


Abstract
The pregnane X receptor (PXR) is a nuclear receptor significantly involved in the transcriptional regulation of drug-metabolizing enzymes and transporters. Interestingly, certain PXR ligands such as rifampin have been shown to readily induce human and rabbit but not rodent members of the cytochrome P450 3A. Because drugs of divergent chemical structures seem to be similarly affected, we hypothesized that specific amino acid residue(s) or domains in rat PXR affect receptor activation by certain human PXR ligands. To identify such a domain(s), an array of human-rat and rat-human chimeric PXR cDNAs in a tandem head-to-tail configuration were created using a random chimeragenesis method. Pharmacological characterization of these chimeras revealed a discreet segment within the ligand-binding domain of rat and human PXR to be essential for the rifampin effect. Within this region, the corresponding residues Leu308 and Phe305 of human and rat PXR, respectively, were found to be important for rifampin activation. Homology modeling derived from the recently determined crystal structure of human PXR indicates that these amino acids are located within or neighboring the flexible loop that forms part of the pore to the ligand-binding cavity. Rifampin, paclitaxel, and hyperforin sensitivity was conferred to rat PXR when Phe305 was converted to leucine, whereas attenuation of sensitivity was observed when Leu308 of human PXR was replaced with phenylalanine. Accordingly, our data provide compelling new insight into the importance of the amino acids comprising the pore to the ligand-binding cavity as a critical modulator of PXR response.
Induction of drug-metabolizing enzymes and transporters is an important physiological adaptation to xenobiotic challenge for enhancing elimination and decreasing exposure to potentially toxic substances. However, for many clinically useful drugs, induction of enzymes and drug transporters can lead to profound reductions in plasma drug levels and loss of therapeutic efficacy. It is now widely recognized that the adopted nuclear receptor, pregnane X receptor (PXR, NR1I2) (Kliewer et al., 1998), plays a central role in the induction process by acting both as a xenosensor and a transcriptional activator (Kliewer and Willson, 2002). Binding of xenobiotics to PXR initiates heterodimerization with the retinoid X receptor (NR2B1) to form a PXR/retinoid X receptor complex which then binds to particular DNA response elements leading to target-gene transactivation (Kliewer and Willson, 2002). Examples of genes regulated by PXR-mediated mechanisms include members of the cytochrome P450 family such as CYP3A (Goodwin et al., 1999; Bertilsson et al., 2001; Burk et al., 2002), CYP2B (Goodwin et al., 2001), and CYP2C (Gerbal-Chaloin et al., 2002) isoforms, which collectively account for a significant proportion of the oxidative enzymes responsible for detoxification of therapeutic agents. Moreover, drug transporters including P-glycoprotein (Geick et al., 2001), multidrug resistance-associated protein 2 (Kast et al., 2002), and organic anion transporting polypeptide 2 (Guo et al., 2002) seem to be induced by PXR-mediated transcriptional up-regulation.
A wide variety of structurally divergent compounds have been identified as ligand activators of PXR. Many PXR ligands such as dexamethasone, spironolactone, RU486, and bile acids (Staudinger et al., 2001; Xie et al., 2001) are steroidal, whereas others such as the HIV-1 protease inhibitors ritonavir (Dussault et al., 2001), amprenavir, and nelfinavir are peptidomimetic in structure. Yet others are large, complex, natural products such as hyperforin, the active component of the St. John's wort (Moore et al., 2000) and the anticancer drug paclitaxel (Synold et al., 2001). A prototypical inducer of CYP3A in humans is the macrocyclic antimycobacterial drug rifampin (Lehmann et al., 1998). Studies examining the regulation of CYP3A expression in cultured hepatocytes from different species indicated that host cellular environment determines enzyme inducibility by compounds such as rifampin (Barwick et al., 1996). Subsequently, it became evident that differences in ligand-dependent induction of CYP3A resulted from the species-dependent differences in the pharmacologic specificities of PXR. Thus, rifampin was found to be an activator of human (Bertilsson et al., 1998; Blumberg et al., 1998; Lehmann et al., 1998) and rabbit (Jones et al., 2000; Savas et al., 2000), rhesus monkey (Moore et al., 2002), dog (Moore et al., 2002), and chicken (Handschin et al., 2000; Moore et al., 2002), but not mouse (Kliewer et al., 1998) or rat (Zhang et al., 1999; Jones et al., 2000) PXR orthologs. Structurally, the DNA binding domains of PXR among these species are well-conserved (>92% amino acid identity); however, their ligand-binding domains are remarkably divergent (rodent and rabbit <85% identical with human PXR) considering the significant sequence conservation usually seen among the nuclear receptor family (Jones et al., 2000). This suggests that interactions with amino acids in the ligand-binding domain confer ligand specificity toward PXR activation.
