


Abstract
Rho1 receptor-channels (ρ1Rs) are GABA-gated chloride channels that exhibit slow kinetics, little desensitization, and inert pharmacology to most anesthetics, except for neuroactive steroids (NSs). NSs differentially modulate ρ1Rs dependent on the steric arrangement of the hydrogen atom at the fifth carbon position. In particular, the NS allotetrahydrodeoxycorticosterone (5α-THDOC) potentiates, whereas 5β-pregnane-3α-ol-20-one (pregnanolone) and 5β-dihydroprogesterone (5β-DHP) inhibit ρ1 GABA currents. Here, we used Xenopus laevis oocytes expressing ρ1Rs as a model system to study the mechanism of NS modulation. The second transmembrane residue, Ile307, was mutated to 16 amino acids. Subsequent testing of these mutants with 5α- and 5β-NSs, at equivalent GABA activity, showed the following paradigm. For 5β-DHP, Ile307 mutation either altered the degree of inhibition or entirely reversed the direction of modulation, rendering 5β-DHP a potentiator. Dependent on the mutation, pregnanolone remained an inhibitor, transformed into a potentiator, or converted to inhibitor and potentiator based on concentration. The extent of mode reversal for both 5β compounds showed a correlation with the side-chain hydrophilicity of the 307 residue. In contrast, Ile307 substitutions did not alter the direction of modulation for 5α-THDOC but caused a significant increase in the level of potentiation. Paradoxical to their impact on the mode and/or the degree of modulation, none of the mutations altered the concentration range producing the response significantly for any of the above NSs. Moreover, preincubation of Ile307 mutants with 5α or 5β alone produced an equivalent effect on the activation time course. Based on the above data, a universal model is presented wherein anesthetic compounds like NSs can potentiate or inhibit the activity of ligand-gated ion channels distinct from interaction with alternative binding sites.
Metabolites of sex and stress hormones, known as neuroactive steroids (NSs), represent a large class of endogenous compounds within the central nervous system (CNS) (Covey et al., 2001; Lambert et al., 2001; Stoffel-Wagner, 2001; Mellon and Griffin, 2002). In vivo, NSs seem to play an important role in the establishment of mood and behavior states. In the clinical setting, administration of NSs can produce a dose-dependent graded effect upon the function of the CNS that ranges from sedation to induction of anesthesia (Purdy et al., 1991; Gray et al., 1992; Frye and Duncan, 1994; Romeo et al., 1998; Girdler et al., 2001; Mellon and Griffin, 2002). NSs mediate their effect by targeting membrane-embedded proteins known as ligand-gated ion channels and thereby alter synaptic transmission within the CNS. One of the most diversified and ubiquitously expressed classes of inhibitory ligand-gated ion channels is the GABAA receptor (GABAAR). NSs are modulators and can be direct agonists of GABAARs. In this regard, their action closely resembles that of intravenous anesthetic barbiturates (Harrison and Simmonds, 1984; Majewska et al., 1986; Barker et al., 1987; Peters et al., 1988; Turner et al., 1989; Morrow et al., 1990; Puia et al., 1990; Majewska, 1992; Twyman and MacDonald, 1992; Woodward et al., 1992; Zaman et al., 1992; Kokate et al., 1994; Shen et al., 2000).
The GABAAR is a pentameric protein complex composed of closely related subunits. Each subunit is postulated to cross the membrane bilayer four times, where the second transmembrane domain is the ion channel (Olsen, 1998; Sieghart, 2000). Closely related to GABAA are GABAC ρ1 receptor-channels (ρ1Rs). Compared with GABAARs, ρ1Rs are insensitive to most anesthetics except in the case of NSs (Shimada et al., 1992; Morris et al., 1999; Chebib and Johnston, 2000; Qian and Ripps, 2001). Chimera studies between the subunits of GABAARs and ρ1Rs and mutagenesis have been essential to mapping the domain and pinpointing the amino acids important for a number of anesthetics. These studies demonstrate that ρ1 Ile307 and Trp328, positioned within the second and third transmembrane domains (TM2 and TM3, respectively), play an important role in anesthetic action (Amin, 1999; Belelli et al., 1999; Walters et al., 2000). Mutation of ρ1 Trp328 to Ala or Met (or any hydrophobic residues), found at the equivalent TM3 position within the α1 and β2 subunits of the GABAA receptor, respectively, confers barbiturate sensitivity to ρ1Rs (Amin, 1999). The residue equivalent to ρ1 Ile307 within the α1 subunit is serine. Mutation of ρ1 Ile307 to Ser also confers barbiturate sensitivity to ρ1Rs (Belelli et al., 1999). Moreover, mutations of both Ile307 and Trp328 to their amino acid counterpart within the α1 and β2 subunit impart benzodiazepine sensitivity to ρ1Rs (Walters et al., 2000).
NSs differentially modulate the ρ1R dependent on the steric arrangement of the hydrogen atom at the fifth carbon position of the cholesterol backbone (Morris et al., 1999). The 5α position of the hydrogen atom significantly reduces the bend within the cholesterol backbone in comparison to 5β position rendering the 5α-NSs (e.g., 5α-THDOC) potentiators, and 5β-NSs (e.g., 5β-DHP and pregnanolone) inhibitors of the ρ1R GABA current. It is interesting that the position of the hydrogen atom at the 5th carbon does not have an effect on the mode of modulation of the NS upon the GABAAR because both 5α- and 5β-NSs are positive modulators of GABAARs.
We used the ρ1R as a model system to study the molecular mechanism of NS action. The ρ1R offers a number of advantages over GABAAR systems including slow kinetics of activation and deactivation, low rate of desensitization (Amin and Weiss, 1994), and 5-fold degeneracy (comparison with GABAAR composed of closely related but not identical subunits). The effect of mutation at position 307 (and others) within the TM2 on the modulation by 5α-THDOC, pregnanolone, and 5β-DHP was determined. The Ile307 mutations impacted the mode and the degree of modulation for 5β compounds, either by entirely reversing the direction of modulation from inhibition to potentiation for both 5β-NS or by producing biphasic activity for pregnanolone. In contrast to 5β compounds, the effect of these mutations on 5α-THDOC was to significantly increase the level of potentiation without altering the mode of modulation. However, the marked impact of Ile307 mutations on direction of the NS action did not parallel a significant alteration in the NS concentration range producing the response. Furthermore, preincubation with 5α or 5β alone produced an equivalent effect on the NS activation time course. Based on these data, we have proposed a unique model for NS-dependent modulation of ligand-gated ion channels.
Materials and Methods
Single amino acid substitutions were created using GeneEditor in vitro site-directed mutagenesis system (Promega, Madison, WI) within the pSp72 plasmid vector (Promega,). The restriction enzyme SspI was used to linearize the plasmid vector containing the mutated or the wild-type human ρ1 cDNA. Linearization with this restriction enzyme leaves a tail on the cRNA of a few hundred base pairs, which may enhance the stability of the cRNA within the oocyte. The cRNA was synthesized from this template using the T7 Megascript in vitro transcription kit (Ambion, Austin, TX). The cRNA was run on a 1% agarose gel containing formaldehyde to determine its quality. The optical density of the cRNA was also determined to assess the quantity of the cRNA.
The Xenopus laevis frogs were anesthetized using 0.1% Tricaine before ovariectomy. After surgery, the oocytes were placed in a calcium-free oocyte Ringer's solution (OR2; 83.5 mM NaCl, 2.5 mM KCl, 1 mM MgCl2, 2 mM sodium pyruvate, 1 mM Na2HPO4, and 5 mM HEPES, pH 7.5). Stage V and VI oocytes were isolated and maintained in OR3 (82.5 mM NaCl, 2.5 mM KCl, 1 mM MgCl2,1 mM CaCl2, 2 mM sodium pyruvate, 1 mM Na2HPO4, 5 mM HEPES, 50 U/ml penicillin, and 50 mg/ml streptomycin, pH 7.5) plus 2% horse serum at 18°C.
The micropipettes for cRNA injection were pulled on a Sutter P87 puller (Sutter Instrument Co., Novato, CA). The tips were cut using microscissors under a dissecting microscope. The cRNA was drawn into the micropipette using negative pressure and then injected into the oocytes with positive pressure from a Picospritzer II device (General Valve Co., Fairfield, NJ).
