


Abstract
Adenosine activates adenosine-induced inwardly rectifying K+ current (IKAdo) and inhibits isoproterenol (100 nm)-stimulated L-type Ca2+ current (β-ICa,L) of guinea pig atrial myocytes with EC50 values of 2.17 and 0.20 μm, respectively. We determined whether this 11-fold difference in potency of adenosine is due to the existence of a greater A1adenosine receptor reserve for the inhibition of β-ICa,Lthan for the activation of IKAdo. Atrial myocytes were pretreated with vehicle (control) or the irreversible A1adenosine receptor antagonist 8-cyclopentyl-3-[3-[[4-(fluorosulfonyl)benzoyl]oxy]propyl]-1-propylxanthine (FSCPX) (10 and 50 nm) for 30 min, and after a 60-min washout period, concentration-response curves were determined for the adenosine-induced activation of IKAdo and inhibition of β-ICa,L. Pretreatment of atrial myocytes with 10 nm FSCPX reduced the maximal activation of IKAdo by 60% (7.9 ± 0.2 to 3.2 ± 0.1 pA/pF). In contrast, a higher concentration of FSCPX (50 nm) was required to reduce the maximal inhibition of β-ICa,L by 39% (95 ± 4% to 58.7 ± 5.6%) and caused a 15-fold increase in the EC50 value of adenosine. Values of the equilibrium dissociation constant (K A) for adenosine to activate IKAdo and inhibit β-ICa,L, estimated according to the method of Furchgott, were 2.7 and 5.6 μm, respectively. These values were used to determine the relationship between adenosine receptor occupancy and response. Half-maximal and maximal activations of IKAdorequired occupancies of 40% and 98% of A1 adenosine receptors, respectively. In contrast, occupancies of only 4% and 70%, respectively, of A1 adenosine receptors were sufficient to cause half-maximal and maximal inhibitions of β-ICa,L. Consistent with this result, a partial agonist of the A1adenosine receptor SHA040 inhibited β-ICa,L by 60 ± 3.5% but activated IKAdo by only 18.1 ± 2.5%. The results indicate that the A1 adenosine receptor is coupled more efficiently to an inhibition of β-ICa,L than to an activation of IKAdo.
Most cardiac actions of adenosine are mediated by cell-surface adenosine receptors of the A1 subtype (1, 2). In sinoatrial nodal, atrial, and atrioventricular nodal myocytes, activation of the A1 adenosine receptor leads to stimulation of IKAdo through a G protein-mediated mechanism (3-6). Activation of IKAdo has been shown to be the underlying basis for the “direct” actions of adenosine, such as slowing of heart rate and atrioventricular nodal conduction and reduction of atrial contractility (3-5). In addition, the A1 adenosine receptor mediates inhibition of adenylate cyclase activity and cAMP formation through a pertussis toxin-sensitive G protein (7, 8). This latter signaling pathway is responsible for attenuation of β-adrenergic receptor-induced increases of cAMP formation and L-type calcium current (and other currents modulated by cAMP) by adenosine and is the mechanism of the “indirect” anti-β-adrenergic effect of adenosine in the heart (9,10).
Activation of K+ conductance and inhibition of cAMP formation by adenosine analogs are blocked by the A1 adenosine receptor antagonist CPX with similarKB values of 8.1 and 9.6 nm, respectively (11, 12). The rank orders of potency of adenosine receptor agonists to activate IKAdo and inhibit cAMP formation are also similar (11, 12). These findings suggest strongly that the same receptor mediates both actions of adenosine to activate IKAdo and attenuate stimulation by isoproterenol of adenylate cyclase and cAMP formation. However, the EC50 value for adenosine to inhibit the β-adrenergic (isoproterenol)-stimulated ICa,Lis 11-fold lower than the EC50 value for adenosine to activate IKAdo (Figs.1 and 2). We hypothesized that this 11-fold difference in potency of adenosine is due to a larger A1 adenosine receptor reserve for the inhibition of isoproterenol-stimulated ICa,Lthan for the activation of IKAdo.

Differential sensitivity of adenosine to activate IKAdo and inhibit isoproterenol (100 nm)-stimulated ICa,L in single atrial myocytes. A, Traces of the holding current at a membrane potential of −40 mV recorded from a single atrial myocyte in the presence of various concentrations (1–300 μm) of adenosine (Ado). The increase in the holding current reflects the activation of IKAdo. The magnitudes of IKAdoactivation caused by 0.1, 1, 10, 100, and 300 μmadenosine in this myocyte were 20, 180, 350, 380, and 370, respectively. B, Traces of ICa,L recorded from a single atrial myocyte in control, during exposure to 100 nmisoproterenol (ISO), and in the presence of both isoproterenol and different concentrations of adenosine. ICa,L was measured in K+-free conditions in response to 200-msec-long depolarizing pulses applied every 6 sec from a membrane potential of −40 mV to a test potential of 10 mV. Amplitudes of ICa,L were 400 pA in control and 1000 pA in the presence of 100 nm isoproterenol. The increase of ICa,L caused by isoproterenol was reduced by adenosine in a concentration-dependent manner to 742, 532, and 395 pA at 0.3, 1, and 10 μm of adenosine, respectively.

Concentration-response relationships for adenosine to inhibit isoproterenol (100 nm)-stimulated ICa,L (β-ICa,L) and activate IKAdo in atrial myocytes. Maximal activation of IKAdo was defined as the increase in peak outward current at −40 mV caused by 300 μm adenosine. Maximal inhibition of β-ICa,L was defined as reduction in β-ICa,L caused by 30 μm adenosine. Magnitude of activations of IKAdo and inhibitions of β-ICa,L by different concentrations of adenosine were normalized to their respective maximal responses. The EC50and n H values are the concentrations of the adenosine that cause half-maximal responses and the Hill coefficients, respectively. The Hill coefficients of the concentration-response relationships for adenosine to mediate the two responses were not significantly different from unity. Values are mean ± standard error of determinations from 6–14 myocytes.
The concept of receptor reserve refers to the phenomenon by which a maximal response to certain drugs, hormones, and autocoids can be achieved at a submaximal receptor occupancy (13, 14). To estimate receptor reserve, it is necessary to determine the response to an agonist as a function of receptor occupancy by the agonist. This relationship between receptor occupancy and response can be determined by using a method developed by Furchgott and Bursztyn (15) that makes it possible to calculate the agonist equilibrium dissociation constant (KA ) and the fractional receptor occupancy from analysis of agonist concentration-response curves obtained before and after irreversible inactivation of a fraction of the receptor population. Application of the method of Furchgott and Bursztyn to estimate A1 adenosine receptor reserve therefore requires an irreversible adenosine receptor antagonist. Recently, we synthesized and reported the pharmacological characteristics of FSCPX, an irreversible antagonist of the A1 adenosine receptor (16). FSCPX attenuated cardiac A1 adenosine receptor-mediated responses in a specific, selective, and irreversible manner (16). In the current study, we used FSCPX and the method of Furchgott and Bursztyn (15) to determine both the equilibrium dissociation constant for binding of adenosine to the atrial A1 adenosine receptor and the A1 adenosine receptor reserves for activation of IKAdo and inhibition of isoproterenol-stimulated ICa,L in single atrial myocytes. As a second goal, we determined whether a partial agonist of the A1 adenosine receptor would selectively activate the response with the higher receptor reserve. For this purpose, we characterized the responses of guinea pig atrial myocytes to SHA040 (2-phenethoxyadenosine), a phenethoxy derivative of adenosine (17), a low efficacy agonist of the A1 adenosine receptor.
