We then computed the ratio of the current evoked by ACh in the presence of 10 μm memantine to the current recorded without this inhibitor. The mean ratio is 0.42 ± 0.01, 0.48 ± 0.05, and 0.42 ± 0.02 for 1, 10, and 100 μm ACh, respectively (three cells). Thus, the EC50 value of ACh for the human α4β2 nAChR seems to not be modified by the presence of 10 μm memantine and suggests a noncompetitive mechanism of blockade for this compound. As observed previously with hexamethonium (Fig. 3A), voltage-ramps recorded in the presence of memantine (or amantadine) show hysteresis depending on the ramp polarity (Fig.7A). In addition, a marked voltage-dependent mechanism of inhibition is illustrated by the current-voltage relationships recorded under increasing concentrations of memantine (Fig. 7B). Similar data were obtained with amantadine (data not shown). The voltage dependence of Ib/Ic for memantine and amantadine is presented in Fig. 7, C and D. Theoretical values are in good agreement with experimental data (see Materials and Methods and Table 2).

Amantadine and memantine are potent OCBs of the human α4β2 nAChR. A, Memantine inhibits human α4β2 nAChRs in a use-dependent manner. Short pulses of ACh (10 μm; 200 msec) were delivered every 8 sec for a control period (left). Memantine (10 μm) then was coapplied with ACh (right). Even with a 8-sec interval of washout between the applications, memantine induces a progressive block of the ACh-evoked current until it reaches a steady state of inhibition (representative of four experiments). B, Memantine blockade is easily reversible. The application of a short pulse of memantine (mem) (open bar, 10 μm, 2 sec) during a prolonged exposure to a low (1 μm) ACh pulse causes a fast and reversible reduction of the evoked current. Although incomplete recovery is observed after a 26-sec washout at −80 mV, full recovery was readily obtained when the same experimental paradigm was repeated at a holding potential of −40 mV. C and D, Dose-inhibition curves (squares) of amantadine and memantine at −100 mV (nine cells for amantadine and five cells for memantine). Continuous lines, values corresponding to the empirical Hill equation (see Materials and Methods).

Blockade of human α4β2 nAChRs by amantadine and memantine is voltage dependent. A, Current-voltage relationships recorded for positive or negative voltage protocols in control and the presence of 10 μm memantine (mem). Hysteresis of memantine blockade is observed clearly in a comparison of the two conditions (thin line, ramp starts at −140 mV;thick line, ramp starts at 40 mV). The current-voltage relationship of the ACh-evoked current is independent of the ramp polarity. The slight difference between the two curves may be attributed to a partial desensitization of α4β2 nAChRs. However, when memantine (10 μm) is coapplied with ACh, the voltage dependence of its block is observed only under a decreasing voltage-ramp protocol. B, Blockade of memantine is concentration and voltage dependent as illustrated by currents evoked by 1 μm ACh during a voltage-ramp protocol from 40 to −140 mV (in 2 sec). A first ramp was performed during exposure to ACh alone and then during coapplication of increasing memantine concentrations (left). C and D, Plot of the current ratios (Ib/Ic) as a function of the cell voltage.Lines through data points, fit obtained with eq. 2 with a K d value of 80 μm and a δ value of 0.76 for amantadine and aK d value of 42 μm and a δ value of 0.45 for memantine.Left, values correspond to the concentration of OCB coapplied with ACh.
Discussion
We investigated the action at the human α4β2 nAChR of compounds that are known to penetrate and block by steric hindrance the ionic pore of LGCs as well as voltage-gated channels.
