


Abstract
We cloned and characterized the canine A3 adenosine receptor (AR) and examined AR-induced degranulation of the BR line of canine mastocytoma cells. Canine A3AR transcript is found predominantly in spleen, lung, liver, and testes and encodes a 314-amino acid heptahelical receptor.125I-N 6-Aminobenzyladenosine binds to two affinity states of canine A3AR withK D values of 0.7 ± 0.1 and 16 ± 0.8 nm, reflecting G protein-coupled and -uncoupled receptors, respectively. Xanthine antagonists bind with similar affinities to human, canine, and rabbit receptors but with 80–400-fold lower affinities to rat A3AR. Although canine BR mastocytoma cells contain A1AR, A2BAR, and A3AR, degranulation seems to be mediated primarily by A2BARs stimulated by the nonselective agonist 5′-N-ethylcarboxamidoadenosine (NECA) but not by the A3-selective agonistN 6-(3-iodobenzyl)adenosine-5′-N-methylcarboxamide. NECA-stimulated degranulation is not prevented by pertussis toxin and is blocked by enprofylline (K i = 7 μm), an antiasthmatic xanthine with low affinity (K i > 100 μm) for A1AR, A2AAR, and A3AR. NECA increases canine mastocytoma cell cAMP, Ca2+, and inositol trisphosphate levels; these responses are antagonized half-maximally by 7–15 μm enprofylline. The results suggest that (i) the cloned canine A3AR is structurally and pharmacologically more similar to human than to rat A3AR; (ii) the A2BAR, and not the A1AR or A3AR, is principally responsible for adenosine-mediated degranulation of canine BR mastocytoma cells; and (iii) the BR cell A2BAR couples to both Ca2+ mobilization and cAMP accumulation. Although A2B receptors play a major role in the regulation of BR mast cell degranulation, multiple AR subtypes and G proteins may influence mast cell functions.
Adenosine exerts numerous physiological effects that were originally thought to be mediated by three adenosine receptors, A1, A2A, and A2B. In the early 1990s, a new adenosine receptor was cloned from rat tissues, first by Meyerhof et al. (1) and then by Zhou et al. (2), who named it A3. More recently, human (3), sheep (4), and rabbit (5) A3 adenosine receptors have been cloned and characterized. Functional expression of A3 adenosine receptors from various species indicates that A1 and A3receptors bind the radioligands [125I]APNEA, [125I]ABA, and [125I]AB-MECA and are negatively coupled to adenylyl cyclase (2, 4, 6). One unusual property of A3 adenosine receptors is a major difference among species in the binding affinity of xanthine antagonists. In particular, the rat receptor is resistant to blockade by xanthines, whereas sheep, human, and rabbit receptors bind certain xanthines with high affinity, although with distinct potency orders (3).
The addition of adenosine to rat basophilic leukemic cells (RBL 2H3 cells; a tumor cell line resembling mast cells) causes facilitation of the release of granules, which is mediated by A3adenosine receptors (7, 8). A3 receptor activation also triggers the degranulation of mast cells surrounding hamster cheek pouch arterioles (9). Based on these results, the observation that the inhalation of adenosine produces histamine release and bronchoconstriction in asthmatics but not in nonasthmatics (10, 11) and the discovery of high levels of A3 adenosine receptor transcript in human and sheep lung, we proposed a role for the A3 adenosine receptor in the pathophysiology of asthma (12). Because rodents may be poor animal models for the investigation of the role of A3 receptors in human allergy and asthma, we decided to clone the canine A3 adenosine receptor as a first step toward characterizing the role of A3 adenosine receptors in canine models of asthma.
Here, we report the cloning, expression, and pharmacological characterization of an A3 adenosine receptor cDNA isolated from BR cells [canine mastocytoma cells (13)]. Low levels of both A1 and A3 adenosine receptors are found on BR cells, but these are not primarily responsible for stimulating degranulation of this canine mastocytoma cell line. Rather, an A2B adenosine receptor causes degranulation via a pertussis toxin-insensitive pathway that mobilizes mastocytoma cell Ca2+ and can be blocked by the antiasthmatic xanthine enprofylline (14).
Experimental Procedures
Materials.
All chemicals were obtained from Sigma Chemical (St. Louis, MO) with the following exceptions. IB-MECA was from Dr. Saul Kadin (Pfizer, Groton, CT). I-ABA and I-ABOPX (also known as BW-A522) were from Dr. Susan Daluge (Glaxo-Wellcome, Research Triangle Park, NC). WRC-0571 [C8-(N-methylisopropyl)-amino-N 6-(5′-endohydroxy)-endonorbornan-2-yl-9-methyladenine] was from Dr. Pauline Martin (Discovery Therapeutics, Richmond, VA). APNEA was from Dr. Ray Olsson (University of South Florida, Tampa, FL). RDC7 (dog A1 adenosine receptor cDNA) was from Dr. Guy Vassart (Brussels, Belgium). Rat A3adenosine receptor cDNA was from Dr. Fereydoun Sajjadi (Gensia, La Jolla, CA). Human A3 adenosine receptor cDNA was from Dr. Marlene Jacobson (Merck, West Point, PA). Rabbit A3 adenosine receptor cDNA was from Dr. Scott Kennedy (Pfizer, Groton, CT). HMC-1 mast cells were from Dr. J. H. Butterfiled (Mayo Clinic, Rochester, MN). NECA, CGS 21680 (2-[4-(2-carboxyethyl)phenethylamino]-5′-N-ethylcarboxamidoadenosine), (R)-PIA, CPA, CPX, XAC, 8-SPT, theophylline, and enprofylline were purchased from Research Biochemicals (Natick, MA). Ro 20–1724 [4-(3-butoxy-4-methoxybenzyl)-2-imidazolidinone] was from BIOMOL Research Laboratories (Plymouth Meeting, PA). Adenosine deaminase was from Boehringer-Mannheim Biochemicals (Indianapolis, IN). Fura-2/AM was from Molecular Probes (Eugene, OR).myo-[3H]Inositol was from Amersham Life Sciences (Arlington Heights, IL). Dowex AG 1- X8 was from BioRad (Richmond, CA). [125I]ABA was synthesized as described previously (15). Cell culture media and supplies were from GIBCO BRL (Gaithersburg, MD).
Cell culture.
COS-7 cells were grown in DMEM with 10% fetal calf serum, 100 units/ml penicillin, and 100 μg/ml streptomycin. Canine BR mastocytoma cells were grown in low-glucose DMEM supplemented with 2% calf serum, 25 mm HEPES, 1.5 mm l-histidine, 100 units/ml penicillin G, and 100 μg/ml streptomycin. The medium was changed every 3 days, and the cells were replated weekly.
Molecular cloning.
To obtain the full-length sequence of the canine A3 adenosine receptor cDNA, a library prepared in λgt10 from BR cell poly(A)+ RNA was screened using a probe generated by RT-PCR of total RNA isolated from dog tissues using the cDNA cycle kit (InVitrogen, La Jolla, CA). Primers for amplification were primer A (sense 132–152), 5′-GACCACCACCTTCTATTTCA-3′; and primer B (antisense 660–680), 5′-GTCTTGAACTCCCGA/TCC-3′. The primers correspond to conserved regions within the first and third intracellular loops of the human, sheep, and rat A3 adenosine receptor cDNAs. Each reaction cycle consisted of incubations at 95° for 1 min, 55° for 2 min, and 72° for 3 min with 0.02 unit/ml of Taq polymerase (Promega, Madison, WI). PCR fragments were subcloned into the TA vector (InVitrogen) and sequenced with Sequenase (United States Biochemical, Cleveland, OH) using modifications for double-stranded sequencing. One fragment from lung RNA was found to be ∼90% identical to the human, rat, and sheep A3 receptor transcripts. This probe was labeled with [α-32P]dCTP (Primit II; Stratagene, La Jolla, CA) and used to screen the BR cell cDNA library. Library screening was carried out by plaque-filter hybridization. Filters were hybridized at 65° overnight in 10% dextran sulfate, 1 m NaCl, 100 μg/ml herring sperm DNA, and 1 × 106 cpm/ml radiolabeled probe and then washed in 0.5× SSC/0.5% SDS at 65°. Recombinants hybridizing to the probe were plaque-purified and reprobed. Recombinant phage DNA isolated by the plaque lysate method were digested withEcoRI and electrophoresed through a 1% agarose gel to determine the insert sizes. Several clones were identified ranging in size from 0.9 to 3 kb. One clone (cA313.1), which was 1.6 kb long, was subcloned into the EcoRI site of the plasmid vector pGEM-7z(−) (Promega). Double-stranded DNA was isolated, and both strands were sequenced in full, first by using T7 and Sp6 primers to get nucleotide sequence information near the 5′ termini, and then with a series of synthetic oligonucleotide primers derived from sequences determined previously.
Radioligand binding studies.
