


Abstract
Chronic morphine treatment has been shown to produce constitutive activation of μ-opioid receptors, and this transition might contribute to the development of tolerance and dependence. The apparent ability of chronic morphine to increase the spontaneous, agonist-independent activation of μ-opioid receptors may be unique, due to its distinct partial agonist properties of possessing a relatively high intrinsic activity coupled with a poor ability to produce desensitization and down-regulation. Therefore, the present study tested the hypothesis that prolonged exposure to morphine would produce greater constitutive activity of μ-opioid receptors than exposure to the full agonist [d-Ala2,N-MePhe4,Gly-ol5]enkephalin (DAMGO). GH3 cells expressing μ-opioid receptors were exposed to chronic morphine, DAMGO, or no opioid under conditions determined to produce maximal desensitization, down-regulation, and cAMP rebound. After chronic treatment, the μ-opioid antagonists naloxone and β-chlornaltrexamine (β-CNA) were evaluated in two assays predictive of inverse agonist activity. Both antagonists produced a concentration-dependent inhibition of [35S]GTPγS binding only in membranes prepared from cells chronically exposed to opioids. This effect was reversed by the neutral μ-opioid antagonist CTAP. Additionally, conditions known to uncouple G protein-coupled receptors from G proteins produced a leftward shift in the competition curve of β-CNA for [3H]DAMGO binding only in membranes prepared from chronically treated cells. In contrast, these conditions produced no shift in the competition curve by the neutral antagonist CTAP in cells exposed to chronic DAMGO. Therefore, prolonged exposure of GH3MOR cells to opioids produced constitutive activation of μ-opioid receptors. Surprisingly, chronic treatment with the more efficacious agonist DAMGO produced greater increases in both measures of inverse agonist activity than did morphine. These observations may lend novel insight into the mechanisms of opioid tolerance and dependence.
Opioid receptors belong to the superfamily of GPCRs that produce their effects by activation of intracellular G proteins (Law and Loh, 1999). The μ-opioid receptor is one of three classes of opioid receptors and it plays an important role in the management of pain (Reisine and Pasternak, 1996) and in opioid tolerance and dependence (Nestler et al., 1993). Activation of μ-opioid receptors leads to the regulation of several intracellular effectors, including the inhibition of adenylyl cyclase activity (Yu et al., 1990), the closing of voltage-gated Ca2+ channels (Piros et al., 1995), and the activation of inwardly rectifying K+channels (Henry et al., 1995). The coupling of μ-opioid receptors to these effectors is achieved by activation of Giα/Goα pertussis toxin-sensitive G proteins. In cellular models, chronic exposure to μ-opioid agonists results in receptor desensitization, down-regulation, and internalization (Keith et al., 1996; Yabaluri and Medzihradsky, 1997). These processes may contribute to opioid tolerance that occurs upon prolonged administration (Collier, 1984). Another adaptive response to sustained exposure to opioids is a sensitization in the cAMP signal transduction system (Yu et al., 1990; Wang et al., 1994). This is manifest by a rebound of cAMP production above basal levels upon administration of a μ-opioid antagonist or the abrupt cessation of the chronic opioid treatment. This may represent a cellular correlate of opioid withdrawal and has been used to define a state of dependence (Sharma et al., 1975; Collier, 1984).
Many GPCRs exhibit constitutive activity, producing spontaneous regulation of effectors in the absence of activation by agonists (Lefkowitz et al., 1993; Samama et al., 1993; Milligan et al., 1995;Charpentier et al., 1996; Arvanitakis et al., 1997). A two-state receptor model has been proposed to account for constitutive activity in which receptors exist in an equilibrium between inactive (R) and active (R*) states. Agonists stabilize the R* state, inverse agonists stabilize the R state, and antagonists have equal preferences for both states (Costa et al., 1992). Therefore, inverse agonists are useful ligands to detect constitutive activity by reducing spontaneous, agonist-independent receptor activity. Constitutive activity of δ-opioid receptors has been demonstrated by the inverse agonist ICI-174,864 (Chiu et al., 1996; Merkouris et al., 1997; Szekeres and Traynor, 1997; Neilan et al., 1999). Recent studies have reported that μ-opioid receptors also display basal signaling activity in the absence of agonist and that the antagonist β-CNA exhibits inverse agonist activity in transfected human embryonic kidney 293 cells (Wang et al., 1999; Burford et al., 2000). More importantly for this study, chronic morphine treatment increases the apparent constitutive activity of μ-opioid receptors (Wang et al., 2000). Therefore, chronic exposure to opioids might result in an increased conversion of μ-opioid receptors from an inactive (R) to a constitutively active (R*) state, and this transition might contribute to the development of tolerance and dependence (Wang et al., 1994).