Two to three days after injection, the oocytes were placed in a recording chamber with a volume of approximately 50 μl. This chamber has an inlet at the top and outlet at the bottom allowing continuous gravity-fed perfusion of the oocytes with either control or test solution. Twenty glass reservoirs are connected to four six-way valves. Each of these valves is also connected to a 2-liter container with OR4 (92.5 mM NaCl, 2.5 mM KCl, 1 mM MgCl2, 1 mM CaCl2, and 5 mM HEPES, pH 7.5). These six-way valves are connected to one four-way valve. In this setup, 20 different test solutions could be applied to the chamber at a given time.
The NSs were purchased from Steraloids (Newport, RI), and stock solutions of 10 mM were made in dimethyl sulfoxide. On the day of recording, test solutions were made by adding these stock solutions to rapidly stirring recording OR2. The hydrophobic nature of these compounds precluded the creation of a concentration greater than 20 μM. The highest concentration of the vehicle solution (0.2%) did not alter the GABA-induced currents from either the wild-type or the mutated ρ1R.
Recording electrodes were pulled on a Narishige pp-83 vertical puller (Narishige Scientific Instrument Lab, Tokyo, Japan). The electrodes were filled with 3 M KCl. Oocytes were clamped at -70 mV using a Turbo TEC-05 npi (Adams and List, Westbury, NY). The output was recorded on a Gould TA 240 chart recorder and digitized using Pulse 8.3 (HEKA Elecktronik, Lambrecht, Germany).
For all mutated ρ1Rs, the concentration-response relationships with GABA were constructed to determine the effective concentration producing 50% (EC50) and 9% of the maximal current (EC9). The calculated EC9 obtained from the concentration-response relationship data were tested by comparing the GABA-induced current at EC9 with the maximal current evoked by a GABA concentration 50 times the EC50 value. If needed, the EC9 was adjusted to a concentration that empirically produced approximately 9% of the maximal response. The standard deviation for EC9 measurements is within 4%, which could result from the level of expression or batch variation within the oocytes. Overall, this apparent variation in EC9 has a greater effect on the level of potentiation than inhibition. For example, a decrease from EC14 to EC4 GABA resulted in an increase in the level of potentiation by 5α-THDOC (10 μM) from 95 ± 7% to 210 ± 16% for wild-type ρ1R (Morris et al., 1999). On the other hand, the level of inhibition is affected less by deviation in EC calculation. For wild-type ρ1R, the level of inhibitions by 5β-DHP (10 μM) were -30 ± 4% and -26 ± 2% in the presence of EC4 and EC14 GABA, respectively (Morris et al., 1999).
For all the NS experiments, the control GABA solution was applied a minimum of two times before the NS application, to assure that the control GABA currents were consistent in amplitude. The time of application for GABA and GABA plus NSs was limited to 3 min to standardize these experiments.
Percentage potentiation and inhibition was calculated using the equation PP or PI = 100 × (INS - IGABA)/IGABA, where INS is the current amplitude in the presence of NS and GABA, and IGABA is the control current amplitude in the presence of EC9 GABA. In this setting, a 100% potentiation would represent a current twice the amplitude of the control current.
Two or more experiments were required to determine the quantitative measurements for the NSs for each mutant, using three or more oocytes per mutant. Determinations of statistical significance were carried out using either the student t test or ANOVA, as appropriate, requiring a p value of less than 0.05. All data are presented as mean ± S.E.M.
Results
The Mutation of ρ1 Ile307 to Ser Reversed the Direction of 5β-DHP modulation. The residue equivalent to the ρ1 Ile307 within the α subunit of GABAAR is Ser. Previous studies have shown that the mutation of Ile307 to Ser in the ρ1R can impart pentobarbital and benzodiazepine sensitivity to ρ1Rs (Amin, 1999; Belelli et al., 1999; Walters et al., 2000). We investigated the effect of the Ile307-to-Ser substitution (ρI307S) on modulation by 5β-DHP. The cRNA corresponding to ρI307S was injected into oocytes; 3 days after injection, the GABA concentration response relationship for the ρI307S receptor-channel (ρI307SR) was established. From these experiments, the effective GABA concentration producing 9% of the maximal current (EC9) was determined (see Materials and Methods). The EC9 was chosen because 1) the activation for ρ1R is slow at such GABA concentrations, facilitating the extraction of the details of the activation kinetics (see below), and 2) the action of the NSs are particularly exaggerated at such submaximal GABA concentrations (see Materials and Methods). The 5β-DHP modulation of ρI307SR in the presence of EC9 GABA was determined next. Figure 1 shows representative current traces of GABA, GABA plus 1, 5, 10, or 20 μM 5β-DHP, and the 5β-DHP concentration-response relationship for wild-type (gray line) and ρI307SR (green line). The control GABA current traces for wild-type and mutant receptor-channels were scaled to the same amplitude to discern the difference between the 5β-DHP actions on the wild-type versus mutant. Single TM2 mutation of Ile307 to Ser reversed the direction of 5β-DHP modulation from inhibition to potentiation. One, 5, 10, and 20 μM 5β-DHP inhibited the wild-type GABA (EC9) currents by -5 ± 2, -27 ± 5, -39 ± 4, and -46 ± 4%, respectively (see also Table 2). In contrast, the GABA-evoked currents for ρI307SR were increased by 9 ± 6, 34 ± 7, 75 ± 28, and 163 ± 34% at the same concentrations of 5β-DHP, respectively. The EC50 value for the 5β-DHP positive modulation of ρI307SR could not be predicted because even at the highest feasible concentration (20 μM; see Materials and Methods), the maximal current could neither be reached nor extrapolated. However, both inhibition and potentiation by 5β-DHP on ρ1 and ρI307SR was initiated at 5 μM.
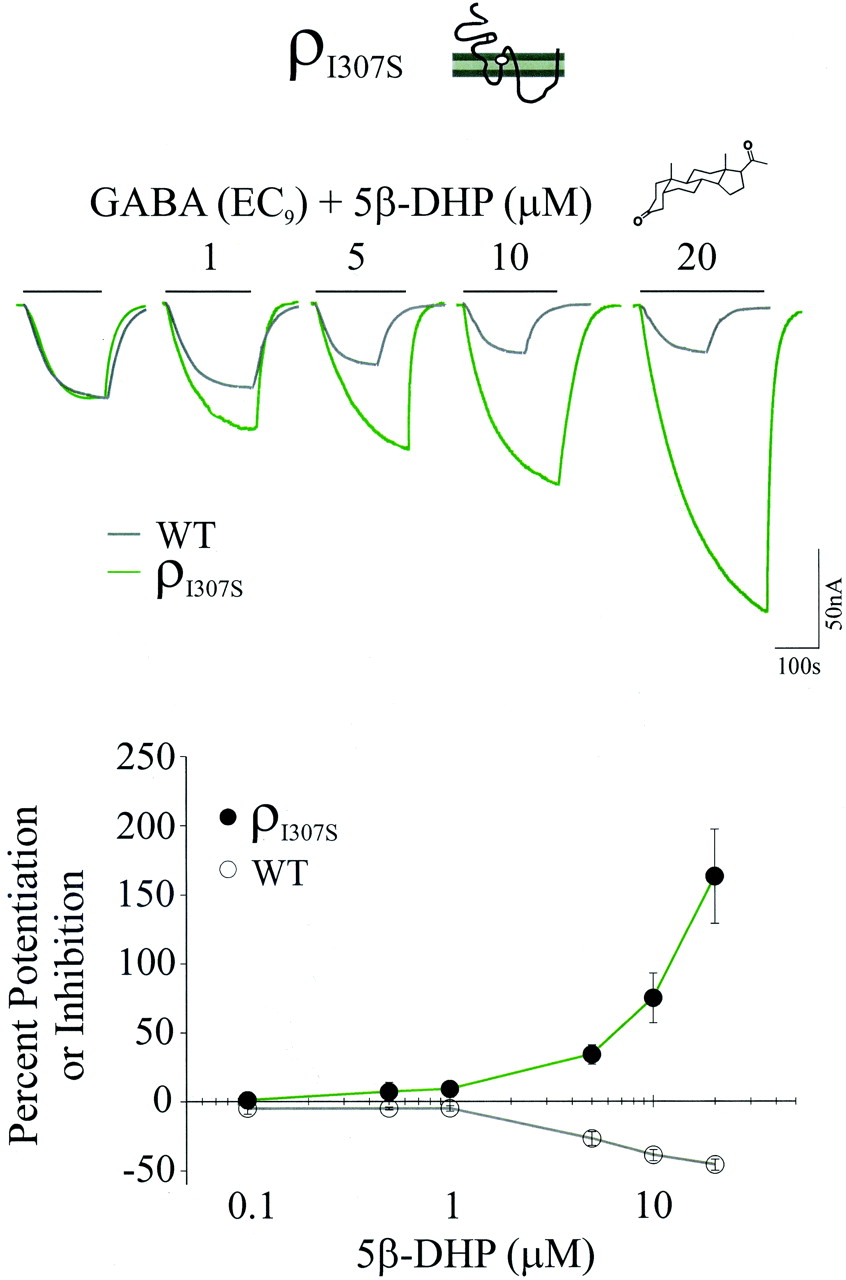
The ρ1 Ile307 mutation to Ser reverses the direction of 5β-DHP's modulation from inhibition to potentiation. Top, representative current traces of GABA (EC9) and GABA plus 1, 5, 10, or 20 μM 5β-DHP for wild-type (gray line) and ρI307SR (green line). The control GABA current traces for wild-type and ρI307SR are scaled to the same amplitude to discern the relative magnitude of 5β-DHP action on wild-type versus mutant. Scale bar representing the current magnitude only applies to mutant current traces. Thick lines above each current trace represent the duration of GABA or GABA plus NS application for ρI307S traces only. Bottom, 5β-DHP concentration-response relationship. Ile307-to-Ser substitution reversed the action of 5β-DHP from an antagonist to a positive modulator. Note that the opposing effect of 5β-DHP on ρ1R and ρI307SR was produced within the same concentration range.