With the recent elucidation of the crystal structures of human PXR ligand-binding domain in its apo- and drug-bound forms, the basis for receptor promiscuity as well as specificity toward drugs is now better understood (Watkins et al., 2001, 2003). The ligand-binding pocket of human PXR is a large, elliptical cavity lined by 28 amino acids, largely hydrophobic, and a small number of polar residues that can accommodate the cholesterol-lowering drug SR12813 in different orientations (Watkins et al., 2001) and active component of the St. John's wort hyperforin in a single orientation (Watkins et al., 2003). Furthermore, the shape of the ligand-binding pocket is not rigid but expands and contracts to conform to specific compounds (Watkins et al., 2003). Although rifampin is a well-recognized agonist of several PXR orthologs, it is significantly larger than either SR12813 or hyperforin (823 compared with 505 and 517 Da, respectively) and the determinants of its species-specific PXR activation remain essentially unknown.
In this report, we took advantage of the species differences in ligand-dependent activation to identify amino acids in rat PXR important for rifampin sensitivity. Using a combination of rat/human PXR chimeras and species-scanning mutagenesis, several amino acids, especially rat PXR residue 305, comprising the pore to the PXR ligand-binding cavity were identified as being critical in permitting rifampin, paclitaxel, and hyperforin receptor activation.
Materials and Methods
Materials. Rifampin, paclitaxel, pregnenolone 16α-carbonitrile (PCN), and nifedipine were obtained from Sigma Chemical (St. Louis, MO). Hyperforin was obtained from Aapin (Oxfordshire, UK). CYP3A4-XREM-Luc luciferase reporter plasmid containing the proximal CYP3A4 promoter, as well as the distal xenobiotic response module (Goodwin et al., 1999), was prepared as described previously (Zhang et al., 2001).
Expression Plasmids. RNA was extracted from human livers with TRIzol reagent (Invitrogen, Carlsbad, CA), and first-strand cDNA was obtained by reverse transcription according to the manufacturer's instruction (Powerscript; BD Biosciences Clontech, Palo Alto, CA) using random hexamers. The hPXR expression plasmid (pEF-hPXR) was prepared by long polymerase chain reaction (Expand Long Template; Roche Diagnostics, Indianapolis, IN) from liver cDNA using forward (5′-GAA CA TGG AGG TGA GAC CCA AAG AAA GC-3′) and reverse primers (5′-TCA GCT ACC TGT GAT GCC GAA CAA CTC-3′) and subsequent cloning of the amplicon into pEF6-V5-His vector (Invitrogen). An optimized Kozak sequence was introduced to improve cellular expression. To prepare the rat PXR expression plasmid (pEF-rPXR), the open reading frame was amplified from rat liver cDNA (BD Biosciences Clontech) by long polymerase chain reaction using forward and reverse primers, 5′-GAG ATG GGA CCT GAG GAG AGG TGG AAC-3′ and 5′-AGC CAC TCA GCC GTC CGT GCT GCT GAA TAA C-3′, respectively. This amplicon was inserted into the cloning vector pCR2.1 (Invitrogen) to produce pCR2.1-rPXR. To produce the pEF-rPXR, the rat PXR cDNA was released from pCR2.1-rPXR with SpeI and XbaI, and the fragment was inserted into the SpeI site of pEF6-V5-His. All plasmids were fully sequenced.