To date, the apparent enhancement of μ-opioid receptor constitutive activity has only been demonstrated after chronic morphine treatment (Wang et al., 2000). Depending on the model system employed, acute morphine exhibits either partial (Zaki et al., 2000) or full (Selley et al., 2000) agonist efficacy at μ-opioid receptors. Chronic morphine treatment also produces less desensitization and internalization of μ-opioid receptors than fully efficacious agonists such as DAMGO (Noble and Cox, 1996; Blake et al., 1997). Therefore, it is possible that the apparent ability of chronic morphine to convert μ-opioid receptors to a constitutively active state is unique, because of its distinct partial agonist properties of possessing a relatively high intrinsic activity coupled with a poor ability to produce desensitization and down-regulation of μ-opioid receptors.
The present study tested the hypothesis that prolonged exposure to morphine would produce greater constitutive activity of μ-opioid receptors than exposure to the full agonist DAMGO. This was accomplished by first developing an appropriate cellular model that accurately reflected the adaptive responses occurring in response to chronic opioid administration (i.e., desensitization, down-regulation, and cAMP rebound). Rat pituitary GH3 cells stably transfected with μ-opioid receptors (i.e., GH3MOR) were selected for these experiments. Once the validity of the model was established, GH3MOR cells were exposed to chronic morphine, DAMGO, or no opioid under conditions determined to produce maximal desensitization, down-regulation, and cAMP rebound. After chronic treatment, the activity of the μ-opioid antagonists naloxone and β-CNA were evaluated in two assays predictive of inverse agonist activity. Prolonged exposure to either morphine or DAMGO converted both antagonists into inverse agonists, indicative of constitutive activation of μ-opioid receptors. Surprisingly, chronic treatment with the more efficacious agonist DAMGO produced greater increases in both measures of inverse agonist activity than did morphine.
Experimental Procedures
Materials.
[3H]DAMGO (56 Ci/mmol), [8-3H]adenine (26 Ci/mmol), [α-32P]ATP (17 Ci/mmol), and [35S]GTPγS (1148 Ci/mmol) were obtained from Amersham Pharmacia Biotech (Piscataway, NJ). DAMGO and CTAP were purchased from Peninsula Laboratories (Belmont, CA) and Phoenix Pharmaceuticals, Inc. (Belmont CA). Morphine was provided by the National Institute on Drug Abuse (Bethesda, MD). Naloxone, GppNHp, GDP, GTPγS, forskolin, and 3-isobutyl-1-methylxanthine (IBMX) were supplied by Sigma Chemical Co. (St, Louis, MO). β-CNA was procured from Sigma/RBI (Natick, MA). Penicillin/streptomycin (10,000 IU/ml and 10,000 mg/ml), geneticin (G418), and Dulbecco's modified Eagle's medium were purchased from Cellgro (Herndon, VA) and fetal calf serum from Summit Biotechnology (Fort Collins, CO). PTX was obtained from Calbiochem (San Diego, CA). All other reagents were purchased from Fisher Scientific (Pittsburgh, PA).
Cell Culture and Drug Pretreatment.
GH3 cells (CCL 82.1) were stably transfected with rat μ-opioid receptor cDNA to produce GH3MOR cells as previously described (Piros et al., 1995). Cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 100 units/ml penicillin, 100 mg/ml streptomycin, and 2.5 mg/ml geneticin in a humidified atmosphere of 5% CO2/95% air at 37°C. For receptor binding and [35S]GTPγS binding experiments, cells were seeded into 175-cm3 flasks. At 70% confluence, cells were incubated with various concentrations of morphine or DAMGO in fresh culture medium for time periods ranging from 0 to 48 h. At the end of drug exposure, cells were detached by incubation with phosphate-buffered saline containing 1 mM EDTA for 5 min and centrifuged at 1000 rpm for 10 min. The cell pellets were then extensively washed three times with 50 volumes of phosphate-buffered saline and finally stored at −80°C until use. For adenylyl cyclase assays, cells were seeded into 17 mm (24 well) culture plates at a density of 8 × 106 cells/plate and cultured in the medium containing the indicated drug pretreatments. For PTX exposure, cells were cultured under the indicated conditions in the presence of 100 ng/ml PTX for 24 h.
