


Abstract
We examined the ligand-binding site of the 5-hydroxytryptamine6 (5-HT6) receptor using site-directed mutagenesis. Interactions with residues in two characteristic positions of transmembrane region V are important for ligand binding in several bioamine receptors. In the 5-HT6receptor, one of these residues is a threonine (Thr196), whereas in most other mammalian 5-HT receptors, the corresponding residue is alanine. After transient expression in human embryonic kidney 293 cells, we determined the effects of the mutation T196A on [3H]d-lysergic acid diethylamide (LSD) binding and adenylyl cyclase stimulation. This mutation produced a receptor with a 10-fold reduced affinity for [3H]LSD and a 6-fold reduced affinity for 5-HT. The potency of both LSD and 5-HT for stimulation of adenylyl cyclase was also reduced by 18- and 7-fold, respectively. The affinity of other N1-unsubstituted ergolines (e.g., ergotamine, lisuride) was reduced 10–30-fold, whereas the affinity of N1-methylated ergolines (e.g., metergoline, methysergide, mesulergine) and other ligands, such as methiothepine, clozapine, ritanserin, amitriptyline, and mianserin, changed very little or increased. This indicates that in wild-type 5-HT6 receptor, Thr196 interacts with the N1 of N1-unsubstituted ergolines and tryptamines, probably forming a hydrogen bond. Based on molecular modeling, a serine residue in transmembrane region IV of the 5-HT2A receptor has previously been proposed to interact with the N1-position of 5-HT. When the corresponding residue of the 5-HT6 receptor (Ala154) was converted to serine, no change in the affinity of twelve 5-HT6 receptor ligands or in the potency of 5-HT and LSD could be detected, suggesting that this position does not contribute to the ligand binding site of the 5-HT6 receptor.
To date, 14 distinct mammalian 5-HT (serotonin) receptors have been identified. The known 5-HT receptors include a ligand-gated ion channel (5-HT3 receptor) and 13 G protein-coupled receptors (1, 2). Unlike the classic 5-HT receptors, the 5-HT6 receptor was first discovered by cloning from rat striatal cDNA but had not been previously identified as a pharmacological entity in physiological or radioligand binding experiments (3, 4). Subsequently, the human 5-HT6receptor was isolated by homology screening (5). The highest levels of 5-HT6 receptor mRNA are present in olfactory tubercle, nucleus accumbens, striatum, and hippocampus (3, 4, 6, 7). In addition to these regions, 5-HT6 receptor-like immunoreactivity was identified in frontal and entorhinal cortex and the molecular layer of the cerebellum (8). The functional significance of this receptor has been investigated using intracerebroventricular injection of 5-HT6 receptor-specific antisense oligonucleotides, which produced a behavioral syndrome, suggesting effects on dopaminergic and/or cholinergic neurotransmission (9, 10).
The 5-HT6 receptor displays a characteristic pharmacological profile, as was shown in competition studies with either 125I-LSD or [3H]5-HT, both of which bind to the 5-HT6 receptor with high affinity (3). Many nonselective compounds, such as tricyclic antidepressant drugs and a large number of antipsychotic agents, tryptamine, and ergoline derivatives, interact with the 5-HT6 receptor (3,11, 12). Because no selective ligands are available, identification of functional 5-HT6 receptors in physiological preparations can be only tentative. A model of the 5-HT6 receptor binding site may suggest modifications of known ligands to improve the affinity and selectivity for this receptor. To verify and adapt models that have been proposed for other G protein-coupled receptors, we attempted to identify residues contributing to the ligand binding site of the 5-HT6 receptor through the use of site-directed mutagenesis.
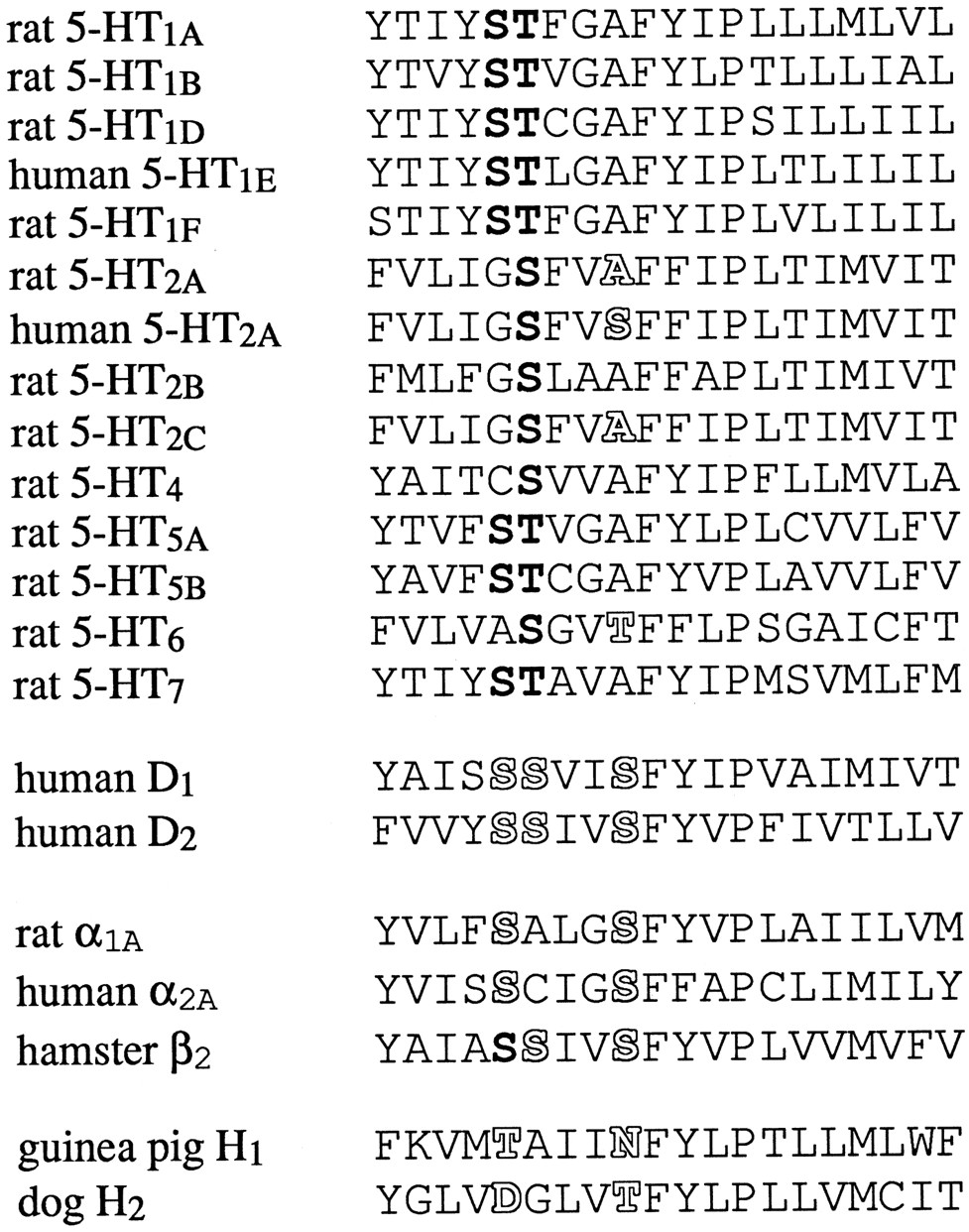