Membranes were prepared from COS-7 cells expressing the canine A3 receptor (cA313.1) or the canine A1receptor (RDC7); HEK 293 cells stably expressing human, rabbit or rat A3 adenosine receptors; or canine BR cells. The full coding region of the receptor cDNAs were subcloned into the expression vector CLDN10B and transiently expressed (60 hr) in COS cells by the DEAE-dextran method (16) or stably expressed in HEK 293 cells after transfection by the Ca2+ phosphate precipitation method (17) and selection in 2 mg/ml G-418. Transfected cells were washed in phosphate-buffered saline; homogenized in 10 mm EDTA, 10 mm Na-HEPES, pH 7.4, and 0.1 mm benzamidine; and centrifuged at 20,000 ×g for 20 min. Pellets were resuspended and washed in 10 mm Na-HEPES, 1 mm EDTA, pH 7.4, and 0.1 mm benzamidine (HE buffer) and resuspended in the same buffer with 10% (w/v) sucrose (sucrose buffer) at a membrane protein concentration of 1 mg/ml. Because [125I]ABA bound poorly to crude BR cell membranes, plasma membranes were enriched by preparing P2 pellets. Cells were homogenized in sucrose buffer and centrifuged at 500 × g for 10 min. The pellet was resuspended in sucrose buffer and centrifuged again at 500 ×g. The pooled supernatants were diluted 3-fold, pelleted, and washed twice by centrifugation at 20,000 × g for 20 min in HE buffer; resuspended; and frozen in sucrose buffer. Protein concentrations were determined using fluorescamine with BSA as standard. Membranes were frozen in aliquots and stored at −80°. For radioligand binding studies, cell membranes were incubated in 0.1 ml for 3 hr at 21° with 5 mm MgCl2 and 5 units/ml adenosine deaminase. For equilibrium binding assays, 6–8 concentrations of [125I]ABA were used in triplicate in tubes, each containing 10–60 μg of membrane protein, and the specific activity of [125I]ABA was reduced 10–20-fold with the nonradioactive compound. Nonspecific binding was measured in the presence of 5 μm I-ABA. [125I]ABA was found to have a higher ratio of specific to nonspecific binding than an alternative radioligand, [125I]AB-MECA. For competition experiments, 0.5–1 nm [125I]ABA was added to tubes, and competing ligands were added over a range of concentrations; the tubes contained 10–50 μg of membrane protein in a final volume of 0.1 ml.
Analysis of binding data.
Specific [125I]ABA binding to A1adenosine receptors was optimally fit to a single site binding model using Marquardt’s nonlinear least-squares interpolation (18). [125I]ABA was found to bind to two affinity states of the recombinant canine, human, and rat A3 receptors. For two-site Scatchard transformation, the relationship between bound/free and bound can be shown to be described by a quadratic equation: bound/free =A ∗ X ∗ X + B ∗X + C, where A = bound;X =Kd 1/Kd 2;B =Kd 1 ∗X +Kd 2 ∗X − B max1 ∗Kd 2 −B max2 ∗Kd 1; C =X ∗ X − B max1∗ X − B max2 ∗X. Optimal parameters for two-site Scatchard plots were generated by using the binomial theorem to solve this equation within each iteration of nonlinear least-squares analysis.
IC50 values of compounds in competition experiments were fit to SBi = Bi − (Bi − NS) [I]/(IC50i + [I]) where i is the number of binding sites, SB is specific binding, and NS is nonspecific binding. Ki values were calculated from IC50,B max, the concentration of [125I]ABA, and itsKd value, as described previously (19). For A3 receptors, the determination of theKi values of competing agonists for an agonist radioligand ([125I]ABA), is complicated by the fact that both the radioligand the competing compounds bind to two affinity states. This is described by four equations: LB = B max1 ∗ (L/Kd 1)/(1 +L/Kd 1+C/Ki 1) + B max2 ∗ (L/Kd 2)/(1 +L/Kd 2+C/Ki 2) + f ∗ L; CB =B max1 ∗ (C/Ki 1)/(1 +L/Kd 1+C/Ki 1) + B max2 ∗ (C/Ki 2)/(1 +L/Kd 2+C/Ki 2) + f ∗ C; LT = L +LB; CT = C + CB; whereLB is radioligand bound, CB is competitor bound,L is free radioligand, C is free competitor, andf is fraction of L or Cnonspecifically bound (assumed to be equal).Kd 1,Kd 2, and the fraction of coupled receptors were derived from equilibrium radioligand binding in the absence of competitor. The other parameters were determined by simultaneously solving these four equations by interpolation within each iteration of nonlinear least-squares analysis. For the analysis of antagonist binding,Ki 1 andKi 2 values were set to be equal based on the assumption that antagonists bind with similar affinities to G protein-coupled and -uncoupled receptors.
Northern blots.
Northern analysis and RT-PCR were used to determine the tissue distribution of A3 adenosine receptor transcript and to identify A2B, A1, and A3 receptor transcripts in BR cells. Total RNA was extracted and poly(A)+ RNA was selected using oligo(dt) cellulose. Five micrograms of poly(A)+ RNA was electrophoresed through 1% agarose gels containing 1% formaldehyde and then transferred to nylon membranes (Genescreen Plus; DuPont). The membranes were hybridized in 10% dextran sulfate, 1 mNaCl, and 100 μg/ml herring sperm DNA with 1 × 106 cpm/ml random-labeled probe at 65° overnight. Filters were washed with 0.5× SSC/0.5% SDS at 65° and then exposed to Amersham Hyperfilm MP for 24–48 hr. The A3 receptor probe consisted of a 600-bp PCR fragment of cA313.1, corresponding to approximately half of the carboxyl-terminal sequence and the 3′ noncoding region. The A2B receptor probe consisted of a 500-bp PCR fragment generated by RT-PCR from BR cell RNA corresponding to transmembrane regions I–IV.
For RT-PCR, 1 μg of poly(A)+-selected RNA was reverse-transcribed and amplified using primers A and B described above. The reactions were electrophoresed and transferred to nylon membranes (Hybond N+; Amersham) using denaturing buffer (0.4 n NaOH) and hybridized with specific probe. Filters were washed under stringent conditions (0.1× SSC/0.5% SDS at 65° for 1 hr). Control reactions were included in which RT or RNA was excluded from the reactions. Some of the PCR fragments were subcloned into the TA vector and partially sequenced.
Mastocytoma cell degranulation.
As an indicator of degranulation of BR cells, we measured the release of β-hexosaminidase (a granule-associated protein that parallels histamine release) using a modification of the method of Schwartzet al. (20). BR cells grown in suspension were washed twice in Ca2+/Mg2+-free Tyrode’s buffer and then resuspended in complete Tyrode’s at a density of 1.2 × 106 cells/ml. Cells were then transferred to a 96-well plate in 250-μl aliquots and prewarmed to 37° for 15 min. Cells were stimulated with agonists added in 50-μl aliquots for 20 min at 37° with shaking. The reactions were stopped by placing the plate on ice for 10 min and then pelleting the cells by centrifugation at 200 × g for 10 min (4°). Two hundred microliters of the supernatant was removed and added to 50 μl of 5 mm p-nitrophenyl-N-acetyl-d-glucosaminide, and 100 mm citric acid, pH 3.8, and incubated at 37° for 2 hr with shaking before the addition of 50 μl of 0.4 mNaCO3. Total cellular β-hexosaminidase was determined by adding 50 μl of lysis buffer (complete Tyrode’s buffer plus 0.6% Triton A-100) to 250-μl aliquots of cells, and 20 μl was removed and assayed. Absorbance was read at 405 nm using a Titertech Multiskan II plate reader. Experiments were performed in triplicate, and release of β-hexosaminidase is expressed as percentage of the total content of unstimulated cells.
cAMP.
BR cells were washed twice and resuspended in serum-free low-glucose DMEM containing 25 mm HEPES, 1 unit/ml adenosine deaminase, and 20 μm Ro 20–1724 and then transferred to polypropylene test tubes (1 × 106 cells/0.2 ml, 21°). Drugs were added in 50-μl aliquots, and the tubes were placed in a 37° shaking water bath for 20 min. Assays were terminated by the addition of 500 μl of 0.15 n HCl. cAMP in the acid extract (500 μl) was acetylated and quantified by automated radioimmunoassay.
Intracellular Ca2+.
BR cells were loaded with 1 μm Fura-2/AM in buffer containing 100 mmNaCl, 5 mm KCl, 1 mmMgSO4, 1 mmKH2PO4, 25 mmNaHCO3, 0.5 mmCaCl2, 2.7 g/liter d-glucose, 20 mm Na-HEPES, pH 7.4, and 0.25% BSA for 45 min. Cells were washed and resuspended in the same buffer without BSA, plus 1 unit/ml adenosine deaminase to a density of 1 × 106cells/ml. Fluorescence was measured with an SLM spectrofluorimeter in a thermostable cuvette (37°).
InsP3.
BR cells were preincubated for 24 hr with 2.5 μCi/ml myo-[3H]inositol in inositol-free low-glucose DMEM supplemented with 2% dialyzed fetal calf serum. The labeled cells were washed and resuspended in low-glucose DMEM with 25 mm HEPES, 1 unit/ml adenosine deaminase, and 100 mm LiCl and then transferred to polypropylene test tubes (4 × 105 cells/0.2 ml) at 37° in a shaking water bath and stimulated by 5× agonists added in 50-μl aliquots for 10 min. Assays were terminated by the addition of 400 μl stop solution (0.5 mHCLO4, 5 mm EDTA, and 1 mm diethylenetriaminpentacetic acid) plus 1 mg/ml phytic acid and placed on ice for 30 min before the addition of 5m K2CO3 to raise the pH to 8–9. After centrifugation, the supernatants were passed through a 0.2-μm filter, applied to 1-ml Dowex AG 1-X8 columns (200–400 mesh), and washed with 5 ml of H2O and 5 ml of 40 mm HCl; then, InsP3 was eluted with 5 ml of 170 mm HCl.