PXR Chimera Plasmids. Rat-human (RH) and human-rat (HR) PXR chimera plasmids were prepared by random chimeragenesis method (Fig. 1) (Buck and Amara, 1994). The master RH-PXR chimera plasmid (pEF-RHPXR) was constructed by releasing rPXR cDNA from pCR2.1-rPXR with EcoRV and BamHI and ligation of this fragment into the KpnI/BamHI site of pEF-hPXR, where the KpnI site was blunted with Klenow enzyme. This clone contains rat and human PXR full-length cDNAs in tandem head-to-tail orientation, respectively. The master HR-PXR chimera (pEF-HRPXR) was constructed by releasing rPXR cDNA from pCR2.1-rPXR with EcoRI, followed by treatment with Klenow enzyme. This fragment was inserted into the XbaI site of pEF-hPXR to produce a clone containing full-length human and rat PXR cDNAs in tandem head-to-tail orientation, respectively. A series of chimeric rat-human PXR expression plasmids were obtained by enzymatic digestion of pEF-RHPXR in the region between the rat and human full-length cDNAs using BamHI and SpeI. After transformation of this linear plasmid into Escherichia coli (TOP10, Invitrogen), monomer-sized clones that contained chimeric rat-human cDNAs in tandem head-to-tail orientation were isolated. Similarly, chimeric human-rat expression plasmids were obtained by transformation of EcoRV and NotI linearized pEF-HRPXR. Overall, 50 chimeric rat-human and 50 chimeric human-rat PXR plasmids were obtained for further testing. To estimate the junction between rat and human PXR, restriction digests of each chimera construct were performed using rat PXR-specific (Van91I and Csp45I) and human PXR-specific enzymes (NarI and NaeI). The exact position of the chimeric junction was determined by sequencing.

Preparation of rat-human and human-rat PXR chimera expression plasmids. cDNAs for rat and human PXR were inserted into the expression vector pEF6 (Invitrogen) in head-to-tail orientation in either rat-human or human-rat orientations. After digestion with two restriction endonucleases and transformation of the linearized constructs, colonies containing an array of monomer-sized, chimeric PXR expression vectors were isolated.
Site-Directed Mutagenesis. pEF-rPXR was used as a template to create F305L, K314Q, M321L, and C325Y mutants (derived from the rat PXR amino acid sequence) using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). Various combinations of mutations were similarly prepared by sequential mutagenesis. The L308F mutant of human PXR was prepared using pEF-hPXR as a template. Mutations were verified by sequencing.
Transient Transfection Assays. HepG2 cells were grown in AlphaMEM (Cambrex Bio Science Walkersville, Inc., Walkersville, MD) containing 10% fetal bovine serum, l-glutamine, and antibiotics. On day 1, cells were seeded onto 12-well plates at 0.6 × 106 cells/well. The next day, cells were transfected with CYP3A4-XREM-Luc (250 ng), chimeric PXR expression plasmid (250 ng), and pRL-tk (Promega, Madison, WI) using Lipofectin reagent in Optimem (both from Invitrogen). Approximately 16 h later, the cells were washed and treated with rifampin or vehicle (dimethyl sulfoxide) diluted in Optimem. After 24 h, luciferase activities were determined using the Dual-Luciferase Reporter Assay System (Promega).
Molecular Modeling. A homology model of rat PXR was built based on the structure of human ortholog (PDB code 1ILH) sharing 76% identity with rat protein. The model was built and analyzed using the program SWISS-PdbViewer (Guex and Peitsch, 1997) and SWISS-MODEL Protein Modeling Server (Peitsch, 1995, 1996). Total energy of the built model was of -9390 KJ/mol as determined by energy computations with GROMOS96 implemented in SWISS-PdbViewer. The model was further optimized by energy minimization in the central nervous system (Brunger et al., 1998) using script for conjugate-gradient minimization with no experimental energy terms. Minimization resulted in a decrease of total energy to -9752 KJ/mol. The final model was used to facilitate the positional analysis of mutations and estimate their functional impact.