Materials and Methods
Chemicals.
Adenosine, adenosine deaminase, and isoproterenol were purchased from Sigma Chemical (St. Louis, MO). CPX, CCPA, and (R)-N6-(2-phenylisopropyl)adenosine were purchased from Research Biochemicals (Natick, MA). N-0861 (N6-endonorbornan-2-yl-9-methyladenine) and SHA040 were gifts from Dr. Noel Cusack (Discovery Therapeutics, Richmond, VA) and Dr. Ray Olsson (University of South Florida, Tampa, FL), respectively. [3H]CPX was purchased from DuPont-New England Nuclear Research Products (Boston, MA). Stock solutions of 10 mm FSCPX and 50 mm SHA040 were prepared in DMSO. The final content of DMSO in the incubation medium was ≤0.2% (v/v).
Isolated atrial myocytes.
Atrial myocytes were freshly isolated from hearts of adult Hartley guinea pigs by a method described previously (18). Briefly, guinea pigs were anesthetized with methoxyflurane, and the hearts were excised quickly and perfused through the aorta for 5–10 min with warm (35°) modified K-H solution containing 127 mm NaCl, 4.6 mm KCl, 2 mm CaCl2, 1.1 mmMgSO4, 2 mm sodium pyruvate, 10 mm glucose, 10 mm creatine, 20 mmtaurine, 5 mm ribose, 0.01 mm adenine, 0.1 mm allopurinol, and 5 mm HEPES, pH 7.4. The hearts were perfused continuously for an additional 10 min with Ca2+-free modified K-H solution and then digested enzymatically by perfusion for 15–20 min with Ca2+-free K-H solution containing 0.4 mg/ml collagenase type 2, 0.04 mg/ml dispase, 0.04 mg/ml trypsin, and 2 mg/ml albumin. The atria were dissected, minced, and incubated at 35° with enzyme solution in a shaker bath. Dissociated atrial cells were collected and stored at room temperature in modified K-H solution containing 0.1 mm Ca2+ until further use.
Electrophysiological measurements.
Myocytes were transferred into a recording chamber and superfused at a rate of 2–3 ml/min and at a constant temperature of 35° with Tyrode’s solution. The composition of the Tyrode’s solution was 118 mm NaCl, 4.6 mm KCl, 1.2 mm CaCl2, 1.1 mm MgCl2, 10 mm glucose, and 10 mm HEPES, pH adjusted to 7.4 with NaOH. In some experiments, K+ was replaced with an equimolar concentration of Cs+. Ionic currents were recorded with glass suction pipettes (Kimax; Kimble Glass, Vineland, NJ) in a whole-cell patch-clamp configuration (19). The recording electrodes had resistances of 2–4 MΩ when filled with pipette solution. Junction potentials between pipette and bath medium were nulled before seal formation. The composition of pipette solution was 10 mm KCl, 130 mm K-aspartate, 4 mmNa2ATP, 1 mmMgCl2, 0.1 mmNa3GTP, 10 mm glucose, 1 mm NaEGTA, and 10 mm HEPES, pH adjusted to 7.2 with KOH. Recordings were made with an Axopatch-1B amplifier (Axon Instruments, Burlingame, CA) and filtered at a bandwidth of 1 kHz. Data acquisition and analysis were performed with pClamp software (Axon Instruments) and an IBM PC 486 computer. Membrane currents were displayed on a storage oscilloscope and recorded simultaneously on a strip-chart recorder.
The activation of IKAdo was measured at a holding potential of −40 mV as an increase in the current observed on application of adenosine. IKAdo was defined as the difference between the magnitudes of the peak outward current elicited by the agonist and the holding current value before exposure to the agonist. Concentration-response curves for adenosine and SHA040 to activate IKAdo were determined by exposure of each cell to several concentrations of adenosine. Applications of successive concentrations of adenosine and SHA040 were separated by ≥4 min. This time interval was necessary to ensure that the cell recovered from desensitization of the response to adenosine. Under these conditions, concentration-response curves were independent of the order of application of various concentrations of agonist. The increase in IKAdo caused by SHA040 was normalized to the increase in current caused by the maximal concentration of adenosine. Thus, after exposure and washout of myocytes to different concentrations of SHA040, a maximal concentration of adenosine was applied, and the increases in IKAdo caused by SHA040 and adenosine were compared. The increases in IKAdo caused by adenosine in control and FSCPX-treated myocytes and SHA040 were expressed relative to the cell capacitance (i.e., pA/pF). The capacitance of atrial myocytes was 20–60 pF.
To elicit ICa,L, 200-msec-long depolarizing pulses applied every 6 sec from a holding potential of −80 mV to a test potential of 10 mV were used. A prepulse to −40 mV for 100 msec was used to inactivate the sodium current, and K+currents were blocked by substituting Cs+ for external and internal K+. ICa,L was defined as the difference between the magnitudes of the peak inward current and the current at the end of a 200-msec depolarizing pulse. The effects of various concentrations of adenosine receptor agonists on β-ICa,L were calculated as the difference in responses (magnitudes of reduction of ICa,L) caused by the agonist in the presence and absence of isoproterenol. The run-down of ICa,Lcan potentially lead to overestimation of the magnitude of the inhibition of ICa,L by adenosine and SHA040; therefore, to exclude the possibility of run-down, the amplitudes of β-ICa,L both before and after exposure to adenosine agonists were measured. When the amplitude of ICa,L elicited by isoproterenol after termination of an exposure to the agonist was <80% of amplitude of the current before the application of agonist, the data were discarded.
Experimental protocol for the measurement of receptor reserve.
Single guinea pig atrial myocytes were pretreated with either vehicle (DMSO plus K-H solution) or FSCPX (10 or 50 nm) for 30 min. After the incubation period, cells were washed repeatedly by changes of the incubation medium for 1 hr to remove unbound FSCPX. Control and FSCPX-pretreated cells were then exposed to increasing concentrations of adenosine, and the magnitudes of activation of IKAdo and inhibition of β-ICa,L caused by adenosine were recorded. Because measurements of both responses in control and FSCPX-treated myocytes were made in different group of cells, the potential confounding effects of desensitization and run-down on the measurement of the receptor reserve were avoided.