Results
Molecular cloning of the canine A3 adenosine receptor.
The screening of a canine mastocytoma cDNA library with an A3 adenosine receptor probe generated by RT-PCR resulted in the identification of several positively hybridizing clones. A clone designated cA313.1 contains a 1.6-kb insert with an open reading frame corresponding to 314 amino acids and 181 and 480 bp of 5′ and 3′ untranslated sequence, respectively (Fig. 1). A hydrophilicity plot of the deduced amino acid sequence predicts seven transmembrane domains, which are indicated in Fig. 2A. Sites found to be conserved within all species of A3 adenosine receptors cloned to date include a putative palmitoylation site at Cys305 of the consensus sequence and two putative N-linked glycosylation sites at Asn4 and Asn162. Several putative phosphorylation sites are conserved among the A3 adenosine receptors, including four potential sites for phosphorylation by protein kinase C (Thr124, Thr125, Ser/Thr215, and Thr230); one potential site for phosphorylation by tyrosine kinases (Tyr120); and one potential site for phosphorylation by cAMP/cGMP-dependent protein kinases (Thr294). The carboxyl tail distal to the palmitoylation site contains several serine/threonine residues that are surrounded by acidic groups that may be sites for phosphorylation by G protein receptor kinases.

Nucleotide and amino acid sequences of cA313.1, the canine A3 adenosine receptor. The sequence has been deposited in GenBank (accession no. U54792).

Deduced amino acid sequence of cA313.1, the canine A3 adenosine receptor. A, Alignment with human, sheep, rat, and rabbit A3 adenosine receptor sequences.Solid lines, putative transmembrane (TM) domains with numbered designations (I–VII). Dashes, sequence gaps. Sites conserved among all species for possibleN-linked glycosylation (glyc); phosphorylation by protein kinase C (PKC), cAMP-dependent protein kinase (cAMP), tyrosine kinases (tyr), and palmitoylation (palm) are designated. B, Dendogram illustrating species and subtype sequence differences among adenosine receptors. Distance along the horizontal axis is proportional to divergence of the amino acid sequences.
The deduced amino acid sequence of cA313.1 is 88%, 86%, 72%, and 77% identical to the human, sheep, rat, and rabbit receptors (Fig. 2B), respectively, and 52% and 47% identical to canine A1 and A2Areceptors, suggesting that cA313.1 is the canine species homolog of the A3 adenosine receptor. Between species, the greatest degree of homology lies within the transmembrane domains, and the least lies within the carboxyl tail. Of the five species of A3 adenosine receptors cloned to date, the human receptor amino acid sequence is most similar to the canine and least similar to the rat. We noted previously that the human and rat A3 adenosine receptors are unusually divergent for species homologs (12). In contrast, canine and human A3 adenosine receptors show a degree of amino acid sequence homology that is similar to species differences among the other adenosine receptor subtypes (Fig. 2B).
Tissue distribution of A3 mRNA.
Northern blots probing for cA313.1 transcripts in several different canine tissues revealed two hybridizing bands of 1.9 and 2.7 kb (Fig. 3). Transcripts were most abundantly expressed in spleen, but high levels also were detected in lung and liver. Two major hybridizing bands were also observed in testes, but the sizes were 1.3 and 2.4 kb. Transcripts were not detected in heart or kidney by Northern analysis. Using the sensitive technique of RT-PCR, trace transcripts were observed in all six tissues studied (data not show). Transcripts for A1, A3, and A2B adenosine receptors were detected by Northern blotting of BR cell poly(A)+ mRNA (data not shown). The size of the A3 transcripts, 1.9 and 2.7 kb, is the same as in dog spleen, lung, and liver. The A2B transcript sizes are 1.6 and 1.8 kb and correspond to transcript sizes noted previously for A2B receptor transcripts in mouse bone marrow-derived mast cells (21).
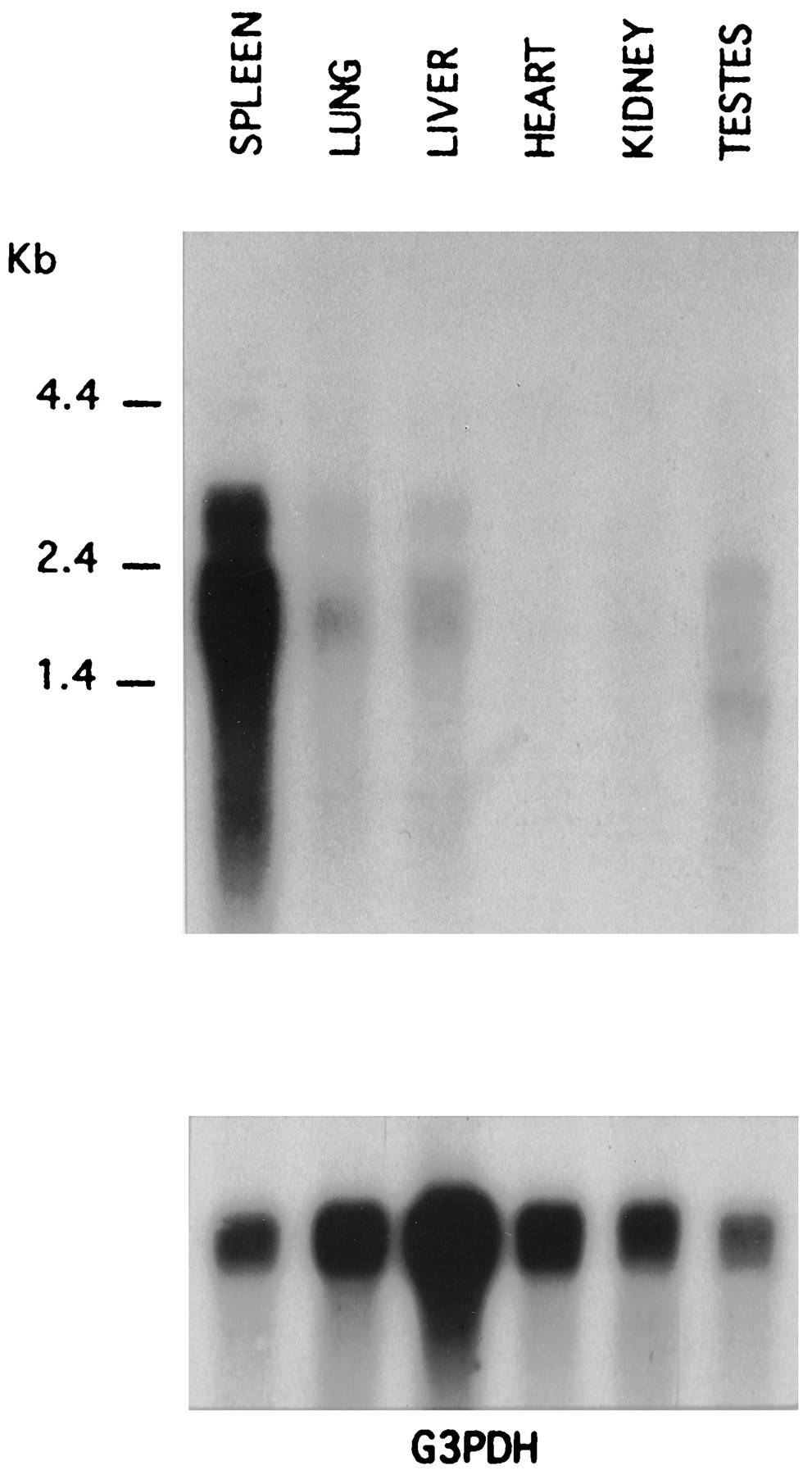
Tissue localization of the canine A3adenosine receptor transcript. Northern blots of poly(A)+RNA (5 μg/lane) from six different dog tissues using a probe corresponding to the carboxyl-terminal tail and 3′ untranslated region of cA313.1. The same blot was stripped and reprobed for glyceraldehyde-3-phosphate dehydrogenase transcript (G3PDH). RNA size markers are indicated.
Pharmacological characterization of canine A3 adenosine receptors.