Results
Identification of a Critical Region in PXR that Confers Rifampin Sensitivity. By using a random chimeragenesis method, an array of HR and RH chimeric PXR expression plasmids were constructed. Sequencing of the constructs indicated that the distribution of interspecies junction sites was equally dispersed throughout the chimeric PXR molecules (Fig. 2). Upon characterization of receptor functions with a cell-based reporter assay, transactivation activities seemed to follow a graded pattern related to the positions of the chimeric junctions for HR and RH constructs. For HR PXR chimeras, as the chimeric junctions moved from the N terminus to the C terminus, rifampin-mediated reporter gene activity increased. A lack of significant rifampin sensitivity was observed when the junction occurred at an amino acid position ≤302 (HR clone 14) (taken from human PXR residues). However, rifampin activated HR PXR chimeras, which had junctions at amino acid positions ≥317 (HR clone 15) (Fig. 2). This transition in rifampin activation indicates that species-specific amino acid(s) between 302 and 317 within the ligand-binding domain of human PXR were important for rifampin sensitivity. For HR chimeras with transitions between amino acids 317 and 401 (HR clones 15-19), similar values for reporter activities were observed. Another transition in reporter gene activity was observed between HR clones 19 and 20, whose chimeric junctions occurred at amino acids 402 and 417, respectively, indicating that species-specific amino acids within this region are important for maximal rifampin-mediated PXR activity.

Identification of amino acid domains affecting rifampin-mediated PXR activation. Twenty-one HR (left) and 21 RH (right) chimeric expression vectors were sequenced to determine junction points (numbering was taken from the human residues). After transfection of individual chimeric PXR expression plasmids into HepG2 cells and treatment with rifampin (5 μM), transactivation of a CYP3A4 luciferase reporter construct was analyzed and expressed as fold activation over vehicle control. Rifampin-mediated transactivation of wild-type rPXR and hPXR are also shown.
For RH PXR chimeras, as the junctions moved from the N terminus to the C terminus, rifampin-mediated reporter gene activation decreased. Similar to the pattern observed in HR PXR chimeras, the pattern of activation by rifampin followed a graded response that was associated with the relative positions of the junctions. Rifampin-dependent transactivation was maintained at a level similar to that of human PXR control when the interspecies junction of RH chimeras occurred within the DNA-binding domain and the 5′ region of the ligand-binding domain (Fig. 2). As the junctions moved toward the 3′ end, rifampin-dependent activation decreased in a stepwise fashion. RH chimeras with junctions at amino acid positions ≤317 (RH clone 15) (taken from corresponding human PXR residues) maintained rifampin sensitivity. By contrast, RH PXR chimeras with junctions at amino acid positions ≥336 (RH clone 16) were devoid of activation by rifampin. Therefore, from reporter gene activation of RH PXR clones, it seems that species-specific amino acids between 317 and 336 in the ligand-binding domain of PXR are critical for imparting rifampin sensitivity.
Specific Amino Acids in the Ligand-Binding Domain of PXR Confer Rifampin Sensitivity. Data from HR chimeras alone indicated that particular amino acids between positions 302 and 317 of rifampin-sensitive human PXR that were different from rifampin-insensitive rodent receptors conferred ligand specificity. After amino acid sequence alignment of available PXR orthologs (Fig. 3A) and comparing their rifampin sensitivities, we deduced from this data set that only the amino acid at position 308 (taken from human residues), which is leucine for human and phenylalanine for rat receptors, could be responsible for rifampin responsiveness. When rat PXR was mutated in this position (denoted as M1) from Phe to Leu, a significant dose-dependent, rifampin-mediated enhancement of reporter gene activity was observed (Fig. 3B) that was clearly absent in the wild-type rat PXR. The estimated EC50 for rifampin activation of rat PXR M1 was 50 ± 12 μM.

Mutations in rat and human PXR affect rifampin responsiveness. A, amino acid sequence alignment of various PXR orthologs in the region critical for rifampin activation. Numbering is taken from human residues. Positions of HR and RH chimeric junctions and residues changed in site-directed mutagenesis are presented. Degree of rifampin activation of PXR orthologs is taken from Moore et al. (2002). B, dose-dependent rifampin activation of rat PXR and its mutated variants (M1, M234, and M1234). VC, cells transfected with blank vector control. C, dose-dependent rifampin activation of human PXR and the L308F mutant. Transactivation assays were performed in HepG2 cells with a CYP3A4 luciferase reporter construct.