The 5β-DHP-dependent modulation of Ile307 mutants in the presence their respective EC9 GABA
All data are presented as mean ± S.E.M.
The Hydrophobicity of Residue 307 Determines the Polarity of Modulation by 5β-DHP. To investigate the role of residue 307 in 5β-DHP-dependent modulation of ρ1Rs, the ρ1 Ile307 was mutated to Val, Cys, Met, Phe, Ala, Thr, Gly, Pro, Ser, Trp, Asn, His, Gln, Glu, Lys, and Arg residues. The side chains of the these residues represent a complete spectrum in size, charge, and hydrophobicity. These mutations were created to determine 1) the amino acid side-chain properties at position 307 that are important for conserving the inhibitory action of 5β-DHP, 2) the key amino acid side-chain requirements in reversing the direction of 5β-DHP's activity from an inhibitor to a positive modulator, and 3) whether any amino acid substitution will shift the 5β-DHP concentration-response range (1 to 20 μM) significantly (i.e., to produce a change within the concentration response curve such that maximal potentiation is reached or no response is seen across the concentrations tested)?
For all mutants, GABA concentration-response relationships were constructed to determine first the viability of these mutated receptor-channels; second, the shift in GABA sensitivity; and finally, the GABA EC9 value for each mutant to test 5β-DHP action at equivalent receptor-channel activity. From the 16 amino acid substitutions, Pro did not yield GABA-gated activity (up to 10 mM), Lys and Phe produced a low maximal, and Phe resulted in receptor-channels that had constitutive activity (Table 1). With the above exceptions, all substitutions at position 307 produced receptor-channels that exhibited similar or higher sensitivity to GABA compared with wild-type (Table 1). The size or the charge of the amino acid side chain at position 307 had little effect on potency. For example, Gly and Trp substitutions (opposite in terms of size) resulted in receptor-channels with GABA EC50 of 0.36 ± 0.019 and 0.53 ± 0.027 μM, whereas for Arg and Glu substitution (opposite in terms of charge), these values were 1.21 ± 0.04 and 2.71 ± 0.45 μM, respectively. Overall, the EC50 for the 307 mutants varied from 0.09 (for ρ1307H, EC50 = 0.09 ± 0.003) to 2.71 μM (ρ1307E, EC50 = 2.71 μM ± 0.45). The mutational tolerance in terms of the GABA potency indicates that position 307 may not be a key component of the GABA-binding domain. However, the maximal GABA-induced current for these mutants ranged from approximately 10% to 100% of the wild type, suggesting that residue 307 may play a role in the assembly, transduction, and/or gating processes of the GABA-dependent activation within the ρ1R.
The EC50 and the Hill coefficient values of mutated ρ1R
The EC50 and Hill coefficient were determined by fitting the concentration-response relationship to the following logistic equation using Sigma Plot 4.0 : I = Imax/[1 + (EC50/[C])n H], where I is the peak current at a given concentration of GABA [C], Imax is the maximal current, EC50 is the concentration of GABA producing 50% of maximal inhibition, and nH is the Hill number. All data are presented as mean ± S.E.M.
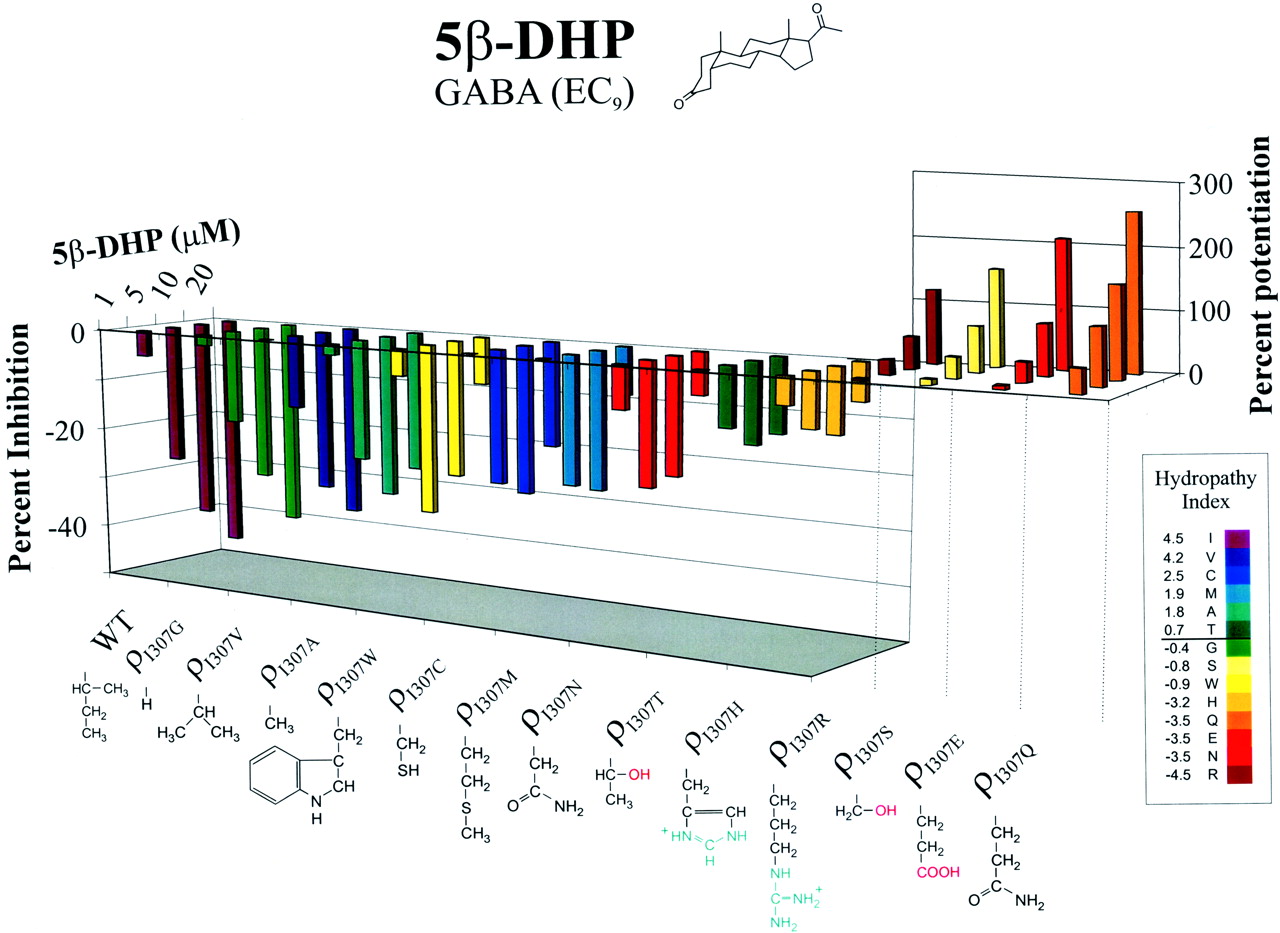
The modulatory effect of 5β-DHP was determined for all 13 Ile307 mutants responsive to GABA (Val, Cys, Met, Ala, Thr, Gly, Ser, Trp, Asn, His, Gln, Glu, and Arg substitution). Figure 2 shows bar graphs representing the 5β-DHP action at 1, 5, 10, and 20 μM for the wild-type and Ile307 mutants (see also Table 2) in the presence of their respective EC9 GABA concentrations (the amplitudes of GABA-elicited currents at EC9 ranged between approximately 20 and 200 nA). The rainbow-like color spectrum in Fig. 2 corresponds to the hydrophobicity index (HI) of the substituted amino acid based on the calculation by Kyte and Doolittle (1982). The color spectrum ranges from violet, symbolizing the most hydrophobic (Ile, HI = 4.5), to red, representing the most hydrophilic residue (His, HI = -4.5). The bar graph represents the degree of inhibition/potentiation by 5β-DHP for the mutant constructs and is arranged by polarity and the relative level of 5β-DHP activity, from the mutant that causes the highest inhibition on the far left to those producing the most potentiation on the far right. Examination of the color spectrum created by these data demonstrated a correlation between the hydrophilicity of the side chain and the direction (from inhibitory to potentiating) of 5β-DHP modulation. The highest level of 5β-DHP inhibitions