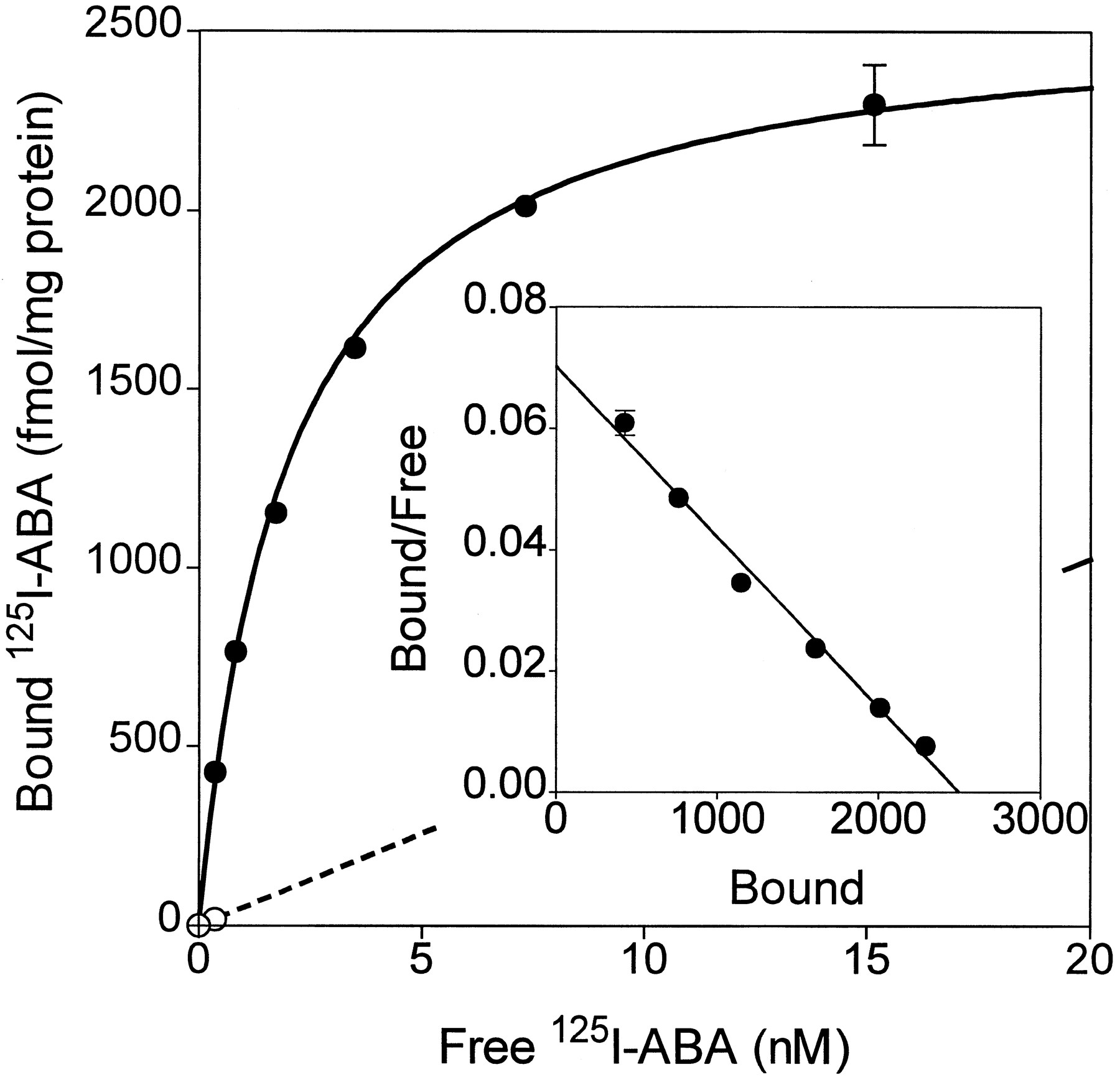
Binding of [125I]ABA was measured to membranes prepared from COS-7 cells transfected with cA313.1 (Fig. 4). Specific binding was absent in untransfected cells and was abolished by 1 μm nonradioactive I-ABA. N-Ethylmaleimide has been reported to alkylate G proteins in the Gi/o family and cause them to become uncoupled from receptors. GTPγS and N-ethylmaleimide both reduced specific binding of [125I]ABA to canine A3 adenosine receptors by ∼60%, indicating that the radioligand is an agonist and that Gi/Go proteins couple to the A3 receptor (Fig. 4A). In equilibrium binding studies, [125I]ABA specific binding was consistently found to fit significantly (p < 0.01) better to a two-site than to a one-site model (22). The respective high and low affinity Kd values of [125I]ABA are 0.53 ± 0.13 and 16.4 ± 0.8 nm, andB max values are 250 ± 9 and 768 ± 123 fmol/mg of membrane protein. In the presence of 50 μm GTPγS, [125I]ABA binds only to the low affinity site, with aKd value of 17.4 ± 0.1 nm and a B max value of 768 ± 123.0 fmol/mg of total protein. The conversion of receptors from two affinity states to a single low affinity state on the addition of GTPγS is most clearly illustrated by Scatchard analysis (Fig. 4C). These results suggest that the high affinity site reflects binding to G protein-coupled receptors and the low affinity site reflects binding to uncoupled receptors. A similar analysis indicates that [125I]ABA also binds to two affinity states of recombinant rabbit A3adenosine receptors with KD values of 1.2 and 34 nm (not shown). In contrast, in filtration assays, [125I]ABA detects only the high affinity state of canine A1 receptors transiently expressed in COS-7 cells (Kd = 2.67 ± 0.50 nm, B max = 1275 ± 52 fmol/mg protein; Fig. 5), and specific binding is almost completely abolished by the addition of GTPγS (data not shown).

Radioligand binding to recombinant canine A3 adenosine receptors. A, Inhibition of [125I]ABA (1.4 nm) binding to COS-7 cell membranes expressing cA313.1 by unradiolabeled I-ABA, GTPγS, and N-ethylmaleimide. B, Equilibrium specific and nonspecific (NS) binding of [125I]ABA to transfected COS-7 cell membranes in the presence and absence of GTPγS. C, Scatchard transformation of the specific binding data shown in B. For B and C, control data were fit optimally to two-site equations as described in Experimental Procedures. Values are mean ± standard error of triplicate determinations (25 μg of membrane protein/tube); where omitted, standard error bars are smaller than the symbols. The results are typical of three experiments. Binding parameters are summarized in Table 1.
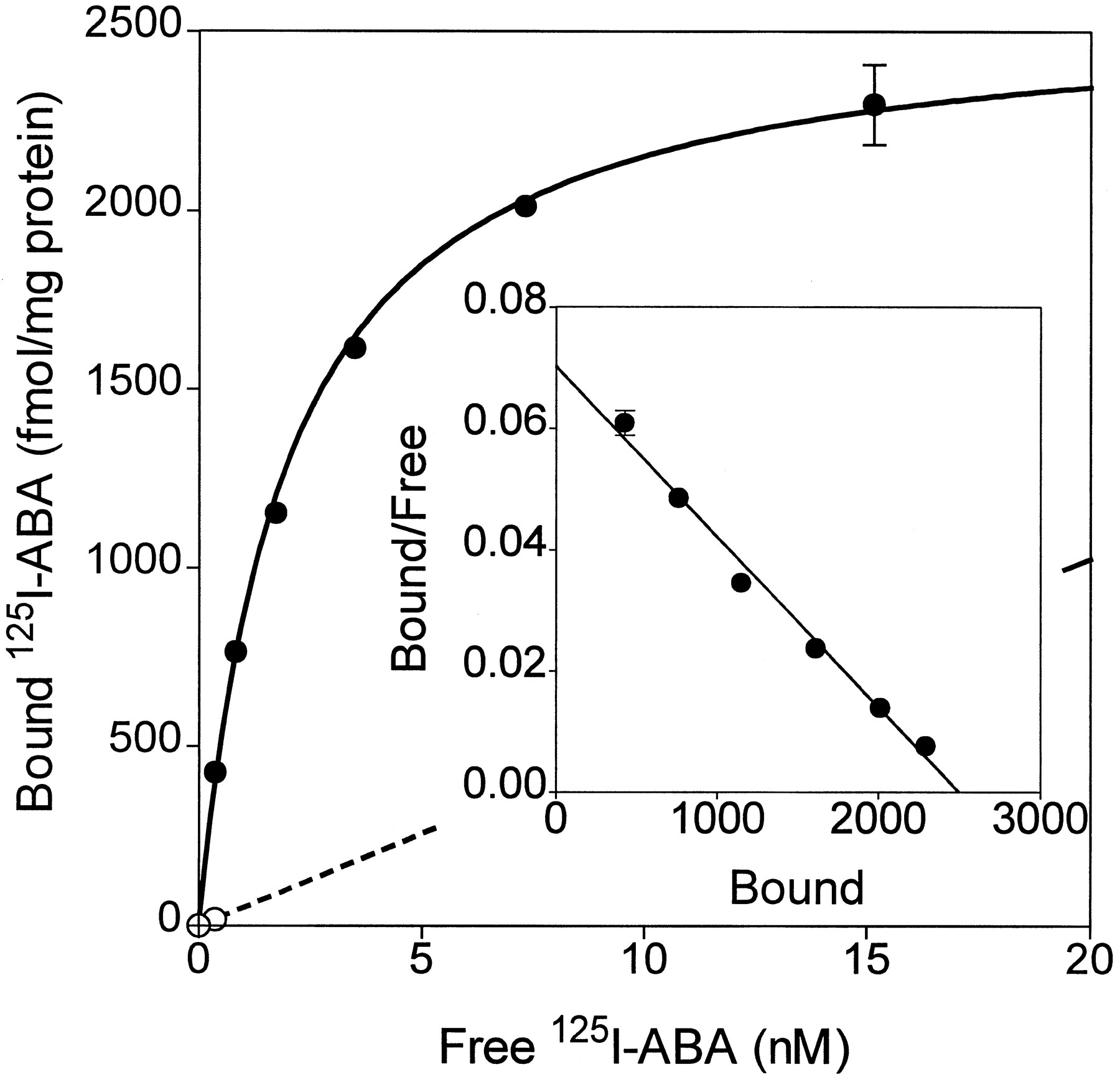
Radioligand binding to recombinant canine A1 adenosine receptors (RDC7). Equilibrium specific (•) and nonspecific binding (○) binding of [125I]ABA to transfected COS-7 cell membranes is plotted. Inset, Scatchard transformation of the specific binding. Values are mean ± standard error of triplicate determinations (10 μg of membrane protein/tube); where omitted, standard error bars are smaller than the symbols. The results are typical of three experiments. Binding parameters are summarized in Table 1.