Focusing on the RH chimera data set alone, it seemed that another region of amino acids between positions 317 and 336 harbored additional species-specific residues involved in rifampin sensitivity. We suspected that residues at positions 317, 324, and 328 (taken from the human PXR sequence) were important for imparting rifampin sensitivity after amino acid alignment of PXR orthologs. Therefore, rat PXR was mutated at each of these positions (designated M2, M3, and M4) to the human residues, and pharmacological function was assessed to determine whether these amino acids also affected rifampin sensitivity. Single mutations of rat PXR at M2 (Lys to Gln), M3 (Met to Leu) or M4 (Cys to Tyr) did not make the receptor rifampin-sensitive, nor did all possible combinations of double mutants (data not shown). However, a triple mutant (M234) had significantly greater rifampin-mediated activation than wild-type rat PXR in reporter assays (Fig. 3B), suggesting that cooperativity exists in these three positions that impact on the rifampin effect. Rat PXR M234 had an estimated EC50 for rifampin activation of 16 ± 2 μM. We further tested the activation of rat PXR with mutations at all four positions (308, 317, 324, and 328) and found that rifampin sensitivity was maintained but was not further enhanced over the M1 or M234 mutants alone (EC50 = 17 ± 1 μM).
To gain further insight into the importance of amino acid 308 in the rifampin-mediated activation of PXR, we mutated human PXR at this position to the rat residue (Leu to Phe). There was a right shift in the EC50 of rifampin-mediated activation from 1.7 ± 0.1 to 3.6 ± 0.2 μM for the wild-type and L308F human PXR receptors, whereas the maximal activation was unchanged. Thus, the apparent affinity but not the capacity for PXR activation was altered by the mutation.
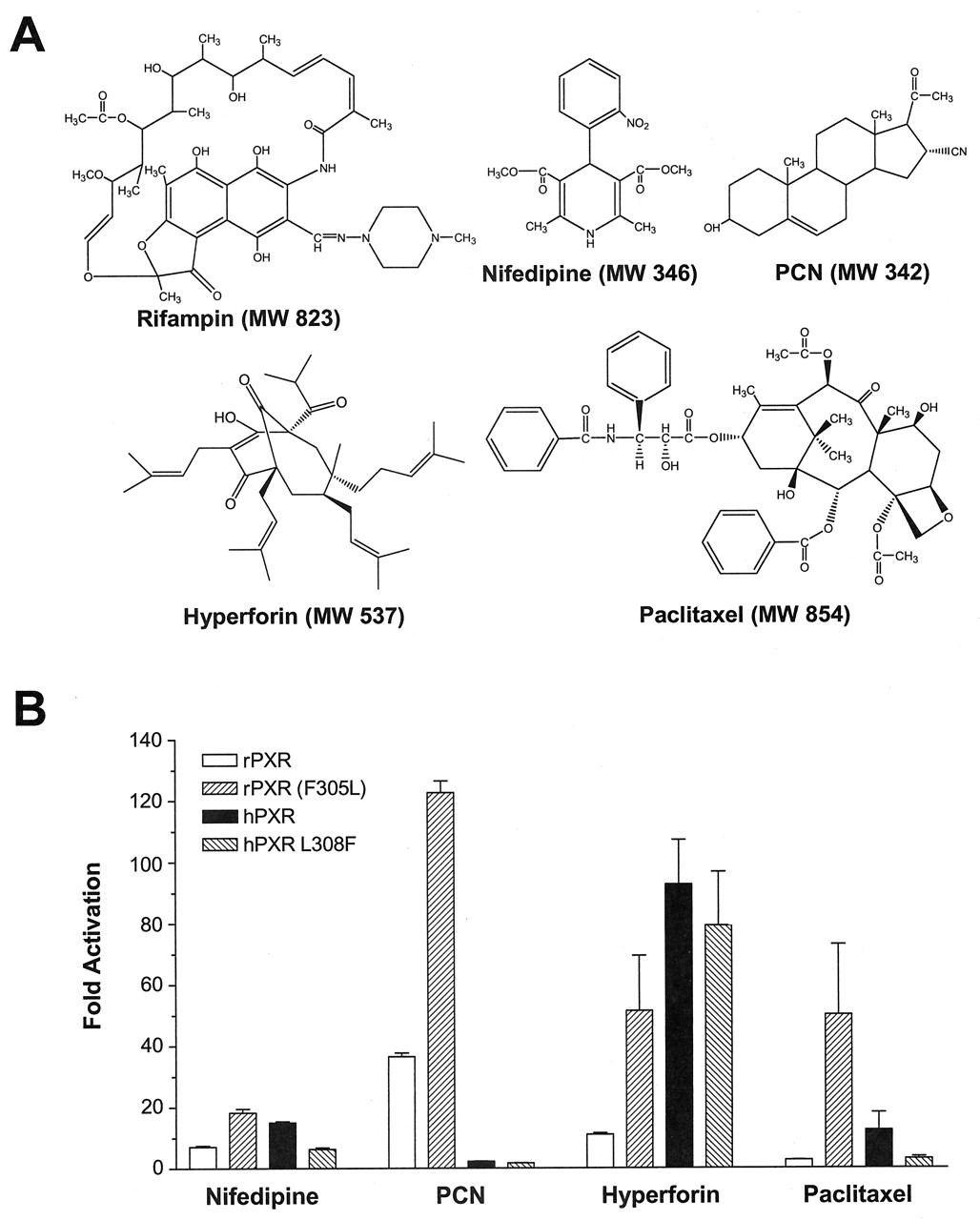