Analysis of concentration-response curves.
The concentration of adenosine and SHA040 that caused a half-maximal response (EC50) and the Hill coefficients of concentration-response relationships were estimated by use of a nonlinear regression algorithm (Marquardt-Levenberg) to fit data to the multiparameter logistic equation:
Estimation of receptor reserve.
The method of Furchgott and Bursztyn (15) was used to estimate the equilibrium dissociation constant (KA
) of adenosine and the fraction of functional receptors (q) remaining after exposure of cells to FSCPX. Pairs of concentrations of agonist were selected that caused equal levels of response (either IKAdo activation or β-ICa,L inhibition) before and after inactivation of a fraction of A1 adenosine receptors with FSCPX. The equieffective concentrations were determined at 12 levels of response of 20–100% of the maximum effect after FSCPX treatment through interpolation from concentration-response curves (see examples in Figs. 2 and 3). These 12 equieffective concentrations were fitted to eq. 2 by linear regression analysis (Table Curve; Jandel Scientific, Sausalito, CA) to yield values of KA
and q (from Ref. 15):

Effect of the irreversible A1 adenosine receptor antagonist FSCPX (10 nm) on the adenosine-mediated activation of IKAdo. Atrial myocytes were incubated with FSCPX (10 nm) or vehicle (DMSO plus K-H solution) for 30 min and then washed repeatedly for 1 hr. A, Concentration-response curves of adenosine to activate IKAdo in vehicle (control)- and FSCPX-treated myocytes. The maximal activation of IKAdoin myocytes pretreated with 10 nm FSCPX was reduced by 60% without a significant change in the EC50 value of adenosine. Values are mean ± standard error of determinations from four to six myocytes. B, Double-reciprocal plot of concentrations of adenosine (A and A′) that caused equal levels of activation of IKAdo in control and FSCPX-treated myocytes. A [control (untreated)] and A′ (FSCPX-treated) values were obtained at 12 levels from the data in A and were used to estimate theK A value of adenosine to bind to receptors that mediate activation of IKAdo.
The KA
values for the binding of adenosine to A1 adenosine receptors to cause either activation of IKAdo or inhibition of β-ICa,L were used to estimate the fractional receptor occupancy by adenosine at any given concentration of adenosine ([A]) on the basis of the law of mass action:
The extent of receptor reserve for each response was estimated from the relationship: percent receptor reserve = 100 − percentage of receptor occupancy required to produce a half-maximal response.
Membrane preparation.
Guinea pig atrial membranes were prepared according to the method of Lohse et al. (20). The atria were minced and then homogenized in ice-cold buffer containing 10 mm imidazole, 5 mm MgSO4, and 300 mm sucrose 300, pH 7.0. The sucrose concentration was then increased to 600 mm. The homogenate was centrifuged at 20,000 × g for 30 min at 4°. The supernatant was diluted with 1.5 volumes of buffer containing 10 mm imidazole, 5 mm MgSO4, and 160 mm KCl, pH 7.0, and centrifuged at 30,000 ×g for 3 hr at 4°. The pellet was resuspended in 50 mm Tris·HCl buffer, pH 7.4, and frozen at −80° until use.
Radioligand binding protocols.
Atrial membranes were prepared as described above and incubated with adenosine deaminase (2 units/ml) for 20 min before assays were carried out. The potencies of CCPA and SHA040 to reduce the binding of [3H]CPX to guinea pig atrial membranes were determined in the absence and presence of 100 μm GTP. Increasing concentrations of each agonist were incubated with [3H]CPX, adenosine deaminase (2 units/ml), and aliquots of atrial membranes (0.2–0.7 mg) in the absence and presence of 100 μm GTP for 3 hr in a 300-μl volume of 50 mm Tris·HCl buffer, pH 7.4. Assays were carried out in triplicate at room temperature. After the incubation period, bound and free radioligands were diluted by the addition of 5 ml of ice-cold Tris·HCl buffer and separated immediately by vacuum filtration of assay contents onto Whatman GF/C filters and washing of trapped membranes with 20 ml of ice-cold Tris·HCl buffer. Filter disks containing membrane-bound radioactivity were placed in 4 ml of Scintiverse (Fisher Scientific, Pittsburgh, PA), and the radioactivity was quantified with a liquid scintillation counter. Specific binding of [3H]CPX was defined as membrane binding displaced in the presence of (R)-N6-(2-phenylisopropyl)adenosine (10 μm).
Data analysis.
All values are expressed as mean ± standard error. Statistical significance of differences between mean values in experiments with multiple comparison groups was determined by analysis of variance. Differences between mean values were considered statistically significant at p < 0.05. The concentrations of an agonist needed to reduce by 50% the specific binding of [3H]CPX in the absence and presence of GTP were determined with the radioligand binding analysis program LIGAND 3.0 (Elsevier-Biosoft, Cambridge, UK) TheKi values were calculated according to the Cheng-Prusoff equation (21).
Results
Concentration-response curves for activation of IKAdoand inhibition of β-ICa,L by adenosine.
The actions of adenosine to activate IKAdo and inhibit β-ICa,L were determined by experiments on single atrial myocytes. Adenosine (0.01–300 μm) activated IKAdo and inhibited β-ICa,L in a concentration-dependent manner (Figs. 1 and 2). However, adenosine inhibited the increase in calcium current caused by isoproterenol at significantly lower concentrations than those needed to activate IKAdo. The EC50 values for adenosine to inhibit β-ICa,L and activate IKAdo were 0.20 and 2.17 μm, respectively. Thus, in guinea pig atrial myocytes, adenosine is 11-fold more potent at inhibiting β-ICa,L than at activating IKAdo.
Effect of FSCPX on concentration-response curves for the adenosine-mediated activation of IKAdo and inhibition of β-ICa,L.
To determine whether the difference in potencies of adenosine to inhibit β-ICa,L and activate IKAdo is due to a difference in coupling efficiencies of A1 adenosine receptors to the two responses, we estimated the A1 adenosine receptor reserve for each response using the method of irreversible receptor inactivation (15). The application of Furchgott’s method requires comparison of concentration-response curves before and after inactivation of a fraction of receptors with an irreversible antagonist. Therefore, atrial myocytes were pretreated with either vehicle or the irreversible A1 adenosine receptor antagonist FSCPX (10 or 50 nm) for 30 min (Figs. 3A and4A). Pretreatment of atrial myocytes with 10 or 50 nm FSCPX reduced the maximal activation by adenosine of IKAdo by 60% and 80%, respectively. The maximal amplitudes of IKAdowere 7.9 ± 0.2, 3.2 ± 0.1, and 1.7 ± 0.3 pA/pF in cells treated with vehicle, 10 nm FSCPX, or 50 nm FSCPX, respectively. The concentrations of adenosine that caused half-maximal increases of IKAdo in control and 10 nm FSCPX-treated cells were not significantly different (1.68 and 2.27 μm, respectively,p < 0.05). In comparison, pretreatment of myocytes with 10 and 50 nm FSCPX reduced the maximal inhibitions of β-ICa,L caused by adenosine by 19% and 39%, respectively. The maximal inhibitions of β-ICa,L in cells treated with vehicle and 10 or 50 nm FSCPX were 95 ± 4%, 77 ± 7.9%, and 58.7 ± 5.6%, respectively. The reduction in the maximal response to adenosine after inactivation by 50 nm FSCPX of a fraction of the adenosine receptor population was accompanied by a 15-fold increase in the EC50 value of adenosine to inhibit β-ICa,L from 0.15 to 2.9 μm in vehicle- and FSCPX-treated myocytes, respectively (p < 0.05). This finding suggests that for adenosine to inhibit β-ICa,L, a large receptor reserve was present.