There is general agreement that for this class of bioamine receptors, the ligand binding site is formed by seven transmembrane helices present in all members of the family (13-15). In these transmembrane regions, the 5-HT6 receptor shows highest homology to other 5-HT receptors (36–41% sequence identity), but the homology to adrenergic, dopamine, and histamine receptors is also quite high (30–35% identity depending on which segments are included in the comparison). In site-directed mutagenesis and modeling studies with other bioamine receptors, several specific regions were consistently found to be involved in ligand binding and receptor activation. In transmembrane helix V of the hamster β2-adrenergic receptor, replacement of either of two serine residues attenuated the activity of catecholamine agonists at the receptor (16) (Fig. 1). Removal of the catechol hydroxyl moieties from the aromatic ring of the ligand mimicked the effects of the mutation, suggesting that in wild-type receptors, hydrogen bond interactions are formed between the serine residues and the meta- and para-hydroxyl group of agonists. These two or three characteristically spaced serine residues (one helix turn or three or four residues apart) were subsequently shown to contribute to agonist binding in α2A- and α1-adrenergic receptors (17, 18) and both D1 and D2 dopamine receptors (19-22). In the histamine receptors, these positions are occupied by threonine and asparagine (H1 receptor) or aspartate and threonine (H2), and mutations of these residues also affect ligand binding (23-26). In all G protein-coupled 5-HT receptors cloned to date, the first of these “hydrogen bonding” positions is occupied by a serine or threonine residue and frequently preceded by another serine (see Fig. 1). In the 5-HT1Areceptor, a serine and a threonine residue are next to each other at this position. Replacement of either residue by alanine reduced the affinity of 5-HT and its ability to stimulate GTPase activity, suggesting the disruption of a normally present hydrogen bond with the hydroxyl-group on the indole ring of 5-HT (27). The second potential hydrogen bonding position in transmembrane region V is occupied by an alanine in 11 of the 13 cloned mammalian G protein-coupled 5-HT receptor subtypes. The only exceptions are the rat and human 5-HT6 receptors (threonine) and the human, pig, and squirrel monkey 5-HT2A receptors (serine), whereas the rat 5-HT2A receptor has an alanine residue in this position (like all other subtypes). This single amino acid variation is responsible for dramatic species differences in the affinity of N1-substituted or unsubstituted ergolines and tryptamines for rat compared with human, pig, or squirrel monkey 5-HT2A receptors, as was demonstrated by reversal of the pharmacology by point mutations in both the human and rat receptor (28-32). This position contributes to the subtype selectivity of ligands because replacement of the wild-type alanine of the human 5-HT2C receptor by serine changes the affinity of several ergoline compounds to values closer to their affinity for the human 5-HT2A receptor, and vice versa (33).

Alignment of transmembrane region V of mammalian 5-HT and related bioamine receptors. Bold, conserved serine and threonine residues in the first potential hydrogen bonding position(s) of the 5-HT receptors. The role of the outlinedresidues in ligand binding has been examined in the receptors listed and/or in species homologues with the help of site-directed mutagenesis. In the current study, Thr196 of the 5-HT6receptor has been mutated to alanine, the residue found in most other 5-HT receptors.
To determine whether this second potential hydrogen bonding position available in the 5-HT6 receptor contributes to ligand binding, we replaced the threonine residue present at the corresponding position of the rat 5-HT6 receptor (Thr196) with alanine (T196A), the residue present in most other mammalian 5-HT receptors.
Using a G protein-coupled receptor model based on the structure of bacteriorhodopsin, Hibert et al. (13) suggested a number of potential interactions of 5-HT with the 5-HT2Areceptor, including a hydrogen bond between the 5-hydroxyl group of 5-HT and a serine in transmembrane V (in the first hydrogen bonding position discussed above) and a second hydrogen bond between a serine residue in transmembrane IV and the indole-nitrogen (N1) of 5-HT (13). This serine residue is conserved in most cloned mammalian 5-HT receptors with the exception of the 5-HT1Areceptors (glycine), 5-HT4 receptor (proline), and 5-HT6 receptors (alanine) (Fig.2). We tested whether the substitution of Ala154 by serine (A154S) (i.e., the introduction of a hydroxyl group) would allow the formation of a new hydrogen bond and thus change the affinity or agonist activity of 5-HT6 receptor ligands.

Alignment of part of transmembrane region IV of mammalian 5-HT receptors. Bold, conserved serine residues. Ala154 of the 5-HT6 receptor (in outline view) has been mutated in the present study.