Amino Acid 305 Affects rPXR Activation by Other Drugs. Because amino acid 305 was found to permit rPXR responsiveness toward rifampin, we examined whether the effects were similar for other PXR agonists with diverse chemical structures (Fig. 4A). In our transactivation system, nifedipine, PCN, and hyperforin were activators of rat PXR (Fig. 4A). Mutation of rat PXR at the M1 position (F305L) further enhanced reporter activity for all drugs examined including paclitaxel, which did not activate the wild-type rat receptor. With regard to human PXR, nifedipine, hyperforin, and paclitaxel were found to be agonists, whereas PCN, as expected, was not an activator (Fig. 4B). Mutation of human PXR at residue 308 to the rat amino acid (L308F) markedly decreased reporter activity for nifedipine and paclitaxel, and to a lesser extent hyperforin, compared with the wild-type human receptor. These results indicate an important role for amino acid 305 in the species-dependent activation of rPXR by various drugs. The degree to which this residue influences the magnitude of PXR activation differs for each compound (Fig. 4B).

Amino acid 308 affects PXR activation by structurally divergent drugs. A, chemical structure of PXR agonists. B, activation of wild-type rat PXR, rat PXR M1 (F305L), hPXR, and hPXR L308F by nifedipine (10 μM), PCN (20 μM), hyperforin (1 μM), and paclitaxel (10 μM). Transactivation assays were performed in HepG2 cells with a CYP3A4 luciferase reporter construct. Data are expressed as fold activation over vehicle control.
Discussion
Species-dependent induction of many drug-metabolizing enzymes and transporters is clearly a consequence of differential activation of orthologous PXRs. This phenomenon has direct implications on drug development because induction studies in preclinical species such as rodents and dogs may not be predictive of effects in humans. Therefore, various in vitro assays using human PXR as well as transgenic mice have been developed as useful tools for predicting drug interactions (Moore and Kliewer, 2000; Xie et al., 2000). However, in silico methods for determination of PXR activation potential by drugs, if they can be developed, may prove to be complimentary, high-throughput, inexpensive screening tools in drug development (Ekins and Erickson, 2002).