Membrane Preparation.
Extensively washed, frozen cell pellets were thawed on ice and resuspended in ice-cold homogenization buffer, pH 7.4, composed of 50 mM HEPES, 1 mM MgCl2, and 1 mM EGTA. Cells were then homogenized with 10 strokes of a glass Dounce homogenizer (Wheaton, Philadelphia, PA) and centrifuged at 40,000g for 10 min at 4°C. Pellets were resuspended in homogenization buffer, homogenized, and centrifuged again as described. This procedure was repeated twice more. The final pellets were resuspended in 50 mM Tris-HCl buffer, pH 7.4, and aliquots were frozen at −80°C until use. Protein concentration was determined using bovine serum albumin as a standard.
Opioid Receptor Binding.
μ-Opioid receptor binding was performed with [3H]DAMGO in 50 mM Tris-HCl buffer, pH 7.4. Competitive inhibition of 3 nM [3H]DAMGO binding by β-CNA (10−12 to 10−6 M) was performed in the presence or absence of the GTP analog GppNHp (25 μM) and NaCl (100 mM). To examine [3H]DAMGO binding after chronic exposure to various concentrations of morphine or DAMGO, membranes prepared from extensively washed pretreated cells were incubated with 3 nM [3H]DAMGO in the presence or absence of a saturating concentration of DAMGO (10 μM). The remaining specific [3H]DAMGO binding was then expressed as a percent of the specific binding in membranes prepared from control cells that had not been exposed to any opioid (i.e., % control). Receptor binding experiments were performed in triplicate, and conducted at room temperature for 90 min, in a volume of 1 ml with 200 μg of protein. The reaction was terminated by filtration through glass GF/B fiber filters using a Brandel 24-well cell harvester. Filters were subsequently washed three times with ice-cold binding buffer, and bound radioactivity was determined 12 h after the addition of 4 ml of scintillation fluid by counting in a Packard Tri-Carb 2100TR liquid scintillation counter (Meriden, CT).
[35S]GTPγS Binding.
[35S]GTPγS binding was performed as previously described (Neilan et al., 1999) with slight modifications. Briefly, membranes (50 μg of protein) were incubated with [35S]GTPγS (0.1 nM) in a binding buffer composed of 20 mM HEPES, pH 7.4, 10 mM MgCl2, 100 mM KCl, and 10 μM GDP. When indicated, DAMGO (1 μM) or varying concentrations of naloxone or β-CNA were added to a final volume of 1 ml and incubated for 1 h at 30°C. Nonspecific binding was defined by the inclusion of 10 μM GTPγS. The reaction was terminated by rapid filtration and bound radioactivity was determined by liquid scintillation counting as described above.
Adenylyl Cyclase Assay.
The effect of opioids on the conversion of [3H]ATP to cyclic [3H]AMP by adenylyl cyclase was determined as described previously (Prather et al., 2000). Briefly, cells were seeded into 24-well plates and cultured for various time periods ranging from 0 to 48 h in the presence or absence of opioid agonists. At the end of agonist exposure, media was removed and washed once with serum-free medium and replaced with an incubation mixture (at 37°C) of Dulbecco's modified Eagle's medium containing the same concentration of the opioid used for pretreatment, 0.9% NaCl, 500 μM IBMX, and 1.25 μCi/well [3H]adenine for 2 h. After incubation, the mixture was removed and cells were washed once with serum-free medium. Each plate was then floated in an ice-water bath for 5 min. During this time, an assay mixture of ice-cold Krebs-Ringer-HEPES buffer, pH 7.4, containing 500 μM IBMX, 10 μM forskolin, and the appropriate concentration of the opioid ligand to be tested was added. Plates were then placed on a water bath at 37°C for 15 min. The reaction was terminated by the addition of 50 μl of 2.2N HCl. An internal standard of [α32P]cAMP was added to each well and radioactive cAMP was separated using alumina column chromatography (Alvarez and Daniels, 1992). Scintillation fluid (10 ml) was added and samples were immediately counted in a Packard Tri-Carb 2100TR liquid scintillation counter (Meriden, CT).