Experimental Procedures
Cloning of the rat 5-HT6 receptor and site-directed mutagenesis.
A cDNA clone representing the 5-HT6 receptor was generated from rat striatal mRNA by reverse transcription-polymerase chain reaction using primers based on the published rat 5-HT6 sequence (3)1 and subcloned into the plasmid pAMP1 using the CloneAmp system (Life Technologies, Eggenstein, Germany). One clone was identified that matched exactly the sequence of Kohen et al. (5)2 with the exception of a single base exchange that did not alter the amino acid sequence. Further details are provided in Boess et al. (12). An EcoRI/XbaI restriction fragment of the pAMP1–5-HT6 construct was ligated into the expression vector pcDNA1.1amp (InVitrogen, San Diego, CA) and into the phagemid pAlter (Promega, Madison, WI). Mutagenesis was performed using the Altered Sites Mutagenesis kit (Promega). Thr196 was mutated to alanine (T196A) using the oligonucleotide 5′-GTCCGGCGTCGCCTTTTTCCT-3′. Ala154 was mutated to serine (A154S) using the mutagenic oligonucleotide 5′-GGTGCCTGGAGCCTCAGCGCGCTTGCCTCCTTC-3′. This oligonucleotide introduced an additional silent mutation, creating a new BssH II restriction site. After transfer into the expression vector pcDNA1.1amp, mutations were confirmed by sequencing the entire coding region using an automated fluorescent sequencing system (ALF; Pharmacia, Vienna, Austria).
Transient transfection of HEK 293 cells.
HEK 293 cells were maintained in DMEM plus 10% FBS containing 100 IU/ml penicillin and 100 μg/ml streptomycin in a humidified atmosphere (5% CO2). HEK 293 cells (80–90% confluent in 75- or 185-cm2 flasks) were cotransfected with the construct pcDNA1.1amp-5-HT6 (8 or 20 μg) containing the coding region of wild-type or mutant 5-HT6receptor according to the Lipofectamine transfection protocol (Life Technologies). After 1 day, the medium was replaced by fresh DMEM plus 10% FBS.
Membrane preparation and receptor binding assays.
Two days after transfection with the pcDNA1.1amp constructs containing mutant or wild-type 5-HT6 receptor coding regions, the cells were detached with ice-cold 50 mm Tris·HCl, pH 7.4, containing 10 mm MgCl2 and 0.5 mm EDTA. The cells were disrupted using a Polytron homogenizer (15 sec at maximal speed) at a concentration corresponding to ∼2 × 106 cells/ml. This homogenate was centrifuged at 50,000 × g for 30 min, resuspended in the same volume, and centrifuged again. At this stage, the resulting pellets were either stored at −20° or used immediately in binding assays. 5-HT6 receptor binding assays were performed using [3H]LSD (specific activity, 82 Ci/mmol; Amersham, Little Chalfont, UK) as radioligand. Membranes were resuspended in assay buffer (50 mm Tris·HCl, 10 μm pargyline, 5 mmMgCl2, 0.5 mm EDTA and 0.1% ascorbic acid, pH 7.4). Binding assays consisted of 100 μl of membranes (corresponding to 4 × 105 cells/assay tube), 50 μl of [3H]LSD, and 50 μl of displacing drug or assay buffer (final assay volume, 200 μl). Nonspecific binding was measured in the presence of 100 μm 5-HT or 10 μm methiothepine. Saturation experiments were performed using eight concentrations of [3H]LSD (final concentrations, 0.163–20 nm for wild-type and mutant A154S and 0.625–80 nm for mutant T196A). Competition assays were performed in the presence of seven concentrations of the displacing ligands (10−10 to 10−4 m) and 1 nm [3H]LSD (5 nm for mutant T196A). Incubations were performed at 37° for 60 min and terminated by rapid filtration through Whatman (Maidstone, UK) GF/B filters pretreated with polyethyleneimine (0.3%). The filters were washed three times with 2 ml of Tris·HCl (50 mm; pH 7.4), and the radioactivity retained on the filters was measured by scintillation spectroscopy in 2 ml of scintillation fluid. All experiments were performed in triplicate and repeated at least three times. Values are given as mean ± standard error. Data were analyzed using the programs EBDA and LIGAND (34, 35). Protein concentrations were determined using the BCA method (Pierce Chemical, Indianapolis, IN).
Adenylyl cyclase measurements.
Two days after transfection, cells grown in DMEM plus 10% FBS (dialyzed) were washed once with DMEM without phenol red (DMEM−), detached with PBS plus 1 mm EDTA, and washed twice with DMEM− (470 × g, 5 min). The final cell density was adjusted to ∼1.25 × 106 cells/ml. Aliquots of 80 μl were transferred to 96-well plates (ca. 105cells/well) and incubated at 37° in a humidified atmosphere for 30 min. 5-HT or LSD, combined with pargyline and the phosphodiesterase inhibitor Ro 20–1724, was added in a volume of 20 μl/well (final incubation volume, 100 μl/well; final concentration of agonists, 10−10 to 10−3 m; pargyline, 20 μm; Ro 20–1724, 100 μm). After 20 min at 37° in a humidified atmosphere (5% CO2), the incubation was terminated by the addition of 200 μl of ethanol/well. After ≥2 hr at −20°, the plates were centrifuged for 5 min at 470 × g (4°), and 75-μl aliquots of the supernatant were transferred to OptiPlates (Packard, Meriden, CT), evaporated under vacuum, and resuspended in 0.05 m acetate buffer. The concentration of cAMP was determined using the BIOTRAK cAMP [125I] Scintillation Proximity Assay system (Amersham) adapted to 96-well plates. The concentration effect curves were analyzed using the equation E = B + Emax *x/(EC50 + x), where E and Emax are the measured and maximum effects (cAMP/well), respectively; B is the basal cAMP level, and xis the concentration of agonist.
Materials.