PXR, unlike other hormone receptors, exhibits promiscuity in binding structurally diverse compounds while being at the same time species-specific. The recently reported crystal structures of human PXR (Watkins et al., 2001, 2003) resulted in better understanding of the molecular basis of the dual nature of this class of receptors. These structures revealed a ligand-binding cavity of 1200 Å3, substantially larger than that of many other nuclear receptors. As a result, the relatively small-molecule cholesterol-lowing drug SR12813, binds in three different orientations, using five critical polar residues spaced evenly throughout the upper portion of the ligand-binding cavity: Ser208, Ser247, Gln285, His407, and Arg410 (Watkins et al., 2001). Most of these residues, with the exception of Ser247, are divergent between species and are believed to determine ligand sensitivity (Moore et al., 2002). Indeed, the mutation of four residues of the mouse PXR “humanized” the receptor in that the previously insensitive receptor could be activated by SR12813 (Watkins et al., 2001). Unlike SR12813, hyperforin binds in a single orientation using a slightly different subset of hydrophobic residues within ligand-binding pocket. Furthermore, hyperforin does not contact one polar side chain, Ser208, which is among SR12813 contacts. Interestingly, binding of SR12813 and hyperforin did not induce significant conformational changes in the protein even among 28 amino acids lining cavity walls, with the exception of Leu209 and His407, which are shifted approximately 4 to 6 Å in the hyperforin structure compared with the apo and SR12813 structures. It allows a gain of approximately 250 Å3 in the volume of the ligand-binding pocket. In addition to these conformational changes, a mobile hydrophobic loop encompassing residues 309 to 321 adopts a helical conformation from 317 to 321 in the PXR-hyperforin structure. High values of temperature factors in this region of the molecule (colored red in Fig. 5) also indicate a possible role of 309 to 321 loop dynamics in the accommodation of much larger ligands, e.g., rifampin. Indeed, binding of a large compound like rifampin is believed to force the solvent-inaccessible pore to open, providing additional residues in the hydrophobic loop for protein-ligand contacts (Watkins et al., 2001). Thus, a “dynamic barrier” might be another strong determinant of species-specificity in pregnane X receptors.

Structural localization of mutants affecting rifampin responsiveness. Ribbon representation of human PXR structure (1ILH) with three experimentally observed positions of SR12813 (26) shown as overlapping van der Waals atomic radii. Ribbons and ligand are colored according to temperature-factor values from blue (lowest) to red (highest) through gray, corresponding to intermediate values. Shown in green are residues corresponding to positions in rat PXR based on the model. The diagram was prepared using SETOR (Evans, 1993).
Rifampin is a potent agonist of human but not rodent PXR. To elucidate the mechanism of human PXR selectivity toward rifampin we used a strategy of combined interspecies random chimeragenesis (Buck and Amara, 1994) followed by targeted site-directed mutagenesis. With functional analysis of an array of HR PXR chimeras, we were able to define a region of amino acids within the ligand-binding domain of PXR (303-317) that was essential for rifampin sensitivity. Within this region, residue 308 seemed important because mutation of human PXR at this position to the rat amino acid (Leu to Phe) reduced the apparent affinity for rifampin-mediated receptor activation. Furthermore, conversion of amino acid at this position in rat PXR to its human counter-part converted the rifampin-insensitive receptor to one that is rifampin-responsive. Functional analysis of RH PXR chimeras indicated another region (amino acids 317-335) is also important for rifampin sensitivity. However, only after the combined mutation of rat PXR at three amino acids to the human residues (K317Q, M324L, and C328Y, taken from human residues) was significant rifampin-mediated receptor activation observed. Therefore, amino acids in these three positions seemed to act coordinately to impart rifampin sensitivity.
The key amino acids critical for PXR sensitivity toward rifampin reside in the flanking regions of the flexible loop 309 to 321 (derived from the hPXR sequence) spanning the space between the β4 and α7. Leu308 lies in the C terminus of the β4, Leu324, and Tyr328 in the N-terminal half of the α7, whereas Gln317 is within the loop itself. Leu308 and Leu324 form a van der Waals contact that is part of the nonsolvent accessible pore (Fig. 5) and locks access into the ligand-binding cavity from the surface. Our results indicate that the presence of phenylalanine at the corresponding rat amino acid (Phe305) to human Leu308, limits receptor activation by large ligands such as rifampin, paclitaxel, and hyperforin. Interestingly, greater efficacy was observed for relatively smaller rat PXR agonists such as PCN and nifedipine when the corresponding phenylalanine residue was converted to leucine. Moreover, substitution of Leu308 with phenylalanine alone in hPXR results in a reduction of rifampin-, hyperforin-, and nifedipine-mediated receptor activation with human PXR (Fig. 4). This suggests that the nature of this particular amino acid influences receptor activation by ligands of varying size.