Hexamethonium is one of the first compounds used to discriminate the ganglionic and muscle nAChRs (Paton and Zaimis, 1951) and later was characterized as being a potent OCB of nAChRs of the rat parasympathetic ganglion cells (Ascher et al., 1979) and of the reconstituted chick and rat α4β2 nAChRs (Bertrand et al., 1990; Charnet et al., 1992). A few years ago,Bencherif et al. (1995) presented biochemical evidence suggesting that the calcium channel antagonist TMB-8 must be a powerful noncompetitive antagonist of peripheral and central nAChRs, with an IC50 value in the low micromolar range, indicating this compound inhibits almost equipotently calcium channels and nAChRs. Although first identified as a specific NMDA noncompetitive antagonist (Wong et al., 1986) with open-channel blockade properties (Huettner and Bean, 1988), (+)-MK-801 later was shown to block nAChRs within the same range of concentrations (Ramoa et al., 1990; Amador and Dani, 1991). Similarly, amantadine and memantine are powerful OCBs of NMDA receptors acting at micromolar concentrations (Chen, 1992; Parsons et al., 1996; Chen and Lipton, 1997) and, as shown more than two decades ago (Albuquerqueet al., 1978; Tsai et al., 1978), amantadine has a blocking effect at the muscle nAChR in the range of 100 μm. Moreover, micromolar concentrations of amantadine inhibit the α-bungarotoxin-sensitive current evoked by ACh in cultured rat hippocampal neurons (Matsubayashi et al., 1997) .
Our results demonstrate that the five compounds listed above inhibit the human α4β2 nAChR in the low micromolar range (Table 1). A low Hill coefficient was observed for all compounds tested. Usually interpreted as indicative of cooperativity, the low Hill coefficient suggests the absence of cooperativity in the blockade processes of the antagonist tested. The discrepancy between the low Hill coefficients observed herein and a predicted value of unity, however, cannot be explained on the basis of the channel blockade. Moreover, no substantial modification of the EC50 value to ACh was observed for the two compounds tested (hexamethonium and memantine) and thus provide further evidence of a noncompetitive mode of blockade. Although surprising at first, the absence of a significant displacement of the EC50, in presence of hexamethonium, which should have been expected based on our knowledge of OCBs (Ascheret al., 1979), can be interpreted as indicating that this compound binds with equal affinity to the resting and active states. Further work is needed to confirm this hypothesis. Full recovery from blockade was observed for the five OCBs tested as illustrated, for example, for hexamethonium and memantine in Figs. 2 and 6. Moreover, it should be noted that these two compounds, which display different voltage-dependent properties (see below), exhibit a significant difference in the recovery time course; hexamethonium is more difficult to wash than memantine, a difference that may indicate that hexamethonium binds more tightly in the pore than memantine.
OCBs generally are known to exhibit a marked voltage-dependent inhibition (Bertrand et al., 1997). One illustration of this property is given by the currents, recorded at different potentials, in the presence of a determined concentration of hexamethonium (Fig. 2B) or memantine (Fig. 6B): the fraction of the inhibited ACh-evoked current increased with more negative holding potential values. This observation is reinforced further on examination of current-voltage relationships (Figs. 3A and 7A). An hysteresis common to all the compounds tested was observed when comparing data obtained with ramps of opposite polarities. This phenomenon is best explained by assuming that compounds enter and block the channel more readily when starting from positive voltages than from negative values. To obtain the best revelation of the channel blockade, all experiments were conducted with voltage-ramps starting at positive values.
In the presence of each of the five compounds investigated in this work, current ratios are fitted adequately using a model adapted from that originally proposed for OCBs (Woodhull, 1973) (eq. 2 and Table 2). The δ parameter represents the fraction of the membrane electrical field that is sensed by the OCB at its binding site. The highest δ values determined for hexamethonium, amantadine, and memantine suggest that the putative channel binding site for these molecules must be located near the middle of the field across the ionic pore (assuming a constant electrical field across the lipid bilayer). According to this model, TMB-8 and (+)-MK-801 should bind to another site located in the upper 10–20% of the pore electrical field (Table 2). Thus, we propose that these molecules inhibit the human α4β2 nAChR via an open-channel block mechanism but may remain trapped at distinct locations within the ionic pore. A comparison of the recovery from open-channel blockade of hexamethonium and memantine reveals that inhibition induced by this second compound recovers more easily than that with hexamethonium. From previous studies performed on chick α4β2 nAChR, it is known that full recovery from hexamethonium blockade is achieved only with a so-called pop-out protocol (Bertrandet al., 1990) that consists of depolarizing the cell in the presence of ACh. Thus, it can be proposed that hexamethonium indeed remains more strongly bound within the ionic pore than memantine. Further evidence for an open-channel block mechanism is given by the use-dependent effect observed for the three compounds tested (hexamethonium, memantine, and amantadine). Compounds termed OCB often can block the receptor even in its closed state, as first illustrated by Adams (1977) for procaine blockade of the ACh receptor at the neuromuscular junction. Currently, however, the complex properties of neuronal nAChRs preclude detailed analysis that would allow discrimination of the possibility of interaction of blocking agents with the closed or desensitized conformations or both.