[3H]LSD (specific activity, 82 Ci/mmol) was obtained from Amersham. 5-HT was from FLUKA (Buchs, Switzerland), and ergotamine was from Sigma (Buchs, Switzerland). Mesulergine, metergoline, methysergide, lisuride, methiothepine, clozapine, amitriptyline, ritanserin, mianserin, and pargyline were purchased from Research Biochemicals (Natick, MA). DMEM, FBS, penicillin, streptomycin, and geneticin were obtained from Gibco Life Technologies (Basel, Switzerland). Ro 20–1724 and LSD were synthesized at F. Hoffmann-La Roche (Basel, Switzerland)
Results
Expression in HEK 293 cells and [3H]LSD binding.
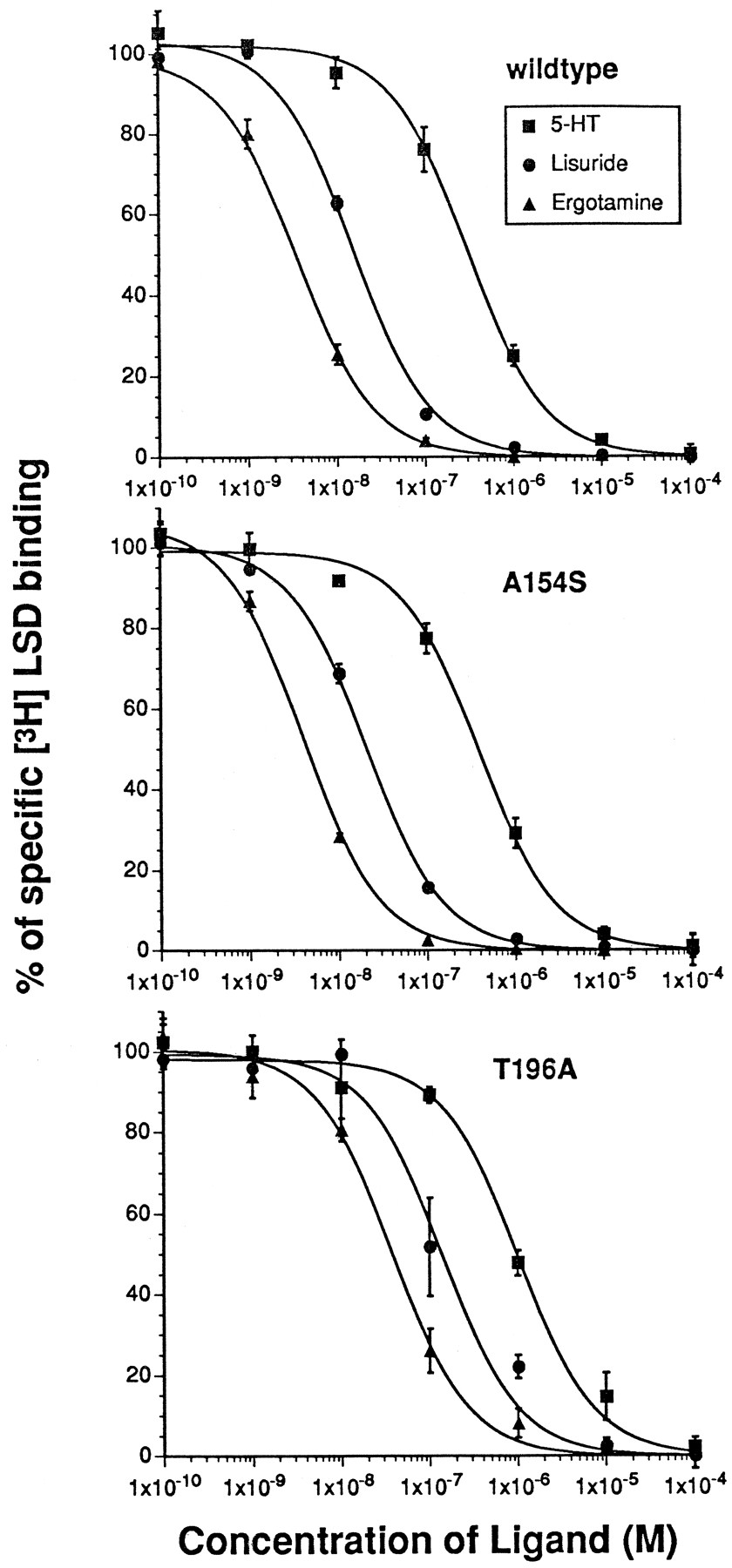
Transient transfection of HEK 293 cells with wild-type 5-HT6 receptor or either mutant receptor construct resulted in high expression levels of 1–3 pmol/mg of protein as determined in [3H]LSD binding assays. When the transfections were performed in parallel using the same batch of HEK 293 cells, the amount of receptor expressed per mg of protein was similar for wild-type and mutant receptors (Fig.3). TheKd values for wild-type 5-HT6 receptor and mutant A154S were not significantly different (1.9 ± 0.3 and 2.1 ± 0.1 nm, respectively). However, the affinity of [3H] LSD for mutant T196A was reduced by a factor of 16 (Kd = 33.9 ± 7.1 nm; p < 0.001).

[3H]LSD binding to HEK 293 membranes expressing wild-type 5-HT6 receptor or mutant receptors A154S or T196A. The examples shown are from HEK 293 cells transfected with the same amount of pcDNAI-5-HT6 receptor wild-type or mutant constructs. Insets, Scatchard transformation of saturation binding data. The Bmax values of the experiments shown are 2.4 (wild-type), 1.8 (A154S), and 1.9 (T196A) pmol/mg of protein, and the Kd values are 1.8 nm (wild-type), 2.3 nm(A154S), and 27.7 nm (T196A).
Comparison of the pharmacological profile of wild-type and mutant receptors.