occurred with the most hydrophobic amino acids, and a decline in inhibitory activity became evident as the hydrophilicity of the substituted residue increased (see also Table 2). For example, 5β-DHP (20 μM) inhibited the ρ1 (Ile, HI = 4.5) and ρI307V (Val, HI = 4.2) GABA currents by -46 ± 4 and -40 ± 6%, respectively. In comparison, 20 μM 5β-DHP displayed little inhibition for ρI307M (-9 ± 14%; HI = 1.9) or ρI307T (-14 ± 5%; HI = 0.7). Finally, with hydrophilic substitutions such as Ser (HI = -0.4), Arg (HI = -4.5), Glu (HI = -3.5), and Gln (HI = -3.5), 5β-DHP was transformed into a potentiator. In the presence of 20 μM 5β-DHP, the GABA current for ρI307S, ρI307R, ρI307E, and ρI307QRs increased by 163 ± 34, 127 ± 8, 213 ± 23, and 254 ± 28%, respectively. The change in the action of 5β-DHP was independent of the size (see Gly and Trp substitutions) or the charge (see Arg versus Glu) characteristics of the substituted residue. However, the change in color spectrum versus activity indicates that the hydrophilicity of the amino acid at position 307 determines whether 5β-DHP acts as a negative or positive modulator.

The relationship between polarity of 5β-DHP's modulation and hydrophilicity of the amino acid side chain at position 307. The modulatory effect of 5β-DHP at 1, 5, 10, and 20 μM for all thirteen Ile307 mutants and wild-type is shown. The rainbow-like color spectrum is a depiction of the hydrophobicity index of the substituted amino acid based on the calculation by Kyte and Doolittle (1982). The color code (right) depicts the hydropathy index and ranges from violet, symbolizing the most hydrophobic, to red, representing the most hydrophilic residue. The bar graph is the degree of inhibition/potentiation by 5β-DHP for the mutant constructs and are arranged (from left to right) by the relative level and the polarity of 5β-DHP activity, with mutants causing the most inhibition positioned on the far left to those producing the highest potentiation on the far right (see Table 2 for S.E.M.). Hydrophilicity of the amino acid at position 307 seemed to be the essential determinant of the polarity of 5β-DHP's modulation. Structures of the amino acid side chains are also depicted to facilitate examination of the data with respect to the size or charge characteristics of the mutated residue.
Independent of potentiation or inhibition, modulation of Ile307 mutants by 5β-DHP first became evident at 5 μM. Therefore, the marked impact on the polarity of the 5β-DHP's modulation paralleled little or no change in the lowest concentration required to produce a response. Moreover, none of Ile307 mutations altered the concentration response curve such that potentiation was maximal for a number of concentration (shift of concentration response to the left) or no response was seen across the concentrations tested (shift of concentration response to the right).
Biphasic Modulation of the ρI307SR by Pregnanolone. The sole structural difference between 5β-DHP and its metabolite, pregnanolone, is a hydroxyl group versus a double-bound oxygen on the third carbon. Although both 5β-DHP and pregnanolone inhibited wild-type ρ1Rs, there were differences between their actions on the wild-type receptor-channel. First, pregnanolone showed a higher potency than 5β-DHP (pregnanolone IC50 = 0.6 ± 0.09 μM; 5β-DHP IC50 = 3.43 ± 0.30 μM). Second, the maximum inhibition for pregnanolone occurred at 10 μM (-46 ± 5), whereas at 20 μM, the inhibitory action was reduced (-34 ± 5). By comparison, 5β-DHP produced a progressively greater inhibition with maximal inhibition at 20 μM (see Figs. 1 and 3).

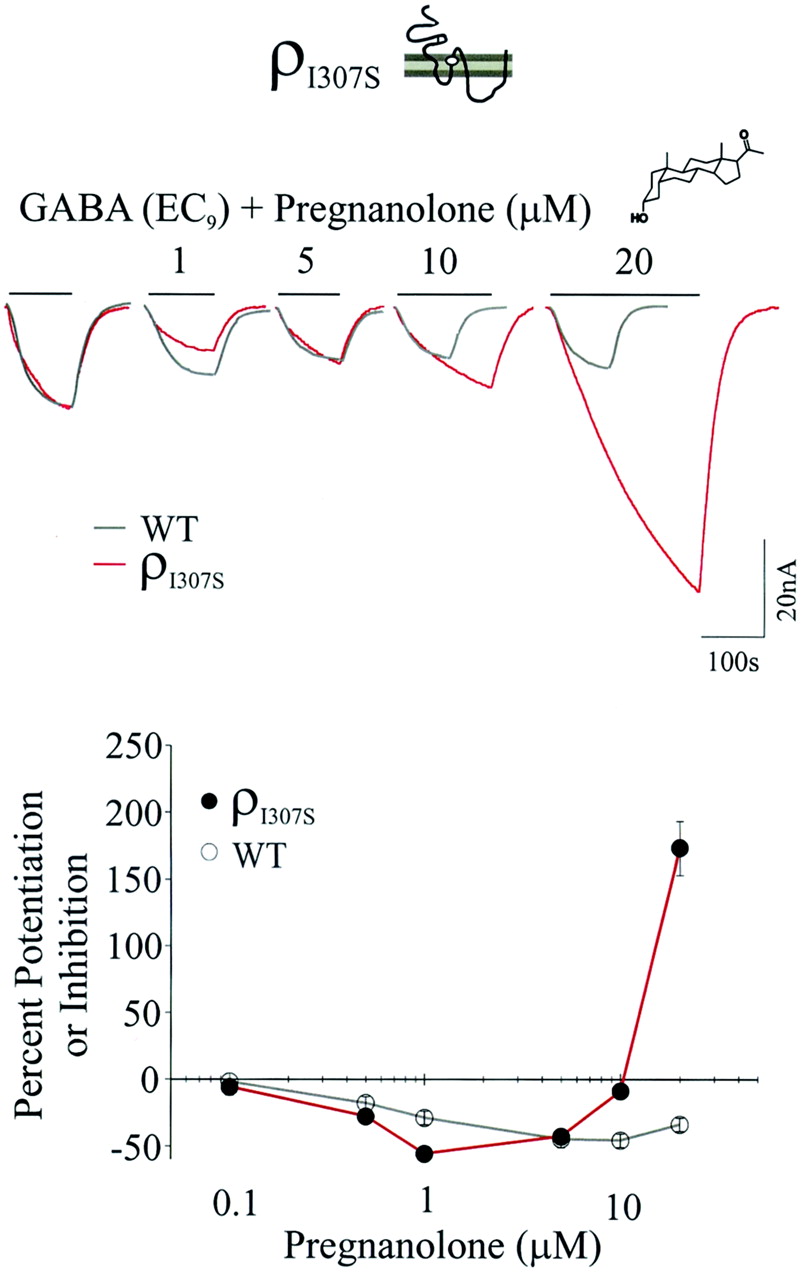
Pregnanolone inhibits and potentiates the GABA currents of ρI307SR dependent on concentration. Top, current trace; bottom, concentration-response relationship for GABA (EC9) and 0.1, 0.5, 1, 5, 10, or 20 μM pregnanolone for wild-type (gray lines) and ρI307SR (red lines). The control GABA current traces (for both wild-type and mutant) are shown at the same amplitude to allow a direct comparison between pregnanolone-dependent modulation of the wild-type and ρI307SR. The scale bar representing the current magnitude applies only to mutant current traces. Thick lines above each current trace represent the duration of GABA or GABA plus NS application for ρI307S traces only. Pregnanolone inhibited the ρI307SR's GABA currents between 0.1 and 5 μM concentrations but had little to no effect at 10 μM. In contrast, 20 μM pregnanolone significantly potentiated the ρI307SR GABA current.