Data Analysis and Statistics.
All statistical and curve-fitting analyses were performed using the computer program Prism v2.0b for Macintosh (GraphPad Software, San Diego, CA). Nonlinear regression analysis was used to determine the best-fit of full concentration-effect curves for adenylyl cyclase experiments, receptor binding and [35S]GTPγS binding assays. The IC50 and IMAX values for each curve were derived from the best-fit analysis. For full dose-response curves, comparison of the effect produced at each drug concentration to that of control values (i.e., in the absence of drug) was accomplished by a one-way analysis of variance followed bypost hoc comparison using Dunnett's test. For statistical comparisons involving three or more groups, differences between means were determined by a one-way analysis of variance followed bypost hoc comparison of all individual groups by Tukey's multiple comparison test. In instances in which only two groups were compared, differences between means were determined by the nonpaired Student's t test. Data are expressed as mean ± S.E.M. and, unless otherwise stated, are represented by a minimum of three separate experiments, performed in duplicate or triplicate.
Results
DAMGO Is a Full Agonist and Morphine Is a Partial Agonist as Measured by Inhibition of Adenylyl Cyclase Activity in GH3MOR Cells.
Previous studies have demonstrated that stably transfected GH3MOR cells express a moderate μ-opioid receptor density of 0.39 pmol/mg of protein (Piros et al., 1995) and that both morphine and DAMGO bind to these receptors with a relatively high affinities of 7.2 and 1.0 nM, respectively (Table 1; Piros et al., 1995). In the present study, both agonists demonstrated potent and efficacious inhibition of the activity of the intracellular effector adenylyl cyclase. However, DAMGO produced significantly greater maximal reduction of cAMP levels (IMAX = 76.2%; IC50 = 18.2 nM) relative to morphine (IMAX = 57.0%; IC50 = 81.0 nM) (p < 0.01). Therefore, as observed in several previous studies (Selley et al., 2000; Zaki et al., 2000), morphine acts a partial agonist relative to the full agonist DAMGO at μ-opioid receptors in GH3MOR cells.
Affinity (K i) values and inhibition of adenylyl cyclase activity by morphine and DAMGO in control GH3MOR cells and cells chronically treated with morphine or DAMGO . The affinity of morphine or DAMGO for μ-opioid receptors in GH3MOR cell membranes was determined by their displacement of 2 nM [3H]diprenorphine. The ability of μ-opioid agonists to reduce 10 μM forskolin-stimulated cAMP accumulation was assessed in transfected GH3 cells as described under Experimental Procedures. Data are presented as the percentage of control cAMP levels (i.e., no opioid). Data points represent the mean ± S.E.M. for three or four experiments performed in triplicate.
Chronic Exposure of GH3MOR Cells to Morphine or DAMGO Produces a Desensitization of μ-Opioid Receptor Inhibition of Adenylyl Cyclase Activity.
The ability of morphine or DAMGO to desensitize μ-opioid receptors was next examined by treating GH3MOR cells with various concentrations of opioid agonists for increasing time periods and monitoring the ability of a maximally efficacious concentration of DAMGO (1 μM) to inhibit forskolin-stimulated cAMP levels. Exposure of cells to a maximally efficacious concentration of morphine or DAMGO (10 μM) showed a time-dependent decrease in the ability of 1 μM DAMGO to reduce cAMP levels, beginning at 6 h and reaching a maximal effect at 24 h (data not shown). Subsequently, when cells were incubated for 24 h with increasing amounts of morphine or DAMGO, the reduction in the efficacy of 1 μM DAMGO was also demonstrated to be concentration-dependent. Specifically, significant desensitization of DAMGO inhibition of adenylyl cyclase activity began when cells were chronically exposed to 100 nM (for DAMGO) or 300 nM (for morphine), reaching maximal effects at concentrations of 10 μM for both agonists (data not shown).