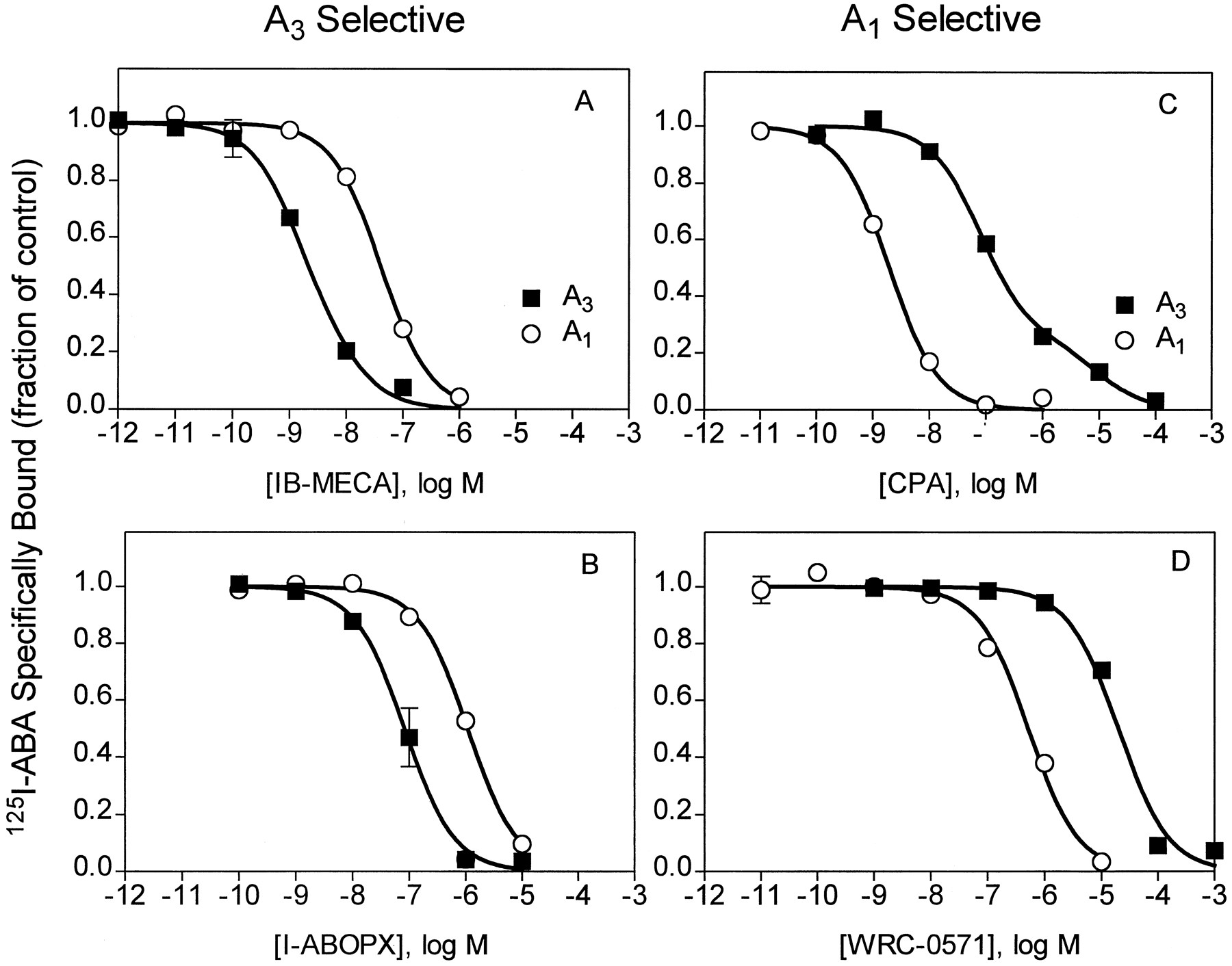
We next compared the binding properties of recombinant canine A1 and A3 adenosine receptors. Fig. 6 shows the results of competition binding studies with compounds found to be the most A3 selective, IB-MECA and I-ABOPX, and those most A1 selective, CPA and WRC-0571.Ki values of these and other compounds are summarized in Tables 1 and2. These tables also summarize the relative affinities of competing compounds for canine A1 and A3 receptors. For the A3 receptor subtype, two dissociation constants for agonists and single dissociation constants for antagonists were calculated as described in Experimental Procedures. The potency order of agonists for canine A3receptors was IB-MECA ≥ [125I]ABA > PIA > APNEA > CPA > NECA > CGS 21680. IB-MECA is 58-fold selective for the A3 over the A1 adenosine receptor (high affinity sites), and WRC-0571 and CPA are 36- and 12-fold selective, respectively, for the A1 over the A3 receptor. The canine A3 adenosine receptor binds antagonists with the potency order of I-ABOPX > CPX > XAC > BWA 1433 > WRC 0571 > 8-SPT (Table 2). Theophylline and enprofylline bind very weakly to canine A3 adenosine receptors; a 100 μm concentration of these compounds reduces specific binding by only ∼40%. I-ABOPX is 16-fold selective for canine A3 over A1 adenosine receptors.

Competition for [125I]ABA binding to COS-7 cell membranes derived from cells transfected with canine A3 or A1 adenosine receptors. Binding is plotted as the fraction of control specific binding (>90% of total binding). Values are mean ± standard error of triplicate determinations from three experiments. Protein, 25 μg/tube (A3) or 10 μg/tube (A1), [125I]ABA, 150,000–300,000 cpm/tube (0.4–0.8 nm). The binding of [125I]ABA and competing ligands was fit to one or two binding sites as described in Experimental Procedures.
Dissociation constants of agonists for the canine A3(cA3 13.1) and A1 adenosine receptors determined in binding assays
Dissociation constants of antagonists for the canine A3(cA3 13.1) and canine A1 adenosine receptors determined in binding assays
Table 3 shows a comparison of the binding affinities of four xanthines with those of recombinant human, canine, rabbit, and rat A3 receptors and confirms that there are marked species differences in the binding affinities of xanthine antagonists. Of the four xanthines examined, the average ratio of binding affinities is 9.6 (canine/human), 5.0 (rabbit/human), and 212 (rat/human). The canine A3 receptor binds CPX with higher affinity than any of the other species examined.
Species differences in the binding of xanthines to A3adenosine receptors
[125I]ABA binding to canine BR mastocytoma cell membranes.
Because both A1 and A3 adenosine receptor transcripts were detected in canine BR cells, we next determined whether [125I]ABA binding to A1and/or A3 receptors could be detected in membranes prepared from these cells. Little specific binding could be detected to crude membranes, but the binding of 0.4 nmradioligand to an enriched P2 membrane preparation was 80% specific (Fig. 7). Of the [125I]ABA binding site on BR cell membranes, 24 ± 3% (three experiments) bind WRC-0571 with low affinity (IC50 = 22 ± 9 μm) characteristic of A3 receptors; the remainder bind WRC-0571 with high affinity (IC50 = 114 ± 38 nm) characteristic of A1receptors (Fig. 7, Table 2). When added at 0.4 nm, [125I]ABA labeled only 2.4 fmol/mg of protein of A3 receptors in the P2 membranes of BR cells, suggesting the density of A3 receptors on BR cells is low.

Competition by WRC-0571 for [125I]ABA binding to enriched plasma membranes derived from BR cells. P2 membranes were prepared as described in Radioligand Binding Studies. Binding was determined in triplicate to membranes containing 156,000 cpm (0.4 nm) [125I]ABA, 60 μg of P2 membrane protein, and various concentrations of WRC-0571. Nonspecific binding (10 μm I-ABA) is 846 ± 18 cpm (dashed line). Standard error bars are smaller than the symbols. The data were fit to a two-site model probably reflecting WRC-0571 binding to A1 and A3 adenosine receptors. Binding parameters from triplicate experiments are summarized in the text.
Characterization of the adenosine receptor that causes degranulation of canine mastocytoma cells.
Adenosine agonists have been shown to enhance A23187 (calcimycin)-stimulated degranulation of several types of mast cells, including murine bone marrow-derived mast cells, human lung mast cells, RBL-2H3 cells, and rat peritoneal mast cells (23, 24). Fig. 8 shows the effect of increasing concentrations of Ca2+ ionophore to elicit β-hexosaminidase release from BR cells when administered alone or in combination with the nonselective adenosine receptor agonist NECA (10 μm). When administered alone, A23187 evokes a maximal release of ∼15.6% of the total cellular content after 20 min of stimulation. NECA (10 μm) alone also stimulates β-hexosaminidase release (5.81 ± 0.59%), and costimulation with NECA and A23187 increases β-hexosaminidase release to 28.2 ± 1.8%. NECA decreases the EC50 for A23187 from 0.32 ± 0.06 to 0.13 ± 0.08 μm.