To examine whether ρ1Ile307 mutation to serine would also produce a reversal in the modulation of another 5β compound, we determined the pregnanolone concentration-response relationship for ρI307SR (at 0.1, 0.5, 1, 5, 10, and 20 μM) in the presence of the EC9 GABA. Figure 3 shows the pregnanolone concentration-response relationship (and the current traces thereof) for wild-type (gray lines) and ρI307SR (red lines). The control GABA current traces (EC9) are shown at the same amplitude to facilitate comparison between pregnanolone-dependent modulation of the wild type and ρI307SR. Pregnanolone inhibited ρI307SR GABA currents between 0.1 and 5 μM (see Table 3); the maximal apparent inhibition occurred at 1 μM (-56 ± 4%). The pregnanolone inhibitory trend showed a reversal at 5 μM (-43 ± 2%) and nearly vanished at 10 μM (-9 ± 6%). In contrast, at 20 μM, pregnanolone became a positive modulator of ρI307S, increasing the GABA currents by 173 ± 20%. Thus, pregnanolone exhibited biphasic activity, producing inhibition at lower concentrations and potentiation at the highest concentration. Although the Ile307-to-Ser mutation produced a marked transformation in pregnanolone mode and degree of modulation, the apparent pregnanolone sensitivity for ρI307SR was not altered. For both mutant and wild-type receptor-channels, pregnanolone initiated a response at approximately 0.5 μM.
Pregnanolone-dependent modulation of Ile307 mutants in the presence their respective EC9 GABA
All data are presented as mean ± S.E.M.
Mono- and Biphasic Modulation of Ile307 Mutants by Pregnanolone. Pregnanolone inhibited or potentiated the ρI307SR GABA currents, dependent on the concentration. To determine the side-chain properties of residue 307 responsible for this dual action, pregnanolone concentration-response relationships were established for each of the above 13 ρ1R mutants. Figure 4 depicts a bar graph representing pregnanolone modulation (at 0.1, 0.5, 1, 5, 10, and 20 μM concentrations) of 13 mutants and the wild type in the presence of their respective EC9 GABA (see also Table 3). The pregnanolone data are presented using the same color-coded hydropathy index as with 5β-DHP experiments. The color spectrum progresses from violet, for the most hydrophobic, to red, representing the most hydrophilic amino acids. Given the bimodal nature of pregnanolone modulation, the data are arranged from left to right based on the number of pregnanolone concentrations producing inhibition for a given mutant. For example, the data for ρI307G is placed to the left of ρI307E, because four concentrations (0.1, 0.5, 1, and 5 μM) of pregnanolone inhibited ρI307G, whereas only three (0.1, 0.5, and 1 μM) inhibited the ρI307ER GABA current. For those mutations that were inhibited by the same number of concentrations, the percentage potentiation at 20 μM pregnanolone was used to assign ranking, from least (left) to greatest potentiation (right). For example, the data for ρI307W is placed to the right of ρI307N because, at 20 μM pregnanolone, the potentiation was greater for ρI307WR than for ρI307NR. Similar to 5β-DHP, the color (hydrophilicity) change versus pregnanolone modulation profile of the mutants showed a correlation between the hydrophilicity of the side chain and the direction of modulation for pregnanolone. Pregnanolone was predominately inhibitory for Ile (wild type, HI = 4.5), Val (HI = 4.2), and Cys (HI = 2.5), the three most hydrophobic residues. With a decrease in hydrophobicity of the substituted residue, pregnanolone showed potentiation starting with the highest concentration (20 μM). For example, for Thr (HI = 0.7) and Ser (HI = -0.4) substitutions, 10 μM pregnanolone decreased the GABA currents by -33 ± 6 and -9 ± 6%, whereas 20 μM increased the GABA currents by 70 ± 6 and 173 ± 20%, respectively. The inhibitory action of pregnanolone was further reduced with Glu, Asn, Trp, and Arg residues. Finally, pregnanolone completely transformed into a potentiator across the concentration range for Gln substitution (HI = -3.5). At 0.1, 0.5, 1, 5, 10, and 20 μM concentration, pregnanolone potentiated the ρI307QR GABA currents by 3 ± 1, 13 ± 5, 23 ± 8, 125 ± 22, 249 ± 31, and 531 ± 66%, respectively. It is interesting to note that pregnanolone is a positive modulator of GABAA receptors, where the equivalent amino acids to the Ile307 within the α, β, and γ subunits are Ser and Asn; these residues are significantly more hydrophilic than Ile. The pregnanolone sensitivity, however, was not altered significantly by any of Ile 307 substitution. Thus, paradoxical to their impact on the mode of modulation, none of the mutations greatly changed the lowest pregnanolone concentration required to initiate a response.

Pregnanolone inhibits, potentiates, or elicits biphasic activity for Ile307 mutants. The bar graph represents the degree of pregnanolone modulation at 0.1, 0.5, 1, 5, 10, and 20 μM concentrations for 13 mutants and the wild-type in the presence of their respective EC9 GABA (see Table 3 for S.E.M.). The hydropathy index of each amino acid is distinguished by the same color code used in the 5β-DHP experiments. The color spectrum progresses from violet, for the most hydrophobic, to red, representing the most hydrophilic amino acid. The data are arranged from left to right based on the concentrations at which pregnanolone modulation switched from inhibition to potentiation and then the degree of potentiation at 20 μM. The color change with respect to pregnanolone activity shows a relationship between the hydrophilicity of the side chain and the reversal of the direction of pregnanolone modulation.
We mutated Ile306, Pro294, and Arg287 within the TM2 of the ρ1R to assess the specificity of position 307 within the TM2 in conferring the biphasic modulation to pregnanolone. The mutation of Ile at position 306 to a serine did not yield a functional receptor-channel. However, the expression of ρP294A and ρR287A cRNA produced receptor-channels with GABA EC50 values of 0.39 ± 0.017 and 2.54 ± 0.19, respectively (Table 1). The pregnanolone concentration-response relationships for both ρP294A and ρR287A were determined at EC9 GABA. Similar to the wild type, pregnanolone inhibited the GABA currents for both ρP294AR and ρR287AR across the entire concentration range (Table 3). Thus, among these residues, mutation of position 307 seems to be specific in imparting biphasic modulation to pregnanolone.
In summary, depending on the Ile307 mutation, pregnanolone remained an inhibitor, transformed into a potentiator, or converted to inhibitor and potentiator of GABA currents based on concentration. The direction of pregnanolone modulation seemed dependent on the hydrophilicity of residue 307. Finally, a lack of mutational effect of other TM2 residues on pregnanolone modulation attested to the notion that the residue 307 may be uniquely positioned in controlling the mode of modulation.