In competition assays with [3H] LSD, the affinity of the endogenous agonist 5-HT for the mutant receptor T196A was reduced 6-fold compared with wild-type 5-HT6 receptor (Fig.4, Table1). The affinities of the N1-unsubstituted ergolines ergotamine and lisuride were reduced 16- and 34-fold, respectively (Fig. 4, Table 1). In contrast, the affinity of N1-methylated ergolines for the mutant T196A did not change (metergoline) or increased 4–7-fold (methysergide, mesulergine) (Table1) (see Fig. 5 for structures). The affinity of several other high affinity 5-HT6receptor ligands that are neither tryptamine nor ergoline derivatives (methiothepine, clozapine, amitriptyline, ritanserin, and mianserin) did not change significantly (Table 1). The Hill slopes of the ligands tested were not significantly different between wild-type and T196A mutant 5-HT6 receptors, with the exception of lisuride and ritanserin, which had lower Hill slopes at the mutant receptor (p < 0.05, unpaired ttest). Neither of the compounds examined showed significant changes in affinity for the second mutant (A154S) (Fig. 4; Table 1).

Interaction of wild-type and mutant receptors with 5-HT and N1-unsubstituted ergolines. [3H]LSD competition experiments were conducted for wild-type 5-HT6 receptor, mutants A154S and T196A with 5-HT (▪), and the N1-unsubstituted ergolines lisuride (•) and ergotamine (▴). Data are mean ± standard error from three experiments performed as described in Experimental Procedures.
Pharmacological profile of mutant 5-HT6 receptors
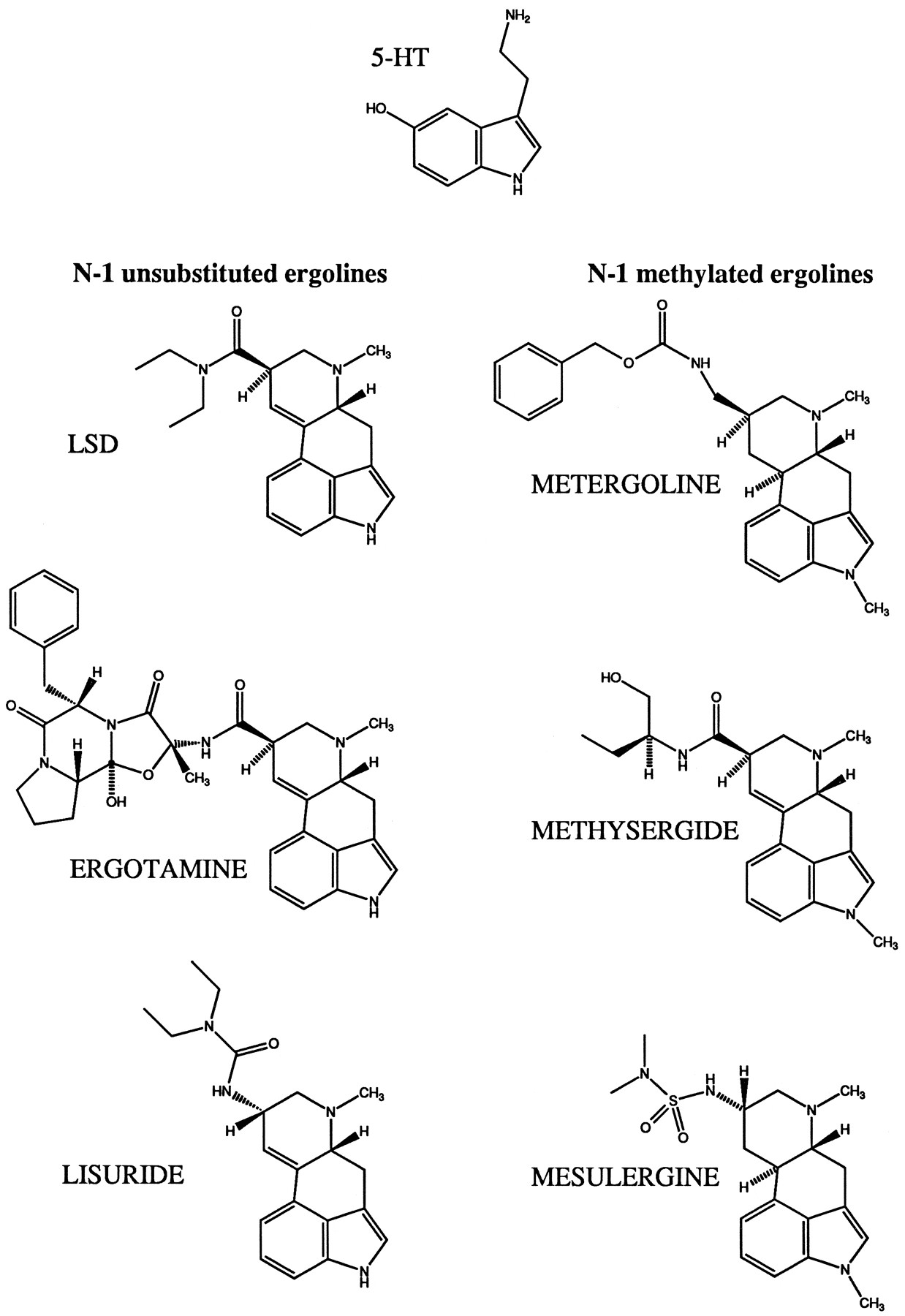
Structure of 5-HT and the ergoline compounds used in the study.
Comparison of the functional properties of wild-type and mutant 5-HT6 receptors.