Stimulation of β-hexosaminidase release from BR cells by A23187 and NECA. A, Cells (300,000/tube) were stimulated with increasing concentrations of A23187 in the presence or absence of 10 μm NECA for 20 min. β-Hexosaminidase released into the supernatant was measured as described in Experimental Procedures. B, Effects on NECA-stimulated β-hexosaminidase release of pretreatment of cells for 15 min with NBTI (1 μm) or XAC (1 μm). Data are pooled from three separate experiments, each assayed in triplicate. ∗, Different from control (p < 0.05).
We next conducted experiments to determine whether NECA acts at receptor sites on the cell surface or at intracellular sites. To test the possibility that NECA may gain access into BR cells via facilitated uptake and influence the degranulation response via an intracellular mechanism, BR cells were preincubated for 15 min with NBTI (1 μm), an inhibitor of nucleoside transport. As shown in Fig. 8B, NBTI did not influence the concentration response of NECA to stimulate β-hexosaminidase release. As additional evidence that NECA acts at cell surface adenosine receptors, we added XAC, a nonselective antagonist of all subtypes of canine adenosine receptors, including A3 (see Table 2). XAC completely abolished the stimulatory effect of NECA (Fig. 8B). Additional experiments were performed with NBTI and XAC during costimulation with a combination of A23187 and NECA. As in the absence of A23187, NBTI had no effect, and XAC abolished NECA-mediated degranulation responses (data not shown).
Pertussis toxin blocks the inhibition of cAMP accumulation in CHO-K1 cells that is mediated by recombinant rat A3adenosine receptors (2). Pertussis intoxication of rats also reduces a putative A3 adenosine receptor-mediated hypotensive response (25). These data suggest that A3 adenosine receptors functionally couple to Gi/Go proteins; therefore, we examined the effect of pretreating BR cells with pertussis toxin on the ability of NECA to stimulate degranulation (Fig.9). For these experiments, BR cells were cultured in serum-free medium with 0.3 or 1 μg/ml of pertussis toxin for 24 hr.1 We found that cells cultured in serum-free medium released a greater amount of β-hexosaminidase in response to 1 μm A23187 (∼25–35% without serum versus ∼15% with serum). Pretreatment of cells with either concentration of pertussis toxin did not prevent NECA-stimulated degranulation. These data suggest that neither A1 nor A3 adenosine receptors are solely responsible for adenosine-mediated degranulation of BR cells.

Effect of pretreatment of BR cells with pertussis toxin (PTX) on NECA-stimulated degranulation. Cells were pretreated for 24 hr with 0.3 μg/ml (A) or 1 μg/ml (B) pertussis toxin and then stimulated for 20 min with 1 μm A23187 and various concentrations of NECA. Data are pooled from three separate experiments, each assayed in triplicate.
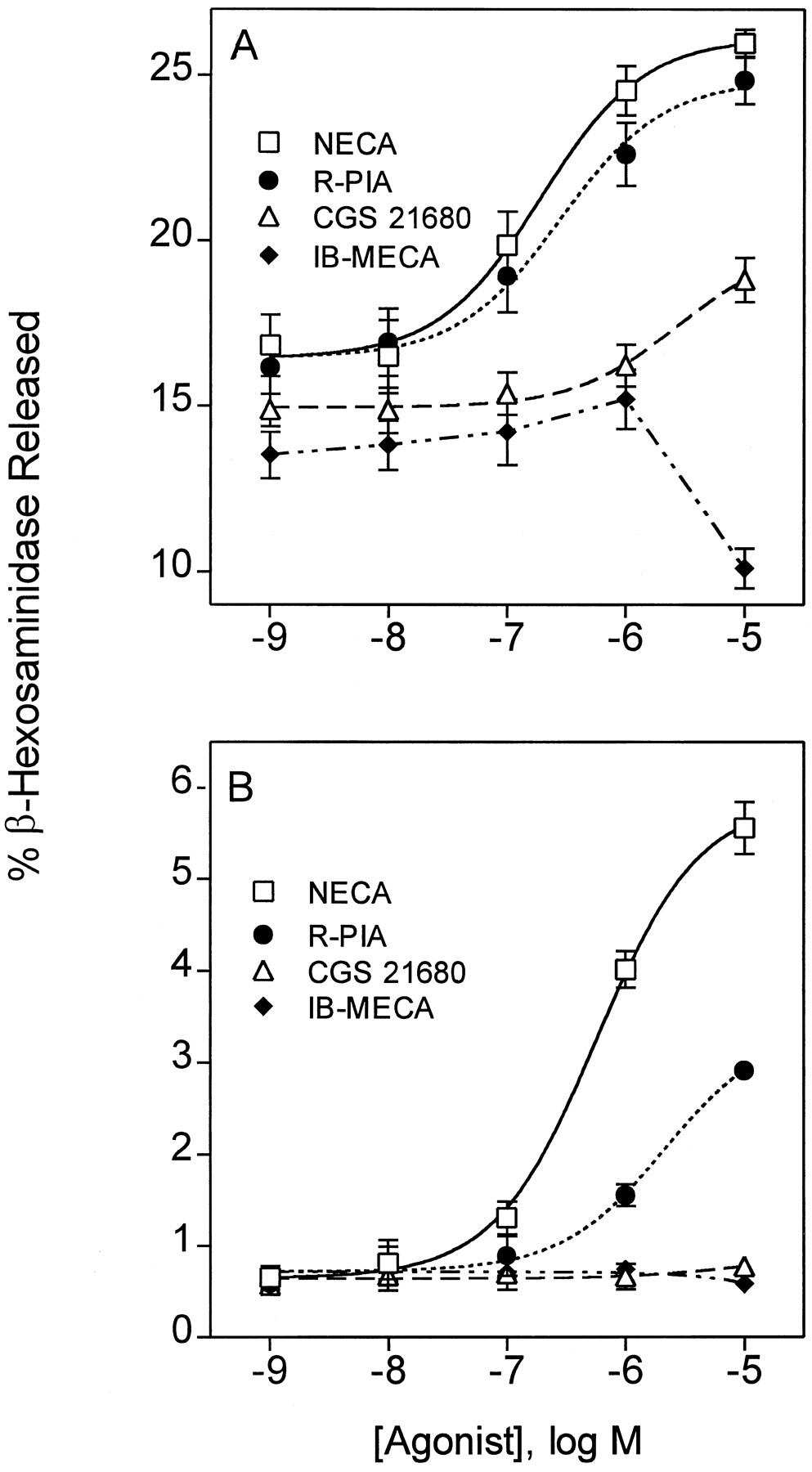
We next determined the potency order of various adenosine analogs to stimulate β-hexosaminidase release from BR cells. In addition to NECA, we examined (R)-PIA (A1selective), CGS 21680 (A2A selective), and IB-MECA (A3 selective). Experiments were performed in the presence or absence of 1 μm A23187 (results are illustrated in Fig. 10 and summarized in Table 4). The potency order of agonists to stimulate canine mast cell degranulation, NECA > PIA > CGS-21680 > IB-MECA, differs from the potency order of these compounds for binding to canine A3adenosine receptors, IB-MECA > PIA > NECA > CGS-21680. IB-MECA has very little stimulatory effect on BR cell degranulation and, when added at 10 μm, IB-MECA inhibits degranulation.

β-Hexosaminidase release from BR cells in response to adenosine receptor agonists. A, Cells were stimulated with adenosine agonists and 1 μm A23187 for 20 min. B, Cells were stimulated with adenosine agonists alone. Values are mean ± standard error of data pooled from three separate experiments, each assayed in triplicate.
Potency of adenosine analogs to stimulate β-hexosaminidase release from canine BR mastocytoma cells mean ± SEM, n = 3
The observed potency order for adenosine analogs to stimulate BR mast degranulation is similar to the potency order reported by Brackett and Daly (26) for activating A2B receptors on NIH 3T3 fibroblasts and the potency order for binding to recombinant human A2B receptors (27). Furthermore, A2B receptors in general have low affinity for agonists, similar to their low affinity for stimulating degranulation of BR cells (Table 4). Because A2B receptor transcript is found in BR cells, we postulated that A2B adenosine receptors are expressed on BR cells and, when activated, stimulate degranulation. To test this hypothesis, we investigated the effects of enprofylline, which we recently identified as a selective antagonist of recombinant human A2B receptors (27). Concentration-response curves for degranulation in response to NECA were generated in the presence of 10, 50, and 250 μm enprofylline. As shown in Fig.11A, increasing concentrations of enprofylline produced parallel rightward shifts of the concentration-response curves for β-hexosaminidase release. Schild regression analysis revealed a slope close to unity (1.12 ± 0.45), suggesting that enprofylline acts as a competitive antagonist at a single receptor subtype. The KD value of enprofylline was estimated to be 7.8 ± 3.3 μm. This value is almost identical to theKI value of enprofylline for binding to human A2B receptors and is well below theKI value of enprofylline for canine A1 or A3 receptors. Interestingly, the Ki value of enprofylline for A2B receptors lies within the therapeutic range of this compound as an antiasthmatic therapeutic agent (28). Because enprofylline also inhibits cAMP phosphodiesterase, we evaluated the effects of another phosphodiesterase inhibitor, Ro 20–1724, on NECA-induced β-hexosaminidase release. Ro 20–1724 is a nonxanthine that does not bind to adenosine receptors. As shown in Fig.11B, Ro 20–1724 has no effect on NECA-induced degranulation. The data are consistent with the possibility that enprofylline blocks BR cell degranulation by binding to A2B adenosine receptors.