Mutation of Ile307 Alters the Degree but Not the Mode of Modulation by 5α-THDOC. Within 5α-THDOC, the 5α positioning compared with the 5β positioning of the hydrogen atom reduces the bend within the cholesterol backbone and thereby increases the overall length. Previous studies have shown that 5α-NSs including 5α-THDOC are positive modulators of wild-type ρ1Rs (Morris et al., 1999). We investigated the effect of Ile307 mutation to Ser on the 5α-THDOC modulation. Figure 5 shows current traces and concentration-response relationships for 5α-THDOC-dependent modulation at 1, 5, 10, and 20 μM (EC9 GABA) for the wild-type (gray lines) and ρI307SR (blue lines). The 5α-THDOC produced a significantly greater potentiation for ρI307SR than wild-type ρ1Rs. At 20 μM concentrations, 5α-THDOC potentiated the ρI307S GABA current (820 ± 79%) greater than four times the wild-type (198 ± 9%). Next, we determined 5α-THDOC concentration-response relationships for ρI307A, ρI307Q, ρI307C, ρI307W, and ρI307G mutants to investigate whether any Ile307 mutation can change the positive direction of 5α-THDOC action. The substituted residues for these mutants represent a large variation in terms of amino acid side chain volume and hydrophobicity. Figure 6 shows the bar graph data for the 5α-THDOC-dependent modulation of these mutants using the same color spectrum as in 5β-DHP and pregnanolone experiments, with violet being the most hydrophobic and red the most hydrophilic (see also Table 4). The 5α-THDOC only potentiated the GABA currents for ρI307G, ρI307C, ρI307A, ρI307W, and ρI307QR across the concentration range. Moreover, the levels of potentiation for all mutants were significantly greater than the wild-type receptor-channels. For example, 5α-THDOC (20 μM) increased the respective EC9 GABA current of ρI307W, ρI307Q, and ρI307CR by 740 ± 80, 1293 ± 43, and 1058 ± 77%, respectively (wild-type = 198 ± 9%). It is interesting that all substituted residues (Gly, Ser, Ala, Cys, Gln, Trp) producing greater potentiation than wild type were more hydrophilic than Ile (native residue).

The 5α-THDOC is a positive modulator of both wild-type and ρI307SR. The representative current traces (top) and concentration response relationship (bottom) for the 5α-THDOC-dependent modulation of EC9 GABA at 1, 5, 10, and 20 μM for the wild-type (gray lines) and ρI307SR (blue lines) are shown. The control GABA current traces (for both wild-type and mutant) are shown at the same amplitude to allow a direct comparison between pregnanolone-dependent modulation of the wild-type and ρI307SR. Scale bar representing the current magnitude only applies to mutant current trace. Thick lines above each current trace represent the duration of GABA or GABA plus NS application for ρI307S traces only. The level of potentiation by 5α-THDOC significantly increased for ρI307SR compared with wild-type ρ1R.

The 5α-THDOC is always a potentiator for Ile307 mutants. The 5α-THDOC increased the GABA currents for ρI307G, ρI307C, ρI307A, ρI307W, and ρI307QR across the concentration range. The levels of potentiation for all mutants were significantly greater than the wild-type receptor-channels. The hydropathy index of each amino acid is distinguished by the same color code used in the previous figures, from violet (most hydrophobic) to red (most hydrophilic). For all tested Ile307 mutants, 5α-THDOC only potentiated the corresponding GABA currents (EC9).
The 5α-THDOC-dependent modulation of Ile307 mutants in the presence their respective EC9 GABA
All data are presented as mean ± S.E.M.
Some of the values obtained for the 20 μM potentiation were greater than the predicted GABA maximal (ρI307Q and ρI307G). If the conductance of the channel was unaltered by the mutation, the maximal potentiation based on EC9 value should not exceed approximately 1100%. Previous studies (Morris et al., 1999) have demonstrated that variation in EC9 calculation can significantly affect potentiation level (but not inhibition; see Materials and Methods). Although the variation in EC9 calculation can be responsible for the above observation, the possibility that for some of these mutants 5α-THDOC can potentiate GABA currents beyond the GABA-induced maximal cannot be ruled out.
The EC50 value for 5α-THDOC modulation could not be predicted, because even with the highest feasible concentration (20 μM; see Materials and Methods), the maximal current could neither be reached nor extrapolated. Nevertheless, the potentiation effect for all mutants and the wild-type became apparent between 1 and 5 μM of 5α-THDOC. Thus, mutation of the Ile307 to a number of different amino acids did not alter the concentration range at which the effects of 5α-THDOC were discernible but caused a marked increase in the level of potentiation by 5α-THDOC compared with the wild-type. However, in contrast to the mutational effect on 5β-NS, none of the tested mutants changed the direction of modulation by 5α-THDOC.
Preincubation with 5α or 5β Alone Produces an Equivalent Effect on NS Potentiation Kinetics. It is postulated that NSs, dependent on being a potentiator or an inhibitor, act on the ligand-gated ion channel via distinct sites (Prince and Simmonds, 1993; Park-Chung et al., 1999; Covey et al., 2000, 2001). Alternative binding sites would probably entail differences in apparent affinity of 5α-versus 5β-NS toward their respective targets. To test the above hypothesis (distinct sites), we investigated the effect of NS incubation alone on the activation time course of GABA plus NS for the mutated ρ1R. The 5α- or 5β-NSs at the highest feasible concentration (20 μM) do not directly gate (are not agonist for) wild-type or the Ile307 mutant receptor-channels (data not shown). In these experiments, t1/2 of activation was measured in the presence of GABA (EC9) and NS (3 min of incubation), with or without a 6-min preincubation of the NS alone. The effects of 5α preincubation on 5α, those of 5β on 5β, and those of 5α on 5β modulation were determined using the above protocol. We chose ρI307SR and ρI307WR because they exhibit differences and similarities with respect to 5β-and 5α-NS action (see above). For example, 5β-DHP is a potentiator of ρI307SR but an inhibitor of ρI307WR GABA current. Pregnanolone modulation is biphasic for ρI307SR and ρI307WR but shows differences in the details of its modulation. At all tested concentrations below 20 μM (0.1, 0.5, 1, 5, 10), pregnanolone inhibited the ρI307SR but at only 20 μM potentiated the GABA current for the ρI307SR. In comparison, pregnanolone inhibited the ρI307WR GABA current only at 0.1, 0.5, and 1 μM, and at higher concentrations was a potentiator of ρI307WR GABA current. On the other hand, 5α-THDOC potentiated the GABA current for both ρI307SR and ρI307WR across the concentration range.
In the first series of experiments, t1/2 of current activation for ρI307SR was measured for coapplication of GABA (EC9) and 10 μM 5α-THDOC (3 min of incubation), with and without 6-min preincubation with 5α-THDOC. Figure 7 shows the current traces for GABA application alone (2 times), GABA plus 10 μM 5α-THDOC, and GABA plus 5α-THDOC (10 μM) immediately after preincubation with 5α-THDOC (10 μM). It is important to note that the holding current did not change during the 6-min preincubation with 5α-THDOC alone (data not shown). Nevertheless, the preincubation with 5α-THDOC decreased the activation t1/2 of GABA plus 5α-THDOC from 70.7 ± 0.9 to 44.7 ± 2.1 s (n = 4), thereby reducing the time interval by 26.0 s (see Table 5).

Preincubation with 5α-THDOC shortens the t1/2 of activation for GABA plus 5α-THDOC. A, two GABA-induced currents (3 min) from an oocyte expressing ρI307SR followed by application of GABA plus 10 μM 5α-THDOC (3 min). Note the slow rise of current induced by GABA plus 5α-THDOC. B, current traces from two applications of GABA alone (3 min) expressing ρI307SR (a different oocyte than that in A). The oocyte was then incubated with 10 μM 5α-THDOC for 6 min followed immediately with application of GABA plus 10 μM 5α-THDOC (3 min). C, the difference in the rate of activation between GABA plus 10 μM 5α-THDOC and GABA plus 10 μM 5α-THDOC after the 5α-THDOC preincubation. D, the 5α-THDOC preincubation effect on the percentage potentiation of the subsequent 5α-THDOC and GABA application.
Pre-incubation with 5α or 5β NS shortens the t½ of activation for the NS and GABA by equivalent intervals
All data are presented as mean ± S.E.M.
Using the above protocol, the effect of pregnanolone pretreatment (20 μM) on the activation t1/2 of GABA and pregnanolone coapplication for both ρI307SR and ρI307WR was examined. The t1/2 of pregnanolone plus GABA was nearly identical for both mutants. The corresponding half-lives for ρI307SR and ρI307WR were 68.0 ± 1.15 and 71.5 ± 2.3 s (n = 4), respectively. The preincubation with pregnanolone (20 μM) for 6 min decreased the t1/2 of activation by very similar intervals for both mutants. After the treatment with pregnanolone alone, the pregnanolone potentiation half-life was reduced to 44.0 ± 1.15 and 44.6 ± 2.7 s for ρI307SR and ρI307WR, respectively.