Effect of the irreversible A1 adenosine receptor antagonist FSCPX (10 nm) on the adenosine-mediated inhibition of β-ICa,L. Atrial myocytes were incubated with FSCPX (50 nm) or vehicle (DMSO plus K-H solution) for 30 min and then washed repeatedly for 1 hr. A, Concentration-response curves of adenosine to inhibit β-ICa,L in vehicle (control)- and in FSCPX-treated myocytes. Pretreatment of myocytes with 50 nm FSCPX reduced the maximal response to adenosine by 39%. This was accompanied by a 15-fold increase in the EC50 value of adenosine. Values are mean ± standard error of determinations from four to six myocytes. B, Double-reciprocal plot of concentrations of adenosine (A and A′) that caused equal levels of inhibition of β-ICa,L in control and FSCPX-treated myocytes. A [control (untreated)] and A′ (FSCPX-treated) values were obtained at 12 levels from the data in A and were used to estimate theK A value of adenosine to bind to receptors that mediate inhibition of β-ICa,L.
Estimation of the equilibrium dissociation constant of adenosine.
Concentration-response curves for adenosine to activate IKAdo and inhibit β-ICa,Lbefore and after treatment of cells with FSCPX (Figs. 3A and 4A) were used to estimate the equilibrium dissociation constants for adenosine (KA ) to bind to A1 adenosine receptors coupled to activation of IKAdo and inhibition of β-ICa,L, according to the method of Furchgott and Bursztyn (15). Concentrations of adenosine, [A] and [A′], that produced equal levels of response (e.g., activation of IKAdo, inhibition of β-ICa,L) in control and FSCPX-treated myocytes, respectively, were determined and used to calculate estimates ofKA and q (see Estimation of Receptor Reserve). Estimates of theKA value for adenosine to bind to receptors coupled to activation of K+ currents and of q in cells pretreated with 10 nm FSCPX were 2.7 and 0.3 μm, respectively (Fig. 3B). The 95% confidence limits of KA and q were 1.2–4.3 μm and 0.2–0.4, respectively. Estimates of the KA for adenosine to bind to receptors coupled to inhibition of β-ICa,L and q in cells pretreated with 50 nm FSCPX were 5.6 μm and 0.03, respectively (Fig. 4B). The 95% confidence limits of KA andq values were 4.0–7.8 μm and 0.02–0.04, respectively. The KA values of 2.7 and 5.6 μm for adenosine to activate IKAdo and inhibit β-ICa,L, respectively, were not significantly different.
Occupancy-response relationships for adenosine to activate IKAdo and inhibit β-ICa,L.
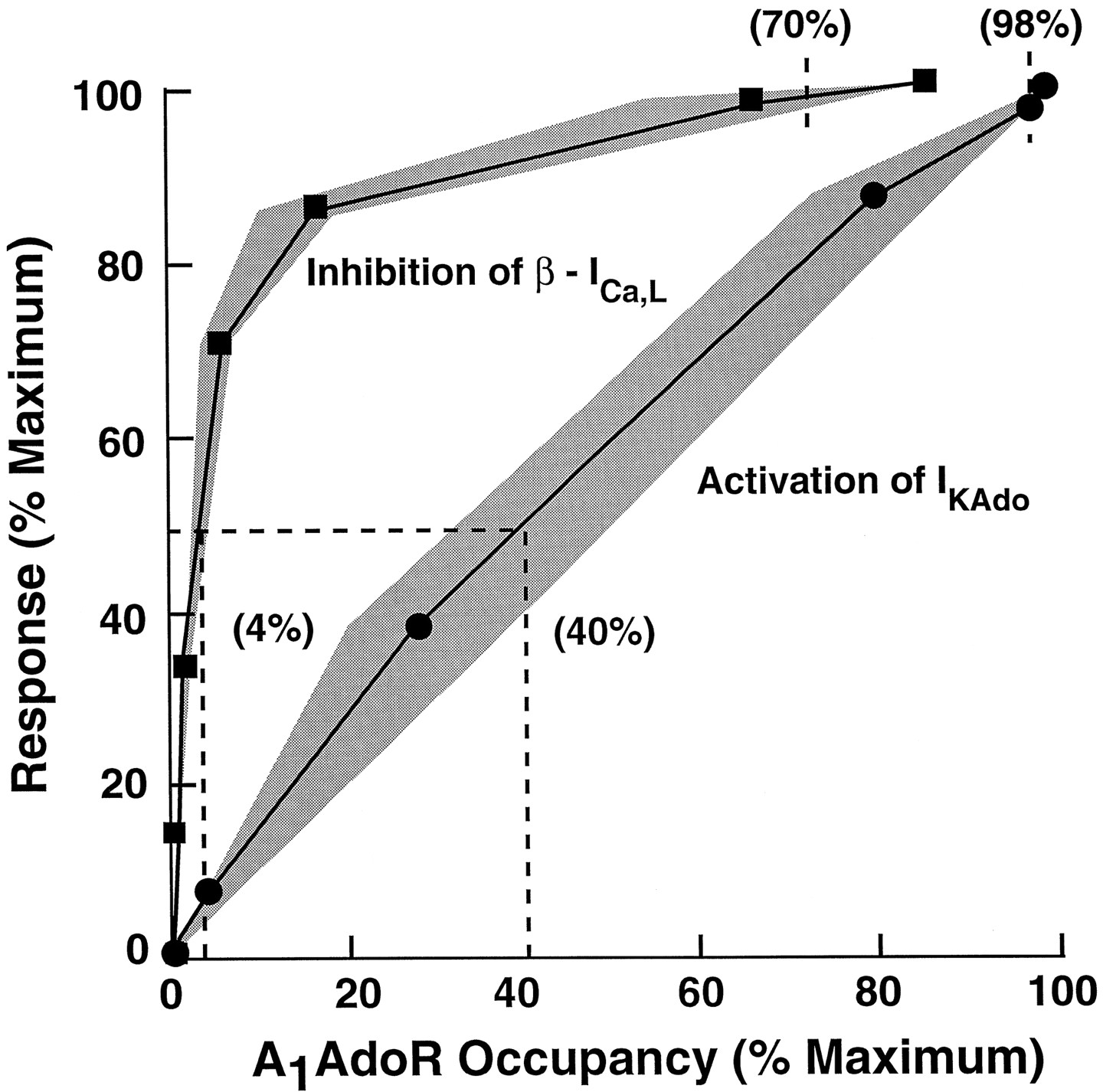