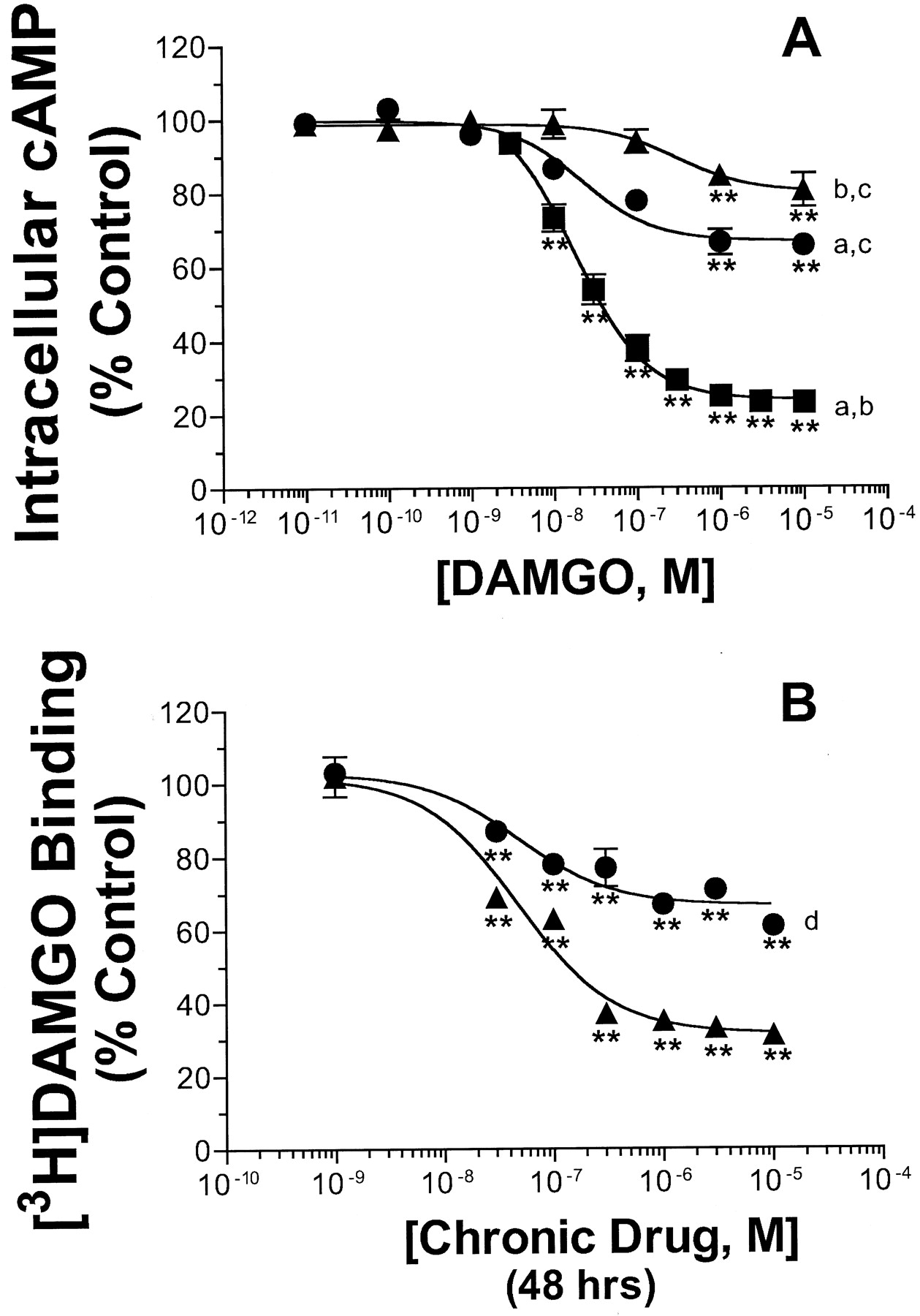
With the knowledge that μ-opioid receptor desensitization in response to chronic agonist exposure in GH3MOR cells was time- and concentration-dependent, the effect of prolonged treatment with maximally desensitizing conditions for morphine or DAMGO (i.e., 10 μM for 24 h) on full DAMGO concentration-effect curves was compared (Fig. 1A; Table 1). Exposure of cells to either 10 μM morphine or DAMGO for 24 h shifted the dose-response curve for DAMGO inhibition of forskolin-stimulated cAMP level to the right compared with the control treatment. The concentration-effect curves for DAMGO inhibition of forskolin-stimulated cAMP accumulation yielded IC50 values of 18.6 ± 4.5, 34.9 ± 22.1, and 325 ± 99.1 nM for control, morphine, or DAMGO pretreated cells, respectively. Chronic pretreatment with DAMGO, but not morphine, resulted in a significantly greater IC50 value for acute DAMGO administration (p < 0.01). In contrast, prolonged exposure to both μ-opioid agonists resulted in a significant reduction in maximal inhibition produced by DAMGO from 76.2% in control cells, to only 34% and 19.8% in cells treated with morphine or DAMGO, respectively (p < 0.01). In addition, chronic exposure to DAMGO produced a significantly greater reduction in the IMAX of acute DAMGO relative to pretreatment with morphine (p < 0.01). Thus, chronic morphine and DAMGO pretreatment resulted in desensitization of the ability of μ-opioid receptors to inhibit adenylyl cyclase activity and the degree of desensitization was directly proportional to the efficacy of the agonist used for pretreatment.

Effect of chronic pretreatment with either DAMGO or morphine on μ-opioid receptor desensitization and down-regulation in GH3MOR cells. A, desensitization of DAMGO (1 μM) inhibition of 10 μM forskolin-stimulated cAMP levels in control (▪) and morphine- (●) or DAMGO- (▴) pretreated GH3MOR cells (10 μM for 24 h). The opioid pretreatment protocol and adenylyl cyclase assay were performed as described under Experimental Procedures. The IC50 and IMAX values are reported in Table 1. B, down-regulation of μ-opioid receptors GH3MOR cells pretreated with varying concentrations of morphine (●) or DAMGO (▴) for 48 h. Membranes