If 5α- and 5β-NSs modulated the receptor-channels through alternate binding sites, pretreatment with one NS (e.g., 5α-THDOC) would be expected to produce a differential effect on the activation kinetics of the other (e.g., pregnanolone). The impact of 5α-THDOC (10 μM) pretreatment on the t1/2 of pregnanolone (20 μM) potentiation was determined using the above protocol for both ρI307SR and ρI307WR (Table 5). Preincubation for 6 min with 5α-THDOC decreased the activation t1/2 for pregnanolone by equivalent intervals for both mutants. Moreover, these shifts in activation time were highly similar to those determined for the preincubational effect of 5α on 5α t1/2 or the effect of 5β on 5β t1/2. Pregnanolone or 5α-THDOC preincubation reduced the activation t1/2 of pregnanolone modulation of the GABA current for either ρI307SR or ρI307WR by approximately 25 s (Table 5).
For the ρI307SR, preincubation with 5α alone increased the percentage potentiation for 5α-THDOC from 348 ± 37 to 433 ± 27%. In comparison, after 5β preincubation, potentiation nearly doubled for pregnanolone-dependent modulation (from 182 ± 46% to 360 ± 29%). Moreover, the significant increase in potentiation level for pregnanolone modulation was independent of whether the preincubation was with a 5α- or a 5β-NS. Thus, preincubation with 5α-THDOC closely mimicked that of pregnanolone upon pregnanolone-dependent modulation, not only in terms of impact on the activation kinetics but also with respect to the effect on potentiation.
Discussion
Within a ρ1R model system, we have demonstrated that a single amino acid mutation at Ile307 can induce a reversal in modulation (inhibition to potentiation) by 5β- but not by 5α-NSs. For Ile307 mutants, 5β-DHP produced a range of modulation from inhibition to potentiation. In comparison, depending on the mutation, pregnanolone remained an inhibitor, transformed into a potentiator, or converted to inhibitor and potentiator of GABA currents based on concentration. The amino acid side chain property/activity plot demonstrated a correlation between 5β-NS direction of activity and side-chain hydrophobicity. On the other hand, the ρ1 307 mutants did not change the mode of modulation by 5α-THDOC but caused a significant increase in the potentiation level. Despite the severe impact on the degree or/and mode of modulation, none of the 307 mutations significantly affected the NS concentration producing the initial response. Moreover, preincubation of Ile307 mutants with either 5α or 5β alone produced an equivalent effect on the activation time course of NS-dependent modulation.
Inconsistency with a Binding Site or Cavity Hypothesis for the NS Action at the Position 307. The prevailing view for molecular actions of NSs and anesthetics is that they interact with defined binding sites or cavities to exert their effects. These current models propose that anesthetics may fill a pocket with specific dimensions and thereby alter the receptor-channel gating and/or agonist binding activity (Wick et al., 1998; Ye et al., 1998; Koltchine et al., 1999; Ueno et al., 2000; Covey et al., 2001; Jenkins et al., 2001).
Our data on NS modulation of ρ1R with respect to position 307 is inconsistent with the binding site(s) or fixed volume cavity hypotheses for the following reasons:
-
The simple mass action principle predicts that subtle impairment of a ligand binding site can be overcome with higher concentrations of ligand; conversely, enhancement of the binding site would require a lower concentration of the ligand to produce an equivalent effect. For example, conservative mutation of the GABA binding domain within the receptor site causes a marked impact on potency. That is to say, mutation of the Tyr157 to Phe (conserving the aromatic ring) or to Ser (conserving the hydroxyl group) within the β2 subunit only of the GABAA receptor (α1β2γ2) impairs the GABA-dependent action by approximately 2 and 3 orders of magnitude, respectively (EC50 of 45.8 ± 3.6 μM for wild type compared with 2570 ± 646 for α1βY157Fγ2 and 43,580 ± 6800 μM for α1βY157Sγ2; Amin and Weiss, 1993). Compared with the mutation effect on the GABA binding site within receptor domain of α1β2γ2, mutations of Ile307 within the ρ1R (and with a 5-fold degeneracy because of the homo-oligomeric nature of ρ1R) to amino acids with opposite charges (Glu versus Arg) or dramatically different sizes (Gly versus Trp) respond to NSs within the same and relatively small concentration range. For instance, the effect of 5β-DHP with respect to inhibition or potentiation is discernible between 1 to 20 μM for all mutants, and the concentration initiating the effect is around 5 μM. If there were a moderate impairment or enhancement within the 5β-DHP binding domain caused by Ile 307 mutation, 5β-DHP would either produce no effect on the GABA currents (shift of sensitivity to higher concentrations) or create nearly maximal modulation (shift of the concentration response to lower concentrations) within such a small concentration range (1-20 μM). Earlier studies (Amin 1999) have shown that the mutation of Trp328 to a number of hydrophobic residues can impart pentobarbital sensitivity to ρ1R. As with Ile307 mutants, Trp328 mutants also exhibit similar sensitivity to pentobarbital direct activation. As a group, these experiments demonstrate a dramatic contrast between the mutational effect on a ligand-binding site like GABA and a key site for NS (or pentobarbital) action.
-
In the last decade, multiple studies have pointed to the importance of residues 307 and 328 within ρ1R (as well as other residues and their equivalents in GABAA receptor subunits) for a number of anesthetics (Walters et al., 2000); Belelli et al., 1997, 1999; Mihic et al., 1997; Amin, 1999). All these residues that are important for anesthetic action are found within the TM domains, yet none is found within the receptor domains. Similar to this study, mutations of these important “anesthetic” residues within the TM domains showed a paradigm completely distinct from the mutational analysis of a typical agonist-dependent activation domain like GABA (see above).
-
A binding pocket(s) or a cavity within a protein structure has defined dimensions and would be constrained by the side chains of its amino acid constituents. Mutations of Ile307 to a more hydrophilic residue at position 307 can markedly affect the level of inhibition or produce potentiation. Pregnanolone can inhibit or potentiate the same mutated receptor-channel depending on the concentration. The biphasic activity by pregnanolone can occur with Ile307 mutations to residues representing opposites within the volume spectrum (Gly or Trp), and opposites within the charge spectrum (Arg and Glu). It seems highly unlikely that amino acids with opposite charge, or of significantly different size, produce equivalent effects within the architecture of a binding site or cavity.
-
Earlier reports (Williams and Akabas, 2002) with the intravenous anesthetic propofol and GABAARs using Cys scanning of the TM3 demonstrated that the profiles of the TM3 movement during GABA-versus GABA and propofol-dependent activation were different. Because the GABAARs transit between a number of open and closed states (Weiss and Magleby, 1989; MacDonald and Twyman, 1992; Maconochie et al., 1994), creation of variable size cavities to accommodate propofol was predicted as channels move from one state to another. Here, preincubation with 5α- or 5β-NS produced equivalent effects on the activation time course of either 5α- or 5β-NS-dependent modulation. The effect of 5α-THDOC preincubation on the pregnanolone activation time course (t1/2) was nearly identical to the effect of 5α-THDOC preincubation on 5α-THDOC t1/2 and that of pregnanolone on pregnanolone t1/2. These effects occurred in the absence of channel activity because the 5α- or 5β-NS does not directly gate the ρ1R. The presence of alternative binding sites for NSs (inhibition versus potentiation) with a fixed volume predicts differential affinity and therefore a disparate impact on the t1/2 of activation. In addition, variably sized binding sites require channel opening to accommodate the ligand. Our findings of nearly identical effects of preincubation with 5α- or 5β-NS on the subsequent channel activity seem inconsistent with alternative or variable binding site hypothesis.
Is Ile 307 Involved in Coupling the NS Binding Event to the Channel Gating? In addition to the impact on NS-dependent modulation, sensitivity of structurally diverse anesthetics such as pentobarbital and benzodiazepines can be imparted upon the ρ1R using the mutation of Ile 307 and/or Trp328. It has been postulated that position 307 may play an essential role in the transduction pathway, between the NS binding and the channel gating. At such a crucial linkage site, one may expect imposing constraints at the level of the amino acid side chain. In addition, it is difficult to reconcile that a single mutation between the binding and the gating can result in reversing the propagation event from inhibiting to facilitating channel gating (from inhibition to potentiation) or act as an `linkage' for both inhibition and potentiation (pregnanolone).