Effects of enprofylline and Ro 20–1724 on β-hexosaminidase release from BR cells. A, Antagonistic effect of enprofylline on NECA-induced β-hexosaminidase release from BR cells costimulated with 1 μm A23187 and various concentrations of NECA. Inset, Schild plot; for enprofylline, pA 2 = 5.1. B, Cells were pretreated with or without 20 μm Ro 20–1724 for 15 min and then stimulated with NECA. Data are mean ± standard error of three determinations; similar results were observed in a replicate experiment.
Second messenger responses evoked by A2B receptor activation in canine mastocytoma cells.
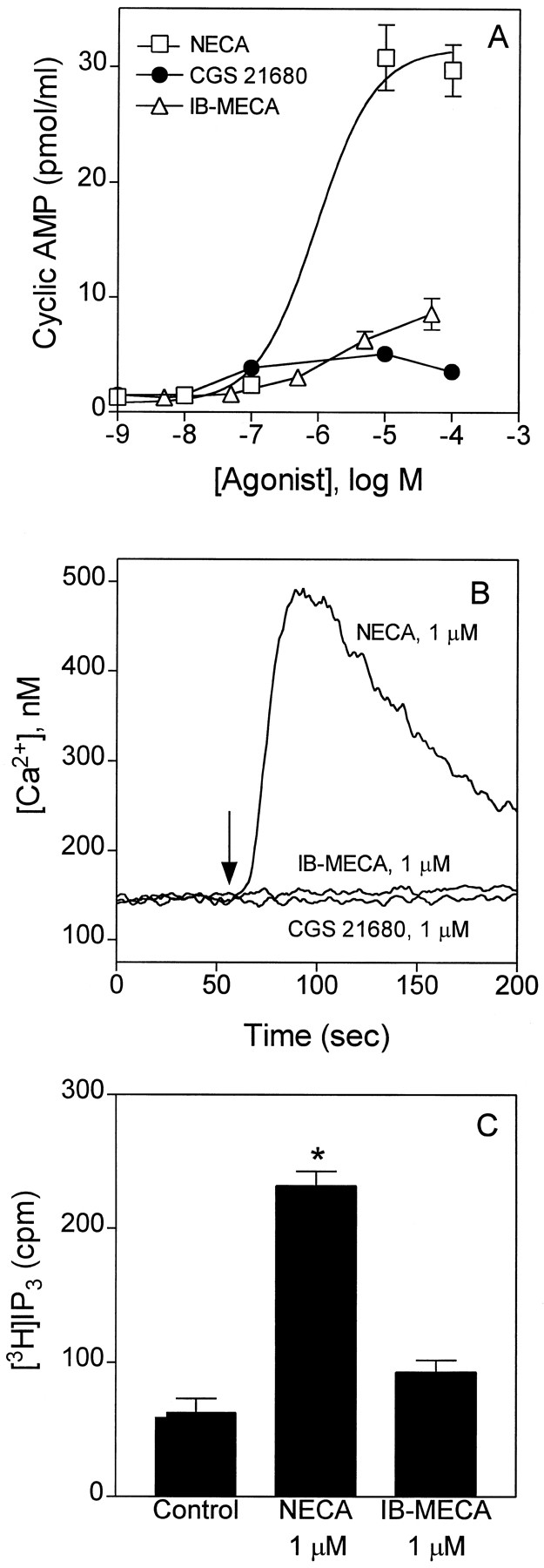
We next measured second messenger responses to adenosine receptor activation in BR cells. Unlike A3 adenosine receptors, which are inhibitory to adenylyl cyclase, A2B adenosine receptors stimulate adenylyl cyclase (26). We measured changes in intracellular levels of cAMP, Ca2+, and InsP3 in response to NECA, CGS 21680, and IB-MECA. As shown in Fig. 12, NECA, but not CGS 21680 or IB-MECA, produces a concentration-dependent increase in intracellular levels of cAMP. The absence of cAMP accumulation in response to CGS-21680 indicates that accumulation of the cyclic nucleotide in response to NECA is not mediated by A2A adenosine receptors. The calculated EC50 for NECA to increase cAMP in BR cells is 0.9 ± 0.2 μm, which is similar to the EC50 value for NECA to stimulate β-hexosaminidase release (0.6 ± 0.3 μm), suggesting that both responses are mediated by the same receptor subtype. NECA (1 μm) also increased intracellular levels of Ca2+ and InsP3, whereas 1 μm CGS -21680 or 1 μm IB-MECA had little effect (Fig. 12). Small responses to CGS 21680 and IB-MECA to influence cAMP, Ca2+, or InsP3suggest that selective activation of A2A or A3 receptors has little effect on cAMP or Ca2+ signaling in canine BR mastocytoma cells.

Changes in intracellular levels of cAMP, Ca2+, and InsP3 in BR cells during stimulation with adenosine agonists. A, cAMP levels were measured in suspended BR cells incubated for 10 min with 20 μm Ro 20–1724. B, Ca2+ levels were measured in suspended cells loaded with FURA-2/AM. C, InsP3 levels were measured in cells prelabeled with myo-[3H]inositol. All incubations included 1 unit/ml adenosine deaminase. Data are mean ± standard error of three experiments. ∗, Greater than control and IB-MECA (p < 0.01).
Enprofylline competitively antagonized the increases in intracellular levels of cAMP and Ca2+ produced by NECA (Fig.13). By Schild regression analysis (slope = 0.81 ± 0.15), theKD value of enprofylline was estimated to be 4.7 ± 3.2 μm from the cAMP response and 15 μm from the Ca2+ response, values similar to theKD value estimated for enprofylline to inhibit NECA-stimulated β-hexosaminidase release (7.8 μm). These results suggest that A2B receptors in BR cells are positively coupled to adenylyl cyclase and phospholipase C.

Antagonistic effects of enprofylline on second messenger responses in canine BR cells. A, cAMP responses to NECA and enprofylline. Inset, Schild plot; for enprofylline, pA 2 = 5.3. B, Concentration-dependence of Ca2 responses to NECA with and without enprofylline (100 μm); the EC50 values for NECA are 760 and 5725 nm in the absence and presence of enprofylline, respectively. The pA 2 value for enprofylline is estimated to be 4.8. Results are typical of three experiments.
Discussion
We cloned and characterized a canine A3 adenosine receptor cDNA designated cA313.1 from canine BR mastocytoma cells. The clone is homologous to A3 receptors cloned from other species. Transcripts for A1 and A2B adenosine receptors also were detected in BR cells, and evidence of A1, A2B, and A3 receptor expression was found on the basis of radioligand binding or functional assays. The A2B receptor predominates in the regulation of BR cell degranulation, as discussed in detail below.
The tissue distribution of A3 adenosine receptor transcript in dog is similar to the human distribution (3), with highest levels expressed in spleen, followed by lung and liver. The tissue distribution is different from rat, in which transcript is much more abundant in testes than in other tissues (1). In terms of sequence homology and pharmacology, the canine A3adenosine receptor is more similar to the human than to the rat A3 receptor.
The results of radioligand binding assays with the A1/A3 agonist [125I]ABA indicate that the canine A3 adenosine receptor binds to both G protein-coupled and -uncoupled receptors withKD values that differ by ∼30-fold. The potency order of agonists for the A3receptor, IB-MECA > R-PIA ≥ NECA > CPA, is consistent among species. In all species, IB-MECA and CPA are A3 and A1 selective, respectively. Specific binding of [125I]ABA to the uncoupled conformation of the canine A3adenosine receptor distinguishes the canine A3receptor from the uncoupled canine A1 receptor, which has too low affinity for [125I]ABA binding to be detected in filtration assays. However, [125I]ABA binds with 10 times higher affinity to bovine than to canine A1 receptors, and the radioligand can detect two affinity states of the bovine A1 adenosine receptor [Kd = 0.09 and 10.4 nm (29)]. The detection in filtration assays of two affinity states of A3 receptors complicates the analysis of competition binding assays because the radioligand binds with two affinities and competing agonists and antagonists bind with two or one affinities, respectively. To calculate theKi values of competing compounds required the derivation of nonstandard analytical procedures (see Analysis of Binding Data). As summarized in Table 1, I-ABA and IB-MECA both bind with high affinity (KD < 1 nm) to the G protein-coupled conformation of canine A3 receptors. Failure to analytically resolve the two agonist affinity states in radioligand binding assays will result in underestimation of high affinity agonist dissociation constants as well as errors in the calculation of the dissociation constants of competing compounds based on the Cheng and Prusoff formula (30). It is notable that in the range of 0.1–1 μm, compounds that often are used as selective agonists of A1 receptors (CPA) or A2A receptors (CGS-21680) also will bind to canine A3 receptors. Hence, caution must be taken in attributing functional responses of these compounds to particular adenosine receptor subtypes.
This study confirms and extends the observation that there are substantial species differences in the binding of xanthines to A3 adenosine receptors. The rat A3 adenosine receptor, the first A3 adenosine receptor to be cloned, was originally reported not to bind xanthine antagonists (2). Subsequent studies have shown that xanthines bind weakly to the rat receptor. The most potent xanthine antagonist, I-ABOPX, binds to the rat A3 receptor with aKI value of 1.5 μm. In contrast, sheep, human, and canine A3 receptors bind I-ABOPX with 80–500 times higher affinity (3, 4).