In the late 1970s, the emergence of the patch-clamp technique led to the investigation of blocking mechanisms of compounds at the single-channel level. Experiments performed with muscle nAChRs confirmed a previous hypothesis that local anesthetics such as QX-222 or QX-314, as well as curare, are entering opened nAChRs and blocking them via a steric hindrance that is revealed through reduction in the mean open time (Neher and Steinbach, 1978). More recently, site-directed mutagenesis and reconstitution experiments allowed the identification of residues lining the ionic pore of the muscle or neuronal α7 nAChRs that interact with QX-222 (Charnet et al., 1990; Revah et al., 1991). These studies confirmed the hypothesis derived from previous macroscopic observations (Beam, 1976; Colquhoun et al., 1979). Although indispensable for the confirmation of the blockade mechanisms at the molecular level, α4β2 single-channel measurements must await study in the outside-out configuration. The substantial run-down observed under these experimental conditions precludes an analysis of single channels under steady state conditions (Buisson et al., 1996) .
Because the α4β2 subtype may be the predominant form of the human brain nAChRs that binds (−)-nicotine with high affinity (Gopalakrishnan et al., 1996) and is responsible for nicotine addiction, it follows that the effect induced by this tobacco alkaloid must be related to this type of nAChR. The addictive properties of nicotine are well documented in rodents, and it was shown that nicotine stimulates dopamine transmission in the nucleus accumbens (Pontieri et al., 1996) in a manner similar to that of cocaine (Merlo Pich et al., 1997). Belonging to the mesolimbic system, the nucleus accumbens is of critical importance in the reinforcing properties of addictive drugs (Nisell et al., 1995). The high affinity nicotine binding sites localized in the nucleus accumbens of the rat are suggestive of expression of the α4β2 subunits in this area (Clarke and Pert, 1985). In the view of these data, it follows that α4β2 nAChR antagonists should constitute potent pharmacological tools in the treatment of smoking cessation. Among the compounds tested in this study, amantadine and memantine display the best inhibition properties at the human α4β2 nAChR, with a use-dependent effect. It is of value to recall that both substances have been used clinically for therapy with persons with Parkinson’s disease for >25 years (Danielczyk, 1995). In the brain of treated patients with Parkinson’s disease, the extracellular concentration of amantadine is estimated to be ∼10 μm(Kornhuber et al., 1995), a value that is close to the IC50 values determined for the human α4β2 nAChR. Thus, in smoking cessation trials, amantadine (or memantine) could be used at concentrations equal to or lower than those used for the treatment of parkinsonism, with good knowledge of the side effects. In conclusion, we propose that hexamethonium, TMB-8, (+)-MK-801, amantadine, and memantine are potent OCBs of the human α4β2 nAChR and that some of the clinical effects observed for amantadine and/or memantine might be related to their action on the neuronal nAChRs.
Acknowledgments
We are grateful to Prof. P. Ascher for his constructive discussions and to S. Bertrand for her constant help. We thank Murali Gopalakrishnan, James P. Sullivan, and Stephen P. Arneric (all from Abbott Laboratories, Chicago, IL) for kindly providing the K177 cell line and for comments on the manuscript.
Footnotes
- Received July 2, 1997.
- Accepted November 21, 1997.
-
Send reprint requests to: Dr. Daniel Bertrand, Dept. of Physiology, CMU, 1, rue M. Servet, CH−1211 Geneva 4, Switzerland. E-mail: bertrand{at}ibm.unige.ch
-
This work was supported by Swiss National Foundation Grant 31–37191.93 and by a grant from the Office Fédéral de l’Education et des Sciences (D.B.).
-
This work was presented in part at the 27th Annual Meeting of the Society for Neuroscience; 1997 Oct 25–30; New Orleans, LA.
Abbreviations
- nAChR
- nicotinic acetylcholine receptor
- ACh
- acetylcholine
- NMDA
- N-methyl-d-aspartate
- TMB-8
- 8-(diethylamino)octyl-3,4,5-trimethoxybenzoate
- LGC
- ligand-gated channel
- OCB
- open-channel blocker
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}