The relationships between A1 adenosine receptor occupancy and atrial myocyte responses to adenosine were determined in two steps. First, both responses (activation of IKAdo and inhibition of β-ICa,L) and fractional receptor occupancy were plotted as functions of adenosine concentration. These plots are shown in Fig. 5. The adenosine concentration-response data are the same as those shown in Figs. 3A and4A for control cells, and the adenosine concentration-fractional receptor occupancy data were calculated by use of values ofKA for adenosine derived from Furchgott analysis (eq. 2) and the law of mass action (eq. 3). The concentration-response relationships for adenosine to activate IKAdo and inhibit β-ICa,Llie to the left of their respective occupancy curves, indicating that amplification of the signal is present. The values of the ratio ofKA and EC50, an index of the extent of amplification, were 1.4 and 34 for adenosine to activate IKAdo and inhibit β-ICa,L, respectively. This result indicates that there is a small receptor reserve for adenosine to activate IKAdo but a large reserve for the nucleoside to inhibit β-ICa,L. Second, to determine the magnitudes of receptor reserve for adenosine to activate IKAdo and inhibit β-ICa,L, the data shown in Fig. 5 were used to calculate myocyte responses as a function of receptor occupancy. This plot is shown in Fig. 6. The occupancy-response relationship for the activation of IKAdo by adenosine was nearly linear. Half-maximal and maximal activations of IKAdorequired occupancies of 40% and 98% of A1adenosine receptors, suggesting little or no receptor reserve for this response. In comparison, occupancies of only 4% and 70% of receptors were sufficient to cause half-maximal and maximal inhibitions of β-ICa,L, respectively.

Dependencies of both A1 adenosine receptor occupancy and response (activation of IKAdo, inhibition of β-ICa,L) on adenosine concentration. A1 adenosine receptor occupancy by adenosine to activate IKAdo and inhibit β-ICa,L was calculated using K A values of 2.7 and 5.6 μm, respectively, according to eq. 3. Responses to adenosine are from data depicted in Figs. 2A and 3A for vehicle (control)-treated cells and are expressed as percent maximal response.
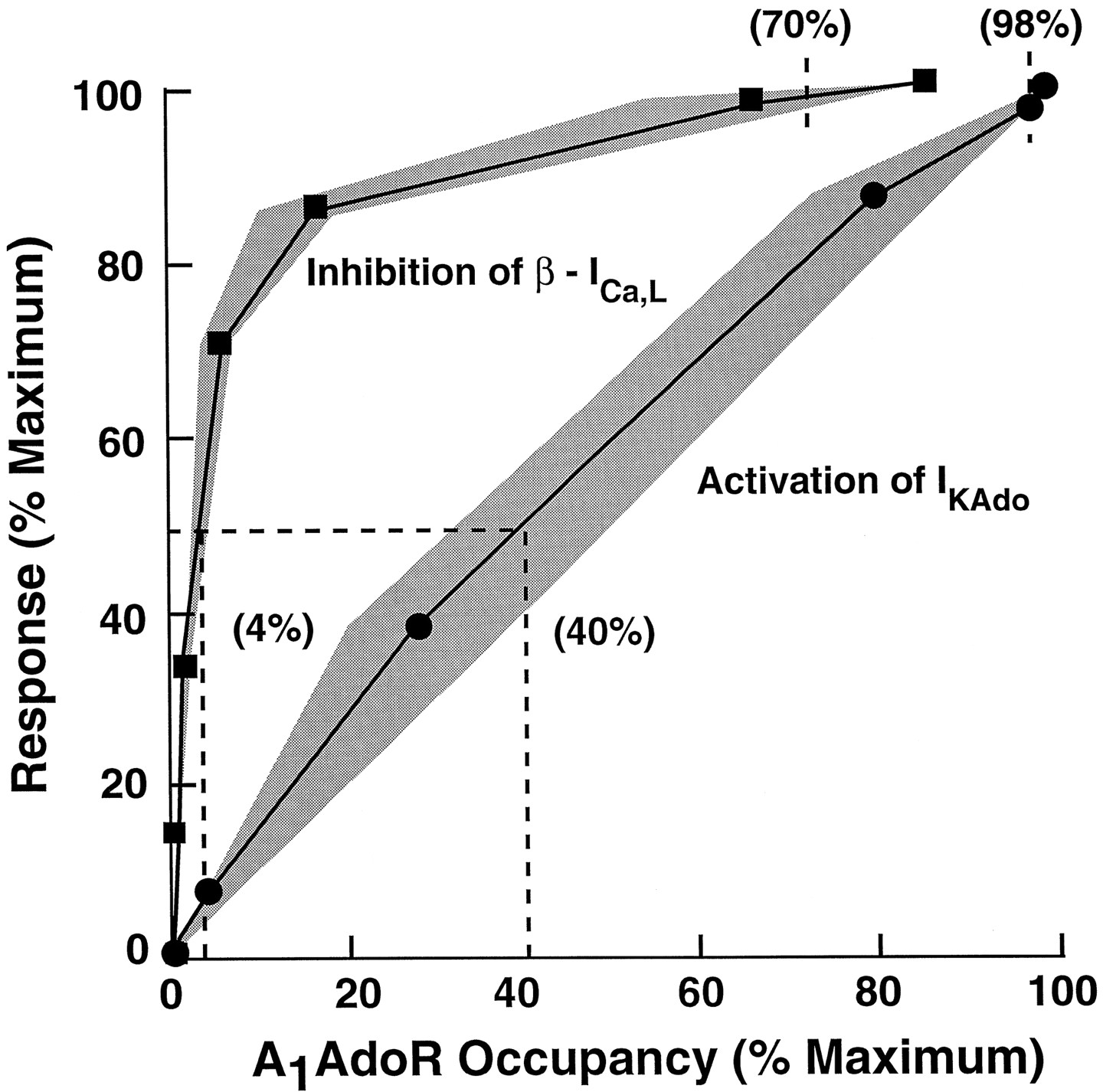
Relationships between A1 adenosine receptor occupancy and responses: activation of IKAdo (•) or inhibition of β-ICa,L (▪). Occupancy of A1 adenosine receptors was estimated by usingK A values as described in the legend to Fig. 4. Shaded areas, error in the estimation of occupancy at a given level of response; this error was determined using the 95% confidence limits ofK A values for adenosine to bind to receptors that mediate activation of IKAdo and inhibition of β-ICa,L. The occupancy-response relationship for activation of IKAdo by adenosine is linear with half-maximal and maximal activations of IKAdooccurring at occupancies of 40% and 98% of A1 adenosine receptors, respectively. In comparison, half-maximal and maximal inhibitions of β-ICa,L required occupancies of only 4% and 70% of receptors, respectively.