CPX is widely regarded as a selective antagonist of A1 adenosine receptors. Although CPX is >250-fold selective for human A1 over A3 receptors (27), this selectivity drops to only 10-fold in the case of canine receptors. This is partly due to the fact that compared with human and sheep A3 receptors, canine A3 receptors bind CPX with relatively high affinity. In addition, canine A1 adenosine receptors have lower affinity than other species for CPX. Consequently, CPX is not particularly useful for discriminating between A1 and A3 receptor-mediated responses in the dog. A preferable compound for this purpose is WRC-0571, an A1-selective nonxanthine antagonist that is >4000-fold selective for human A1 over A3 receptors (31). Although WRC-0571 binds with much lower affinity to canine A1 receptors (KI = 484 nm) than to human A1 receptors (KI = 3 nm), it still is 35-fold selective as an antagonist of canine A1 over A3 receptors. Species differences in binding affinity also are significant for BW-A1433 [8-(4-carboxyethenylphenyl)-1,3-dipropylxanthine], which is sometimes used as a A3 receptor antagonist on the basis of its moderate affinity for sheep and human A3 receptors (3, 4). BW-A1433 is a relatively weak and nonselective antagonist of canine A3receptors, binding with 10-fold lower affinity to canine than to human A3 receptors.
Enprofylline, an antiasthmatic agent that has moderate affinity for human A3 receptors [KI = 156 ± 110 μm (27)], binds poorly to the canine A1 and A3 receptors (KI > 100 μm). Because enprofylline binds to the human A2B adenosine receptors with aKI value of 7 μm (27), the compound was evaluated in this study to discriminate between canine A2B and A1 or A3 adenosine receptor-mediated responses. Inasmuch as the canine A3 receptor clone was isolated from a canine mastocytoma cDNA library, we anticipated that the A3 receptor subtype would be responsible for stimulating the release of granule-associated mediators. However, A2B and A1 as well as A3 transcript were found in BR cells, and low levels of A1 and A3receptor binding sites could be detected on enriched plasma membranes prepared from the canine mastocytoma cells. Nevertheless, several lines of evidence indicate that BR cell degranulation requires activation of the A2B but not the A1 or A3 adenosine receptor: (i) degranulation of BR cells is not prevented by pretreatment of cells with pertussis toxin; (ii) the response is blocked by enprofylline with a pA 2 value near 5, an affinity similar to that of human A2B receptors and higher that the affinity of enprofylline for canine A1 or A3 receptors; (iii) the potency order of agonists to stimulate degranulation, NECA > PIA > CGS-21680 > IB-MECA, differs from the potency order of these compounds to bind to recombinant canine A3 adenosine receptors; and (iv) NECA, but not CGS-21680, elevates cAMP, which is consistent with the existence of functional A2B receptors on BR cells.
It was somewhat unexpected that A2B adenosine receptors seem to couple to Ca2+ mobilization in canine mastocytoma cells inasmuch as A2Badenosine receptors have been shown to couple to stimulation of cAMP accumulation (26). Apparent dual coupling to cAMP and Ca2+ has also been noted in HEK 293 cells stably transfected with recombinant human A2Breceptors,2 and it is significant in this regard that the expression of recombinant rat A2B receptors in Xenopus laevisoocytes results in the appearance of adenosine-mediated Ca2+-dependent Cl− current (32). Dual coupling of G protein coupled receptors to Gs and Gq/11 is not unprecedented. For example, the human prostacyclin receptor also displays such dual coupling (33). Coupling of A2Badenosine receptors to a Ca2+-mobilizing G protein resistant to pertussis toxin (Gq/11) may be essential for triggering BR cell degranulation because agents that elevate cAMP in various kinds of mast cells, including agonists of A2A adenosine receptors, either have no effect or are inhibitory to degranulation (34, 35).
The conclusions of previous studies have been inconsistent regarding the adenosine receptor subtype that mediates mast cell degranulation. Recent DNA antisense experiments suggest that activation of A1 adenosine receptors may contribute to bronchoconstriction in a rabbit model of asthma (36). However, the low potency of various A1-selective xanthines to block histamine release from asthmatic human lung fragments (10) and the low potency of enprofylline to block human A1adenosine receptors (27) are consistent with the participation of A2B and/or A3 receptors in human disease. The A3 adenosine receptor has been implicated in the degranulation of RBL 2H3 rat mast cells and in triggering vascular responses3 secondary to degranulation of mast cells in the hamster cheek pouch (9, 37) and the pithed rat (25). A2B adenosine receptors seem to mediate the degranulation of murine bone marrow-derived mast cells (21), and although pretreatment of RBL 2H3 rat mast cells with pertussis toxin abolishes NECA-mediated degranulation, Ca2+ mobilization in these cells requires activation of Gi3 or Gq(38). Moreover, activation of phosphoinositide breakdown in RBL 2H3 cells is not well correlated with the affinity of adenosine analogs for A3 adenosine receptors (39). The treatment of murine bone marrow mast cells with pertussis toxin produces a decrease in the potency of adenosine to enhance degranulation in response to A23187, similar to the result in the current study with canine mastocytoma cells. Pretreatment of murine mast cells with pertussis toxin fails to reduce adenosine-mediated Ca2+mobilization, which is consistent with an A2B-mediated, but not an A3-mediated, response (40). In the human HMC-1 mast cell leukemia line, the ability of 300 μmenprofylline to block NECA-stimulated interleukin-8 release was taken as evidence that this response also is mediated by A2B adenosine receptors (41). The current study tends to substantiate previously published reports that suggest the receptor subtype primarily responsible for adenosine-mediated mast cell degranulation is variable. It is not yet clear whether this variability depends on species, tissue source, or environmental factors that affect signaling cascades and/or the phenotype of various mast cells. The results of this study indicate that A2B receptors play a major role in the regulation of mast cell degranulation but are consistent with possible participation of multiple adenosine receptor subtypes and multiple G proteins.
The canine A3 adenosine receptor described in this study is structurally and pharmacologically more similar to human A3 receptors than are A3receptors from rodent species. This finding, along with the fact that it seems that murine bone marrow mast cells, canine mastocytoma, and human HMC-1 leukemic mast cells are regulated by adenosine, primarily via A2B receptors, raises the possibility that canine models of asthma may be better predictors of human disease than rodent models. It will be important to determine which adenosine receptor subtype or subtypes are responsible for facilitating mast cell degranulation in the asthmatic human lung. Once the predominant receptor or receptors are identified, novel antagonists superior to theophylline and enprofylline for the treatment of asthma may be developed.
Acknowledgments
cAMP determinations were made at the University of Virginia Radioimmunoassay Core. We are grateful to Fereydoun Sajjadi (Gensia), Marlene Jacobson (Merck), and Scott Kennedy (Pfizer) for their gifts of A3 adenosine receptor cDNAs and to J. H. Butterfield (Mayo Clinic, Cleveland, OH) for HMC-1 cells.
Footnotes
- Received June 17, 1997.
- Accepted July 31, 1997.
-
Send reprint requests to: Joel Linden, Ph.D., Box MR4 6012, Health Sciences Center, University of Virginia, Charlottesville, VA 22908. E-mail: jlinden{at}virginia.edu
-
↵1 Serum-free medium was used to avoid the potential for neutralization of pertussis toxin by anti-toxin antibodies that exist within some lots of serum.
-
↵2 X. Jin and J. Linden, unpublished observations.
-
↵3 Mast cell mediators such as histamine can produce vasoconstriction or vasodilation. Microvascular vasoconstriction is mediated in part by histamine and thromboxane acting on vascular smooth muscle cell receptors (9). Systemic vasodilation and hypotension secondary to A3 adenosine receptor activation are mediated in part by circulating histamine, which triggers nitric oxide release from endothelial cells.
-
This work was supported by National Institutes of Health Grants RO1-HL37942 and T32-HL07284.
Abbreviations
- [125I]APNEA
- N6-2-(4-amino-3-[125I]iodophenyl)adenosine
- [125I]ABA
- N6-(4-amino-3-[125I]iodobenzyl)adenosine
- [125I]AB-MECA
- N6-(4-amino-3-[125I]iodobenzyl)-adenosine-5′-N-methylcarboxamide
- IB-MECA
- N6-(3-iodobenzyl)-adenosine-5′-N-methylcarboxamide
- GTPγS
- guanosine-5′-O-(3-thio)triphosphate
- XAC
- 8-(4-[(2-aminoethyl)aminocarbonylmethyloxy]-phenyl)-1,3-dipropylxanthine
- CPA
- N6-cyclopentyladenosine
- AM
- acetoxymethyl ester
- (R)-PIA
- (R)-N6-phenylisopropyladenosine
- NECA
- 5′-N-ethylcarboxamidoadenosine
- I-ABOPX
- 3-(4-amino-3-iodobenzyl)-8-oxyacetate-1-propyl-xanthine
- 8-SPT
- 8-sulfophenyltheophylline
- NBTI
- nitrobenzylthioinosine
- RT
- reverse transcription
- PCR
- polymerase chain reaction
- BSA
- bovine serum albumin
- SSC
- standard saline citrate
- SDS
- sodium dodecyl sulfate
- DMEM
- Dulbecco’s modified Eagle’s medium
- HEK
- human embryonic kidney
- InsP3
- inositol trisphosphate
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}