The basal levels of cAMP accumulation (after 20 min of incubation without agonist) were 1.3 ± 0.3, 0.9 ± 0.2, and 0.9 ± 0.2 pmol/105cells for HEK 293 cells expressing wild-type, mutant A154S, and mutant T196A 5-HT6 receptors, respectively. HEK 293 cells expressing the wild-type 5-HT6 receptor or either of the mutant receptors responded to 5-HT and LSD with increased cAMP production (Fig. 6), whereas nontransfected cells did not respond in the concentration range tested. The maximum level of cAMP accumulation obtained after stimulation with 5-HT was 27.5 ± 6.9 pmol/105 cells for wild-type 5-HT6 receptor, 25.1 ± 4.7 pmol/105 cells for mutant A154S, and 21.7 ± 3.6 pmol/105 cells for mutant T196A (mean ± standard error, three experiments). Therefore, the mutations did not influence the efficiency of receptor/G protein interactions. The maximum levels of cAMP accumulation obtained with LSD were 67 ± 9% (wild-type), 66 ± 7% (A154S), and 69 ± 12% (T196A) of the maximum observed with 5-HT. This indicates that LSD is a partial agonist at the 5-HT6 receptor and the changes introduced in the mutants do not alter the partial agonist character of this ligand. In cells expressing wild-type 5-HT6receptor, 5-HT stimulated cAMP accumulation with an EC50 value of 74 ± 22 nm. LSD was more potent, with an EC50 of 16 ± 7 nm. Replacement of Ala154 by a serine residue in transmembrane region IV (mutant A154S) did not significantly change the potency of either 5-HT (EC50 = 89 ± 13 nm) or LSD (EC50 = 17 ± 8 nm). However, in cells expressing the mutant T196A, the agonist potency of 5-HT was reduced 7-fold (EC50= 540 ± 73 nm, n = 3,p < 0.01), and that of LSD was reduced 18-fold (EC50 = 290 ± 74 nm,p < 0.05) (Fig. 6).
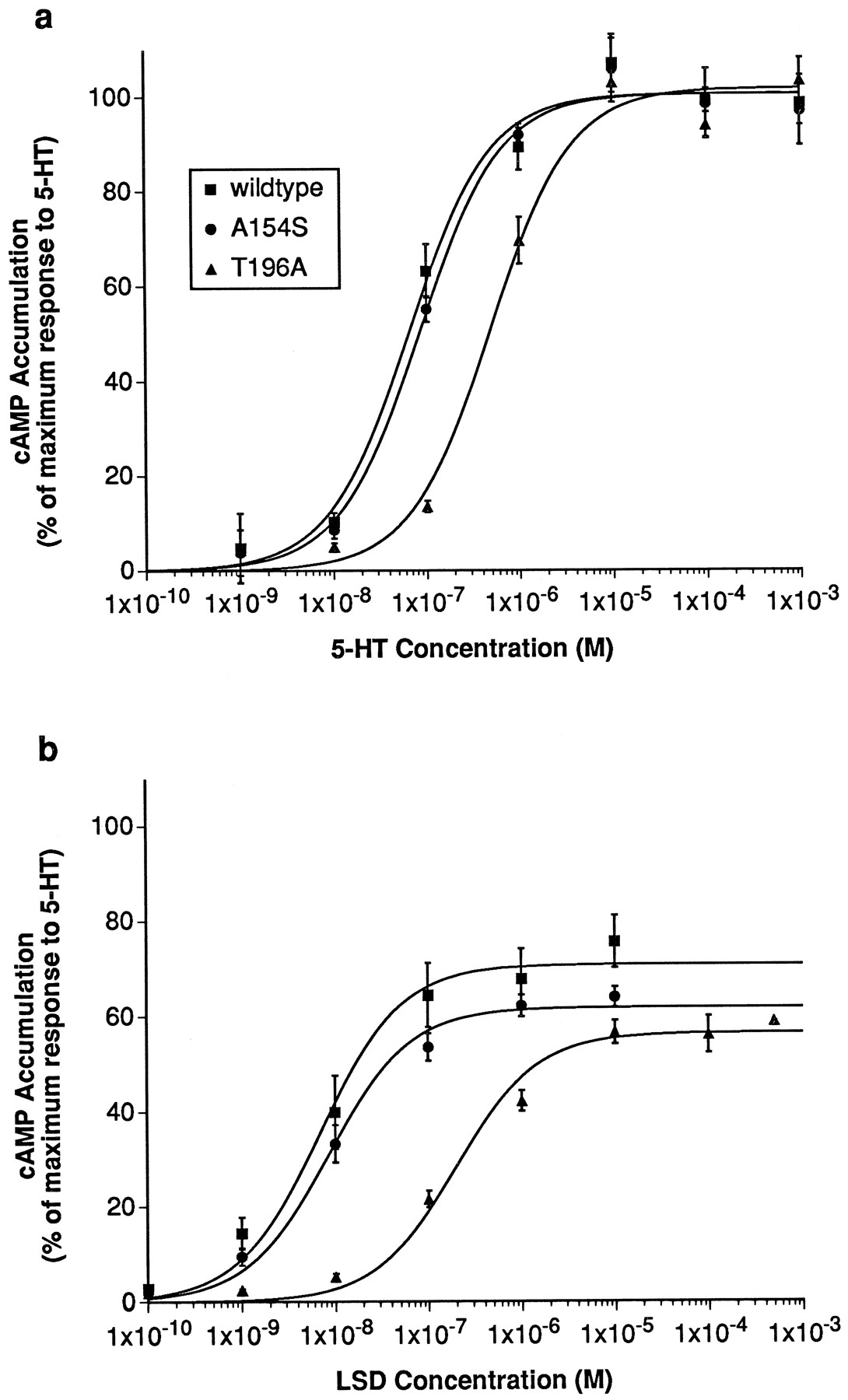
Functional responses of wild-type and mutant 5-HT6 receptors. Stimulation of cAMP accumulation in HEK 293 cells expressing wild-type 5-HT6 receptor (▪), mutant A154S (•), or mutant T196A (▴) by 5-HT (a) and LSD (b). Values are expressed as percentage of maximum cAMP accumulation obtained with 5-HT at the wild-type 5-HT6 receptor. Data are the mean ± standard error of three experiments performed as described in Experimental Procedures.
Change in free energy.
For LSD and 5-HT, the change in binding energy Δ(ΔG) introduced by the mutation T196A could be calculated both from the EC50 values determined in adenylyl cyclase stimulation assays and theKd andKi values determined in [3H]LSD binding experiments (Table2). The values calculated for 5-HT (5.1 and 4.6 kJ/mol) were lower than those observed for LSD (7.5 and 7.3 kJ/mol). The change in free energy observed for ergotamine (7.2 kJ/mol) and lisuride (9.1 kJ/mol) calculated from theKi values was also higher than for 5-HT.