The location of the critical rifampin-sensitizing hot spots are right beside and within the flexible loop, suggesting that the amino acid substitutions mentioned above might also affect loop dynamics and the ability of this region to rear-range in response to ligand binding. Indeed, binding of hyperforin induces helical conformation in the 317 to 321 region (Watkins et al., 2003), which is coincident with the fact that residues in adjacent positions 317, 324, and 328 act coordinately to impart rifampin sensitivity to rodent PXR, probably via the maintenance of certain structural propensities of the 309 to 321 loop.
Another finding from functional analysis of PXR chimeras was the apparent graded rifampin-mediated response observed as the chimeric junctions moved from the N to the C terminus. These graded responses suggest positional specificity of other important amino acid domains necessary for maximal rifampin-mediated PXR activation. Thus, from the functional analysis of HR PXR chimeras, the region of amino acids between residues 402 and 417 is important for maximal rifampin sensitivity. This same region is found to be important for conformability of the PXR ligand-binding pocket to hyperforin. Residue His407 within this region shifts by 5.8 Å between the apo and hyperforin structures (Watkins et al., 2003). Additionally, a stepwise activity pattern in RH PXR chimeras identifies other key regions (residues 280-301 and 178-268) that are also essential for full rifampin response. These two rifampin-sensitive domains correspond to residues in PXR, which form the ligand-binding cavity. Interestingly, residues within the second region also mediate an induced-fit mechanism to accommodate hyperforin (Watkins et al., 2003). In comparison to the apo form, the main chain moves by up to 1.6 and 3.3 Å in the 207 to 210 and 230 to 235 regions, respectively, to conform to hyperforin (Watkins et al., 2003). In contrast, region 280 to 301 does not undergo conformational changes when hyperforin binds, although Phe281 and Trp299 directly contact the hydrophobic C-24 arm of hyperforin. Thus, although the substitution of key amino acids at the entry of the ligand-binding cavity in rat PXR was able to produce rifampin responsiveness, the extent of receptor activation was lower than that of the wild-type human receptor. Therefore, species-dependent amino acid differences within the ligand-binding cavity seem to determine the extent of receptor activation. Determination of X-ray crystal structure of rifampin bound to PXR will ultimately be required to identify amino acids that influence rifampin specificity via direct contacts with the ligand.
In conclusion, we have identified amino acid residues that modulate the rifampin responsiveness of rPXR. In particular, residue 305 in rPXR seems to be the key amino acid residue that modulates receptor activation by large molecules such as rifampin, paclitaxel, and hyperforin. Indeed, data from the current study would suggest that activation of PXR is not simply a function of the amino acids that form the ligand-binding cavity but also is critically dependent on the amino acids that form the flexible loop and pore to the ligand-binding cavity. Accordingly, data presented in this study relating to the functional analysis of human/rat PXR chimeras along with available structural information significantly enhances our understanding of the structural basis of PXR “directed promiscuity” (Watkins et al., 2001) and may aid in the development of models that better predict PXR activation and drug-drug interactions in humans.
Footnotes
-
This work was supported in part by United States Public Health Service grants GM31304 (to R.B.K.), GM54724 (to R.B.K.), and P30 ES00267 (to L.M.P).
-
ABBREVIATIONS: PXR, pregnane X receptor; HR, human-rat; RH, rat-human; rPXR, rat pregnane X receptor; hPXR, human pregnane X receptor; PCN, pregnenolone 16α-carbonitrile; RU486, mifepristone; SR12813, 3,5-di-tert-butyl-4-hydroxystyrene-β,β-diphosphonic acid tetraethyl ester.
- Received June 5, 2003.
- Accepted September 26, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}