Actions of SHA040 on atrial A1 adenosine receptors.
A1 Adenosine receptor agonists with low intrinsic efficacies may be expected to cause a greater inhibition of β-ICa,L without causing significant activation of IKAdo. Therefore, the actions of a phenethoxy derivative of adenosine (20), SHA040, to inhibit β-ICa,L and activate IKAdo were investigated. Recent results indicated that SHA040 was a low efficacy full agonist of A1adenosine receptors in DDT1MF-2 cells.1 We hypothesized that SHA040 may be a partial agonist at atrial A1 adenosine receptors because the density of A1 adenosine receptors in atrial myocytes is lower than in DDT1MF-2 cells.
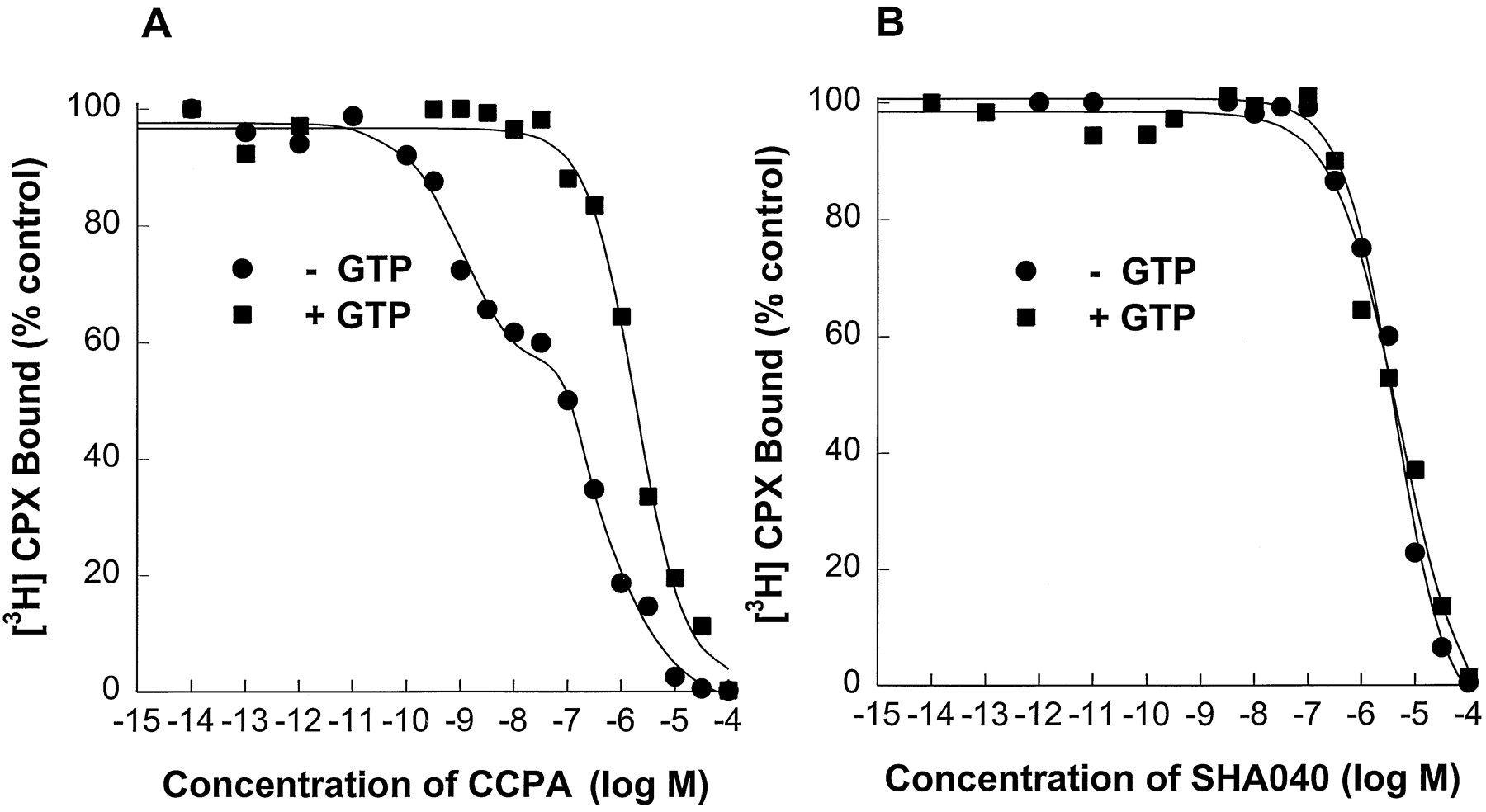
To demonstrate that SHA040 is a partial agonist at atrial A1 adenosine receptors, the inhibition of specific binding of [3H]CPX caused by SHA040 was determined and compared with that caused by the full A1 adenosine receptor agonist, CCPA. Because the magnitude of the rightward shift of competition curves of agonists observed in the presence of guanine nucleotides is considered to be an index of the intrinsic efficacy of the agonists (14), the effect of 100 μm GTP on reductions by a full A1adenosine receptor agonist, CCPA, and by SHA040 of [3H]CPX binding to atrial membranes was investigated (Fig. 7). Reduction of the specific binding of [3H]CPX by CCPA in the absence of GTP was best fit by a two-site model, indicating the presence of both high and low affinity binding states. Inclusion of 100 μm GTP in the binding assay medium caused a rightward shift of the CCPA competition curve and converted all receptors to a low affinity binding state (Table 1). In comparison, even though SHA040 completely inhibited the binding of [3H]CPX, the inhibition by SHA040 was not affected by the presence of GTP (Fig. 7). The reduction of specific binding of [3H]CPX caused by SHA040 in the absence and presence of GTP was best fit to a one-site model. TheKi values for the reduction of specific binding of [3H]CPX by SHA040 in the absence and presence of GTP were not significantly different (Table 1). This result is consistent with hypothesis that SHA040 is a partial agonist of atrial A1 adenosine receptors.

Reductions by CCPA (A) and SHA040 (B) of the specific binding of the A1 adenosine receptor antagonist [3H]CPX to guinea pig atrial membranes in the absence and presence of 100 μm GTP. Reduction in the specific binding of [3H]CPX caused by CCPA was best fitted by a two-site model indicating the presence of both high and low affinity agonist binding sites. In contrast, the reduction by SHA040 of the specific binding of [3H]CPX was best fitted by a one-site model. Inclusion of GTP caused a reduction of the affinity of CCPA, but not the affinity of SHA040, for [3H]CPX binding sites.