Change in binding energy caused by the mutation T196A
Discussion
The 5-HT6 receptor has a characteristic pharmacological profile that distinguishes it from other 5-HT receptors, but so far, no selective 5-HT6receptor ligands have been described. We attempted to identify specific interactions between the 5-HT6 receptor and nonselective ligands to allow the adaptation of existing G protein-coupled receptor models to the binding site of the 5-HT6 receptor. Eventually, such models could be used to suggest structural modifications that might improve ligand affinity and selectivity. Several groups have proposed three-dimensional models of the ligand binding site of other G protein-coupled 5-HT receptors (13, 36-38). As a first step, we looked for characteristic differences in the amino acid sequence of the 5-HT6 receptor compared with other 5-HT receptors in positions that had been suggested to form part of the ligand binding site.
One of the interactions of 5-HT with the 5-HT2Areceptor suggested by Hibert et al. (13) was a hydrogen bond between a serine residue in transmembrane region IV and the indole-nitrogen (N1) of 5-HT. This serine residue is conserved in the majority of cloned mammalian 5-HT receptors, with the exception of the 5-HT1A receptors (glycine), 5-HT4 receptor (proline), and 5-HT6 receptors (alanine). We tested whether substitution of Ala154 by serine (i.e., the introduction of a new hydroxyl group at this position) would allow the formation of a new hydrogen bond, thus changing the affinity or agonist activity of 5-HT6 receptor ligands. However, neither 5-HT- or LSD-induced adenylyl cyclase stimulation nor the affinities of a range of compounds tested in [3H]LSD binding assays were altered. This postulated interaction is apparently not possible in the case of the 5-HT6 receptor. Whether it actually takes place in the 5-HT2A or any other serotonin receptor has not yet been experimentally verified to our knowledge.
In contrast, removal of a potential hydrogen bond forming site in transmembrane helix five of the 5-HT6 receptor by changing Thr196 to alanine (the residue present in most other mammalian 5-HT receptors) selectively reduced the affinity of the natural agonist 5-HT (a N1-unsubstituted indoleamine) and that of several N1-unsubstituted ergolines (LSD, lisuride, ergotamine) while not affecting the affinity of the N1-methylated compound metergoline and increasing the affinity of the N1-methylated ergolines mesulergine and methysergide. The potency of 5-HT and LSD in cAMP accumulation experiments was reduced by the same factor as the affinity determined in binding experiments. The change in free energy [Δ(ΔG)] for the interaction with 5-HT calculated from the ratio of either the EC50 values (5.1 kJ/mol) orKi values (4.6 kJ/mol) was less than that for LSD (7.5 and 7.3 kJ/mol, respectively). The other N1-unsubstituted ergolines tested in [3H] LSD binding assays, ergotamine (7.2 kJ/mol) and lisuride (9.1 kJ/mol), also showed larger changes in their free energy than 5-HT. However, all values were in the range expected for hydrogen bonds (2.1–7.5 kJ/mol = 0.5–1.8 kcal/mol) (39). This is consistent with the existence of a hydrogen bond between the indole N of 5-HT and N1-unsubstituted ergoline compounds and the hydroxyl group of Thr196 in the wild-type 5-HT6 receptor (Fig.7). The efficacy of adenylyl cyclase coupling was not affected by exchanging Thr196 for alanine because the maximum levels of adenylyl cyclase stimulation obtained for this mutant and wild-type receptor were similar and the partial agonist character of LSD was unchanged. The decreased potency of both 5-HT and LSD observed in the cAMP accumulation experiments with mutant T196A presumably is a direct consequence of the reduction in binding affinity that is detected in the [3H] LSD binding assays. The expression levels reached for wild-type and mutant receptors were similar, so effects due to changes in the ratio of receptor to G proteins can be excluded. The affinities of metergoline and those of several antagonists with nonergoline structures were not significantly decreased. This shows that the overall structure of the receptor has been well preserved and the observed reduction of the affinity of 5-HT and N-1 unsubstituted ergolines must be due to the elimination of a specific interaction of the side chain of Thr196 and these ligands. The increased affinity of mesulergine and methysergide could be the result of the elimination of an unfavorable steric interaction between the methyl group in the N1 position of these ligands and the hydroxyl and methyl group of Thr196 in the wild-type receptor that are removed in the T196A mutant (Fig. 7). Metergoline may bind to the receptor in a slightly different orientation that is not influenced by the size of the side chain of residue 196.