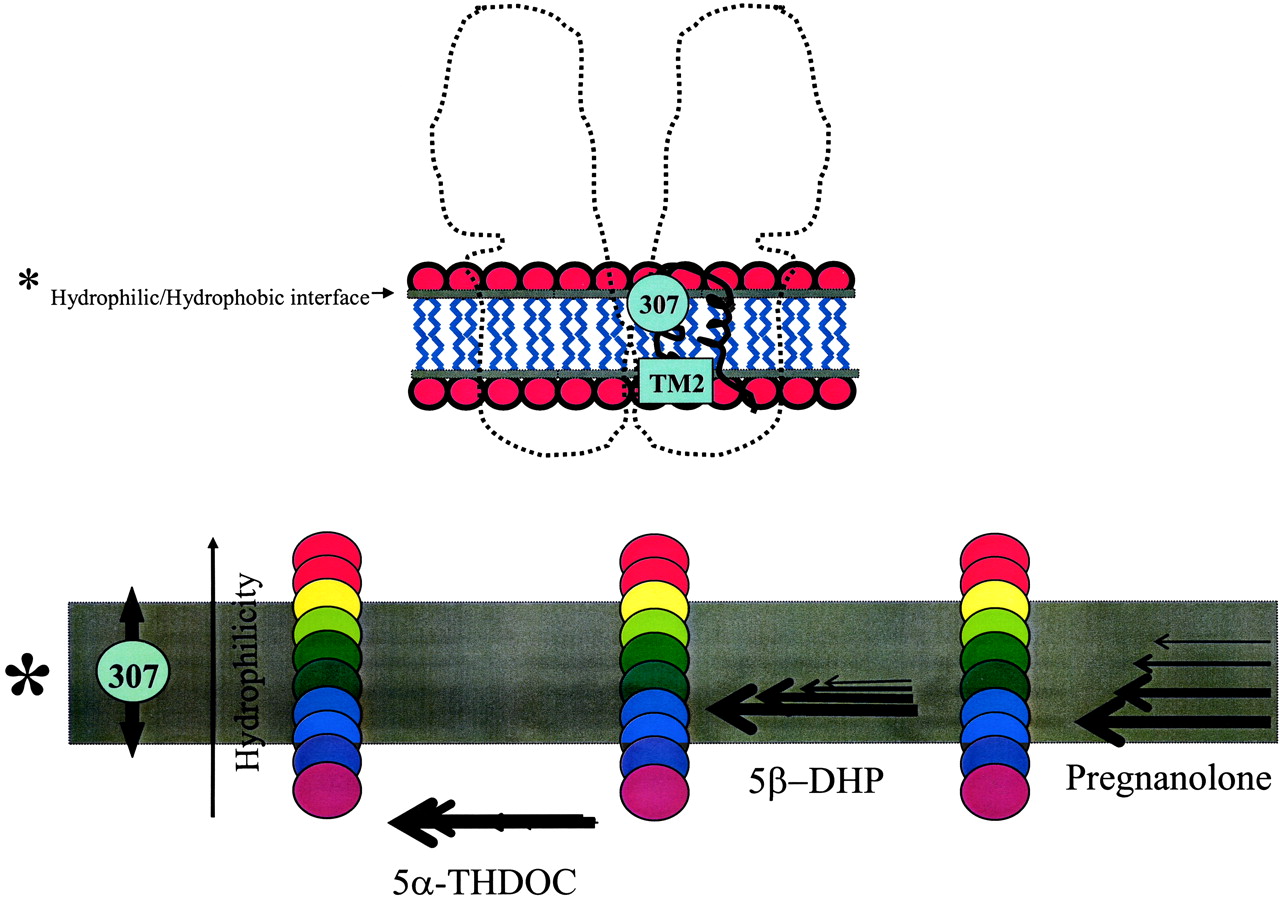
A Proposed Model for NS Action. How does a single mutation reverse polarity for modulation of 5β-DHP but not for that of 5α? What is the mechanism for pregnanolone biphasic modulation? We propose a model in which 1) the mutation of Ile307 can influence the positioning of a sensor site and 2) each NS exerts a unique three-dimensional force dependent on minute structural differences as well as concentration (Fig. 8). The TM2 domain contains important gating features for the ion channel, and Ile307 is an agile component of this module (Unwin, 1995; Horenstein et al., 2001). Amino acid 307 is located five amino acids from the extracellular surface, at the interface of the hydrophobic/hydrophilic boundary of the membrane bilayer (Fig. 8, asterisk). Within the wild-type ρ1R, Ile307 favors the hydrophobic domain (Ile has the highest hydrophobicity index). However, mutations of residue 307 to any residue more hydrophilic than Ile may support the movement of this sensor site toward the energetically more favorable hydrophilic environment. Transmembrane domains of the receptor-channel (crossing the membrane) can induce gross perturbation (disorder) within the lipid bilayer. NSs, in this scenario, can incorporate within these perturbed membrane domains closely surrounding the receptor-channel at or near the protein/membrane interface and dependent on minor structural differences and concentration induce a unique three-dimensional energy profile on the sensor site. The position and length of the arrow within the model represents the distinct depth and the pressure that a given NS may exert upon the gating component. If the sensor site is above the arrow, the forces acting upon the channel gate will translate into potentiation. On the other hand, if the sensor site is below the arrow, the action of the NS on the ion channel will be inhibitory (see Cantor, 1997 for another rendition). This model can account for the mutational effect reversing the action of 5β-DHP. Furthermore, it explains how pregnanolone can inhibit or potentiate a given mutant based on its concentration. Finally, such a model foresees the presence of a neutral zone for the sensor site where the NS window of impact may have little effect on the GABA-dependent modulation. In accordance with this view, 5β-DHP showed little effect upon certain Ile307 mutants such as ρI307T (see Results).

A model representation of 5α and 5β-NS action upon a ligand-gated ion channel. In this model, the position 307 is a sensor site and each NS exerts unique force dependent on minute structural differences and concentration. Amino acid 307 is positioned at the interface of hydrophobic/hydrophilic boundary of the membrane bilayer. Within the wild-type ρ1R, Ile307 may favor the hydrophobic domain (because Ile has the highest hydrophobicity index). The amino acid color code ranges from violet (most hydrophobic) to red (most hydrophilic). All residues within the 307 mutational series are more hydrophilic than the Ile. Thus, mutations of residue 307 to any residue may support the movement of this position toward the hydrophilic side to reduce free energy. Transmembrane domains of the receptor-channel crossing the membrane can induce gross perturbation (or disorder) within the lipid bilayer. NSs, in this scenario, can incorporate within these perturbed membrane domain closely surrounding the receptor-channel at or near the protein/membrane interface and dependent on minor structural differences and concentration induce a unique three-dimensional profile on the sensor site. The position and length of the arrow represent the distinct depth and the pressure that a given NS may exert upon the gating component. If the sensor site is above the arrow, the forces acting upon the channel gate will translate into potentiation. On the other hand, if the sensor site is below the arrow, the action of the NS on the ion channel will be inhibitory.
In a homo-oligomeric ρ1R model system, we demonstrated that mutation of a key residue within TM2 can induce a profound effect on the NS mode and degree of modulation yet produce little change in the apparent sensitivity of the receptor-channel to the NS. Preincubation with either a 5α- or 5β-NS produced an equivalent effect on the potentiation time course. Future experimentation, by means of single channel analysis as well as high-resolution structural mapping of the NS interaction site with its target, will be needed to further test this model.
Acknowledgments
We thank J. Harper and Dr. M. A. Pacheco for editing, Drs. E. S. Bennett and C. Doupnik for critical reading of the manuscript, and S. Hadley for technical assistance.
Footnotes
-
This work was supported by a grant from the National Eye Institute (EY11531) and The Established Investigator Award from the American Heart Association.
-
ABBREVIATIONS: NS, neuroactive steroid; CNS, central nervous system; GABAAR, GABAA receptor; ρ1R, ρ1 receptor-channel; TM, transmembrane domains; OR2, oocyte Ringer's 2; DHP, dihydroprogestrone; 5α-THDOC, allotetrahydrodeoxycorticosterone; pregnanolone, 5β-pregnane-3α-ol-20-one; HI, hydrophobicity index.
- Received December 18, 2003.
- Accepted April 1, 2004.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}