Potencies of adenosine receptor agonists to compete for [3H]CPX binding sites in atrial membranes, in the absence and presence of 100 μm GTP
Differential effects of SHA040 on IKAdo and β-ICa,L.
The actions of SHA040 to activate IKAdo and inhibit β-ICa,Lwere determined in experiments using single atrial myocytes. SHA040 (0.3–100 μm) activated IKAdo and inhibited isoproterenol (100 nm)-stimulated ICa,L (β-ICa,L) in a concentration-dependent manner with Hill coefficients close to unity (Fig. 8). Both activation of IKAdo and inhibition of β-ICa,L caused by SHA040 were blocked by the A1 adenosine receptor-selective antagonists CPX (100 nm) and N-0861 (5 μm). The magnitudes of IKAdo activated by 50 μm SHA040 were 80 ± 5 and 10 ± 1 pA in the absence and presence of 100 nm CPX, respectively, and 70 ± 3.2 and 6 ± 2 pA in the absence and presence of N0861, respectively. Peak ICa,L was increased by 100 nmisoproterenol from a control value of 490 ± 19 to 1265 ± 25 pA. In the continued presence of isoproterenol, 50 μmSHA040 reduced ICa,L from 1265 ± 25 to 890 ± 60 pA. The decrease in β-ICa,Lcaused by SHA040 was attenuated by 100 nm CPX to 1175 ± 55 pA (i.e., by 88 ± 6%). Similarly, 5 μmN-0861 attenuated the inhibition of β-ICa,Lcaused by SHA040 by 90 ± 6%. Thus, the data indicate that both activation of IKAdo and inhibition of β-ICa,L caused by SHA040 were mediated by A1 adenosine receptors.

Concentration-response relationships for SHA040 to inhibit isoproterenol (100 nm)-stimulated ICa,L(β-ICa,L) and activate IKAdo in atrial myocytes. SHA040 inhibited β-ICa,L by 60% but did not significantly activate IKAdo (<20%).Points, mean ± standard error of determinations from four to six atrial myocytes.
The maximal activation of IKAdo and inhibition of β-ICa,L caused by SHA040 were lower than those observed with adenosine, which suggests strongly that SHA040 is a partial agonist for each of the responses (Fig. 8). The EC50 values of SHA040 to activate IKAdo and inhibit β-ICa,Lwere 7.8 ± 0.5 and 6.5 ± 0.9 μm, respectively. More importantly, the inhibition of β-ICa,L caused by SHA040 was markedly greater than the activation of IKAdo. As shown in Fig. 8, the maximal inhibition of β-ICa,L caused by SHA040 was 60 ± 3.5% of that caused by 100 μmadenosine, whereas the maximal amplitude of IKAdocaused by SHA040 was only 18.1 ± 2.5% of that caused by 100 μm adenosine.
Discussion
In this study, we show that there is a lower A1 adenosine receptor reserve for activation of IKAdo than for inhibition of β-ICa,L of guinea pig atrial myocytes. Consistent with this finding, the relative magnitude of inhibition of β-ICa,L (equal to 60% of that caused by adenosine) caused by a partial agonist of the atrial A1 adenosine receptor, SHA040, was markedly greater than the magnitude of activation of IKAdo(equal to 18% of that caused by adenosine). This result is consistent with the postulate that the extent of receptor reserve is an important determinant of the magnitude of response caused by a partial agonist (14, 22, 23).
We estimated the receptor reserve for adenosine to inhibit β-ICa,L and activate IKAdo by use of the method of Furchgott and Bursztyn (15) and an irreversible A1 adenosine receptor antagonist, FSCPX (16). Our results can explain the observation that adenosine inhibits β-ICa,L at lower concentrations than those needed to activate IKAdo. Determinations of receptor reserves for muscarinic, adrenergic, and dopaminergic receptor-mediated responses have been used to explain differential potencies of agonists at these receptors (24-27). Thus, our findings are consistent with results of the above studies on other G protein-coupled receptors and provide further evidence for the importance of the concept of receptor reserve in understanding the differential potencies with which agonists can elicit distinct functional responses.
Alternatively, the differential potency of adenosine to activate IKAdo and inhibit β-ICa,Lcan be explained by the existence of distinct subtypes of A1 adenosine receptor (e.g., A1a and A1b) subserving each of these responses. Our study was not designed to investigate the possibility that different subtypes and/or affinity states of the A1 adenosine receptor mediate activation of IKAdo and inhibition of β-ICa,L. However, a review of the literature suggests strongly that in guinea pig atria, the same A1 adenosine receptor subtype mediates both the direct and indirect actions of adenosine. For example, the A1 adenosine receptor antagonist CPX blocked the increase in 86Rb efflux (which reflects IKAdo activation) caused by CCPA with aKB value of 8.1 nm (11). Similarly, the inhibition of isoproterenol-stimulated cAMP formation by CCPA in the same preparation was antagonized by CPX with a KB value of 9.1 nm (12). Furthermore, the rank order of potency of adenosine analogues to inhibit isoproterenol-stimulated cAMP and increase 86Rb efflux were identical. In addition, as shown in Figs. 3B and 4B, the agonist equilibrium dissociation constants of adenosine (KA ) to activate IKAdo and to inhibit β-ICa,L were 2.7 and 5.6 μm, respectively; these values were not significantly different. Consistent with these results, only one splice variant of the A1 adenosine receptor is expressed in the heart (2). The similarities in antagonistKB values, in rank order of agonist potency profiles, and in dissociation constants of adenosine argue against the existence of different A1 adenosine receptor subtypes in guinea pig atria. Regardless, future studies will be necessary to determine the possibility that two subtypes of A1 adenosine receptor and/or different affinity states of the same receptor subtype are coupled to activation of IKAdo and inhibition of β-ICa,L.
The finding of a difference in receptor reserve for activation of IKAdo and inhibition of β-ICa,L suggests that the coupling efficiencies of A1 adenosine receptors to the two responses are different. Because G proteins and their interactions with effectors are important determinants of the amplification of a signal transduction process, the contribution of these proteins to the differences in receptor reserve may be significant (28-30). It is possible that differences in receptor reserve for activation of IKAdo and inhibition of β-ICa,L may be due either to participation of different G proteins or participation of different subunits of the same G protein in mediating each response or to distinct differences in the relative affinities and deactivation rates of activated subunits to K+ channels and adenylate cyclase. However, the results of our studies do not allow us to distinguish among these possibilities. The differential receptor reserve for adenosine to activate IKAdo and inhibit β-ICa,L may also be due to differences in the number of elements (steps) in the two signal transduction pathways. Inhibition of β-ICa,L by adenosine involves a Gi protein, adenylate cyclase, cAMP, and dephosphorylation of L-type calcium channels (1, 8, 9). Thus, the initial stimulus, occupancy of A1 adenosine receptors by an agonist, may be considerably amplified at each step of this biochemical cascade, resulting in the 34-fold difference we observed between the KA and EC50 values of adenosine to inhibit β-ICa,L (Fig. 5). Contrariwise, activation of IKAdo by adenosine occurs primarily through a membrane-delimited pathway involving βγ subunits (29) and does not seem to involve second messengers, resulting in a small (1.4-fold) difference between the KA and EC50 values (Fig. 5). This lack of signal amplification in the pathway for activation by adenosine of IKAdo is probably caused by the small number of transduction steps and/or the absence of enzyme-catalyzed reactions in this pathway.