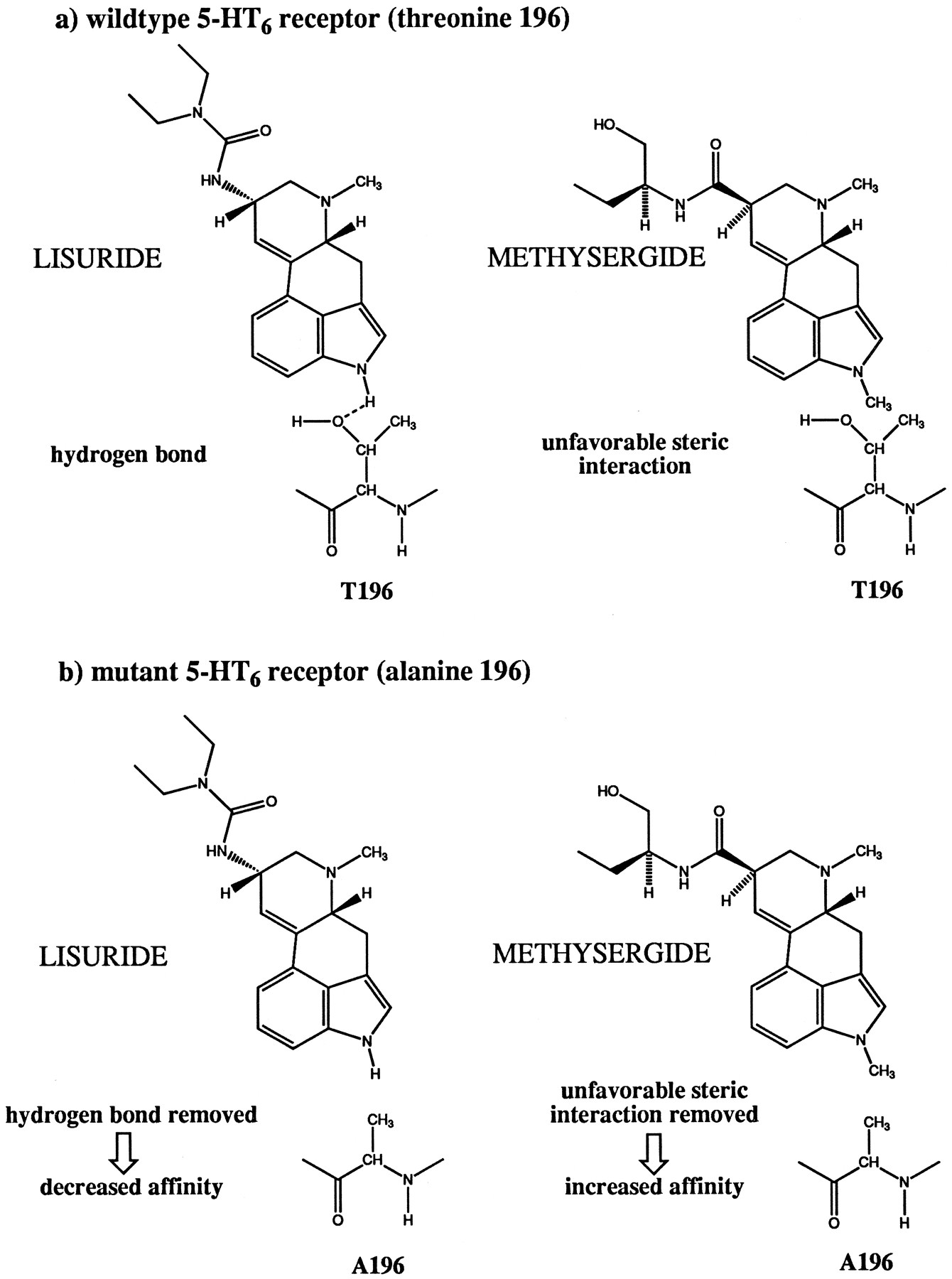
Proposed interactions of a N1-unsubstituted ergoline (lisuride) and a N1-methylated ergoline (methysergide) with the side chain of residue 196 in wild-type 5-HT6receptor and mutant T196A.
The hydrogen bond that can be formed between the N1 of ergoline compounds and the hydroxyl group of Thr196 is analogous to the interaction of these compounds with the amino acid present in the corresponding position in human, pig, and squirrel monkey 5-HT2A receptor (Ser242) (28-32). All other cloned mammalian 5-HT receptors, including the rat 5-HT2A receptor, contain an alanine residue at this position (Fig. 1). N1-Unsubstituted ergolines and tryptamines have a higher affinity for human, pig, and squirrel monkey 5-HT2A receptors because they can form a hydrogen bond with the serine residue present in these species. N1-Substituted ergolines have a higher affinity for the rat 5-HT2A receptor and lower affinity for human, pig, and squirrel monkey 5-HT2A receptors, presumably because of unfavorable steric interactions with the hydroxyl group present in the serine-containing species subtypes. Replacement of the serine residue present in the human 5-HT2Areceptor by alanine results in a pharmacology similar to that of the rat receptor, and replacement of the alanine in the rat sequence by serine did the reverse (28, 29). This position also contributes to the selectivity of ligands between the human 5-HT2Areceptor and the human 5-HT2C receptor (33).
Among 5-HT receptors, the interaction between agonists and the second hydrogen bonding site in transmembrane region V is unique to the 5-HT6 receptor and the species subtypes of the 5-HT2A receptor mentioned above. In contrast, all 5-HT receptors cloned to date contain either a serine or a threonine residue one helix turn (three residues) closer to the extracellular surface (Ser193 in the 5-HT6 receptor). Mutation of the corresponding residue in the 5-HT1Areceptor (a threonine) to alanine decreased the affinity and agonist activity of 5-HT (27). This hydrogen bond with the 5-hydroxyl-group of the indole ring of 5-HT may thus be present in the binding site of all 5-HT receptors. In the related cationic amine receptors (i.e., α1A-, α2A-, and β2-adrenergic receptors; D1 and D2 dopamine receptors; and H1 and H2histamine receptors), both positions are occupied by potential hydrogen bond-forming residues, and in most cases, both positions seem to contribute to the ligand binding site. Of particular interest with respect to our results is the suggestion that the asparagine residue present at the position corresponding to Thr196 in the histamine H1 receptor may interact with the τ-nitrogen of the imidazole ring of histamine (24). This τ-nitrogen can assume the same relative position to the positively charged ω-nitrogen of histamine as the indole (N1) nitrogen of 5-HT to the ω-nitrogen of 5-HT. The histamine H2 receptor contains a threonine residue at this position, and mutation of this residue also affects ligand binding and agonist potency (23).
The combination of site-directed mutagenesis (guided by knowledge obtained for related receptors) with a series of related ligands differing in a particular structural feature has allowed the identification of a specific interaction between Thr196 in transmembrane region V of the 5-HT6 receptor and the indole nitrogen of N1-unsubstituted ergolines and tryptamines. These results provide information that can be used to improve models of 5-HT6 receptor/ligand interactions and may contribute to the design of selective drugs for this receptor type.
Footnotes
Abbreviations
- 5-HT
- 5-hydroxytryptamine
- HEK
- human embryonic kidney
- LSD
- d-lysergic acid diethylamide
- DMEM
- Dulbecco’s modified Eagle’s medium
- FBS
- fetal bovine serum
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}