The method developed by Furchgott and Bursztyn has been used successfully to estimate the equilibrium dissociation constant (KA ) of agonists and determine the receptor reserve for a variety of receptors (24-27). The application of Furchgott’s analysis to concentration-response curves of adenosine yielded KA values of 2.7 and 5.6 μm for the binding of adenosine to A1 adenosine receptors that mediate activation of IKAdo and inhibition of β-ICa,L, respectively. A comparison of these functional KA values with those obtained independently from radioligand binding studies would be useful for establishing the validity of theKA values we report; however, the rapid degradation of adenosine by adenosine deaminase (31) as well as the formation of endogenous adenosine in membrane preparations (32) has made it difficult to obtain accurate estimates of theKA value for adenosine using the radioligand binding method. Regardless, theKA value for binding of adenosine to A1 adenosine receptors of mammalian brain membranes calculated using [3H]adenosine was estimated to be 0.7–9.4 μm (31). The functional KA values we report (2.7 and 5.6 μm) fall within this estimated range. Thus, regardless of the method used to estimate theKA values, it seems that adenosine binds with a low affinity to the A1 adenosine receptor.
Actions of SHA040.
SHA040 inhibited β-ICa,L by up to 60%, as shown in Fig. 8, but activated IKAdo with a relative efficacy of only 18.1% of that of adenosine. The magnitude of responses to SHA040 (inhibition of β-ICa,L, activation of IKAdo) correlated with the extent of A1 adenosine receptor reserve for each response. Receptor reserve for inhibition by adenosine of β-ICa,L was 70%, whereas receptor reserve for activation by adenosine of IKAdo was only 2%. Thus, the response with the greater receptor reserve was more activated by the partial agonist SHA040.
The large receptor reserve for the inhibition of β-ICa,L raised the possibility that partial agonists may be used to selectively inhibit the stimulatory effects of catecholamines on the heart without producing the direct, depressant effects on cardiac functions that are observed with full A1 adenosine receptor agonists. As a first test of this hypothesis, we determined whether a partial agonist selectively inhibited β-ICa,L. The results show that at high concentrations, SHA040 but not adenosine was a selective inhibitor of β-ICa,L. However, at lower concentrations, SHA040 (2–100 μm) and adenosine (<10 μm) exhibited similar degrees of selectivity for inhibition of β-ICa,L. For example, concentrations of SHA040 that inhibited β-ICa,L by 30% and 60% activated IKAdo by only 6% and 18%, respectively. Likewise, concentrations of adenosine that inhibited β-ICa,L by 30% and 60% activated IKAdo by 10% and 12%, respectively. Thus, these findings provide partial support for the hypothesis that SHA040 was better able than the full agonist adenosine to selectively inhibit β-ICa,L
Implications.
The differential potency of adenosine to activate IKAdo and inhibit β-ICa,L may have important physiological implications. In contrast to the constitutive inhibitory actions of adenosine in the kidney and adipose tissue (33, 34) the actions of this nucleoside on the heart seem to be limited to situations in which the balance of oxygen supply and demand is greatly disturbed. The estimated range of interstitial concentrations of adenosine in isolated guinea pig, rat, and rabbit hearts is 0.1–0.3 μm (35, 36). Our results show that adenosine at concentrations of 0.1–0.3 μm causes a significant inhibition of β-ICa,L but only a small activation of IKAdo of atrial myocytes. Thus, one might expect that endogenous adenosine tonically inhibits the stimulatory effects of β-adrenergic receptor activation in the atria. This may also be true of specialized tissues of the heart, in which adenosine at concentrations of ≤0.3 μm does not activate IKAdo of atrioventricular nodal myocytes but markedly inhibits β-ICa,L in these cells.1 Consistent with our results in single atrial myocytes, adenosine deaminase and adenosine receptor antagonists do not increase heart rate or shorten atrioventricular nodal conduction time (37, 38) but do enhance the stimulatory effects of β-adrenergic receptor agonists on both in vitro and in vivocardiac preparations (39, 40)
The concentration of isoproterenol used in this study, 100 nm, is sufficiently high to elicit a near-maximal increase in ICa,L. The use of lower concentrations of isoproterenol (10 nm) may lead to an overestimation of the inhibitory action of adenosine, whereas higher concentrations (e.g., 1 μm) of this β-adrenergic receptor agonist are known to facilitate calcium overload and trigger spontaneous activity in cardiomyocytes. Although not measured in the current study, it is predictable that the magnitude of receptor reserve for adenosine is inversely related to the concentration of the β-adrenergic receptor agonist and is influenced by experimental conditions that affect the cAMP/protein kinase A pathway (e.g., presence of a phosphodiesterase inhibitor). However, because there was a large receptor reserve for adenosine to inhibit a near-maximal increase of ICa,L caused by 100 nm isoproterenol, it is likely that adenosine would be able to inhibit responses to higher levels of β-adrenergic receptor stimulation.
The results of the current study are potentially relevant for the development of adenosine receptor-based therapies and for understanding the regulation by adenosine of cardiac functions. Whether differences in receptor reserve can be exploited to achieve organ and response selectivity for A1 adenosine receptor agonists and account for the tonic effects of adenosine in some organs remains to be demonstrated.
Footnotes
- Received April 17, 1997.
- Accepted July 2, 1997.
-
Send reprint requests to: L. Belardinelli, M.D., Professor of Medicine, University of Florida, P.O. Box 100277, Gainesville, FL 32610. E-mail: ramsey.med{at}shands.ufl.edu
-
↵1 M. Srinivas, J. C. Shryock, D. M. Dennis, S. P. Baker, and L. Belardinelli, unpublished observations.
-
This work was supported by National Institutes of Health Grant HL56785.
Abbreviations
- CPX
- 8-cyclopentyl-1,3-dipropylxanthine
- FSCPX
- 8-cyclopentyl-3-[3-[[4-(fluorosulfonyl)benzoyl]oxy]propyl]-1-propylxanthine
- CCPA
- 2-chloro-N6-cyclopentyladenosine
- DMSO
- dimethylsulfoxide
- K-H
- Krebs-Henseleit
- EGTA
- ethylene glycol bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- IKAdo
- adenosine-induced inwardly rectifying K+current
- ICa,L
- L-type Ca2+ current
- β-ICa,L
- isoproterenol-stimulated L-type Ca2+current
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}