prepared from extensively washed pretreated cells were incubated with 3 nM [3H]DAMGO in the presence or absence of a saturating concentration of DAMGO (10 μM). [3H]DAMGO binding determined after each opioid pretreatment concentration was expressed as a percent of the specific binding in membranes prepared from control cells that had not been exposed to any opioid (i.e., percentage control). Each data point on both graphs represents the mean ± S.E.M. of three or four independent experiments conducted in triplicate. ∗∗p < 0.01; significantly different from control (i.e., no opioid), (Dunnett's test). Significantly different from maximal inhibition determined for chronic DAMGO (a), chronic morphine (b), or control group (c),p < 0.01 (Tukey's test). d, significantly different from [3H]DAMGO binding observed after chronic treatment with 10 μM DAMGO, p < 0.01 (unpaired Student's t test).
Chronic Exposure of GH3MOR Cells to Morphine or DAMGO Produces a Down-Regulation of μ-Opioid Receptors.
The effect of chronic morphine and DAMGO exposure on μ-opioid receptor density was also determined by measuring the amount of [3H]DAMGO binding remaining in membranes prepared from pretreated cells after extensive washing to remove residual agonist (Fig. 1B). To assure maximal effects of chronic drug exposure, cells were incubated with opioids for 48 h. Treatment of cells with increasing concentrations of both opioid agonists decreased [3H]DAMGO binding to membranes prepared from these cells with similar IC50 values of 48.8 ± 1.0 and 48.8 ± 0.82 nM for morphine and DAMGO, respectively. All concentrations of morphine or DAMGO of 30 nM and higher produced significant decreases (p < 0.01) in μ-opioid receptor binding. As observed above for desensitization, the degree of decrease in binding (i.e., down-regulation) was also directly associated with the efficacy of the agonist used for chronic pretreatment. For example, the maximal percent of the reduction of [3H]DAMGO binding of 68.7% by DAMGO was significantly greater than that produced by morphine of only 33.4% (p < 0.01).
Chronic Exposure of GH3MOR Cells to Morphine or DAMGO Produces a Rebound in cAMP Levels above Basal Levels in Response to the μ-Opioid Antagonist Naloxone.
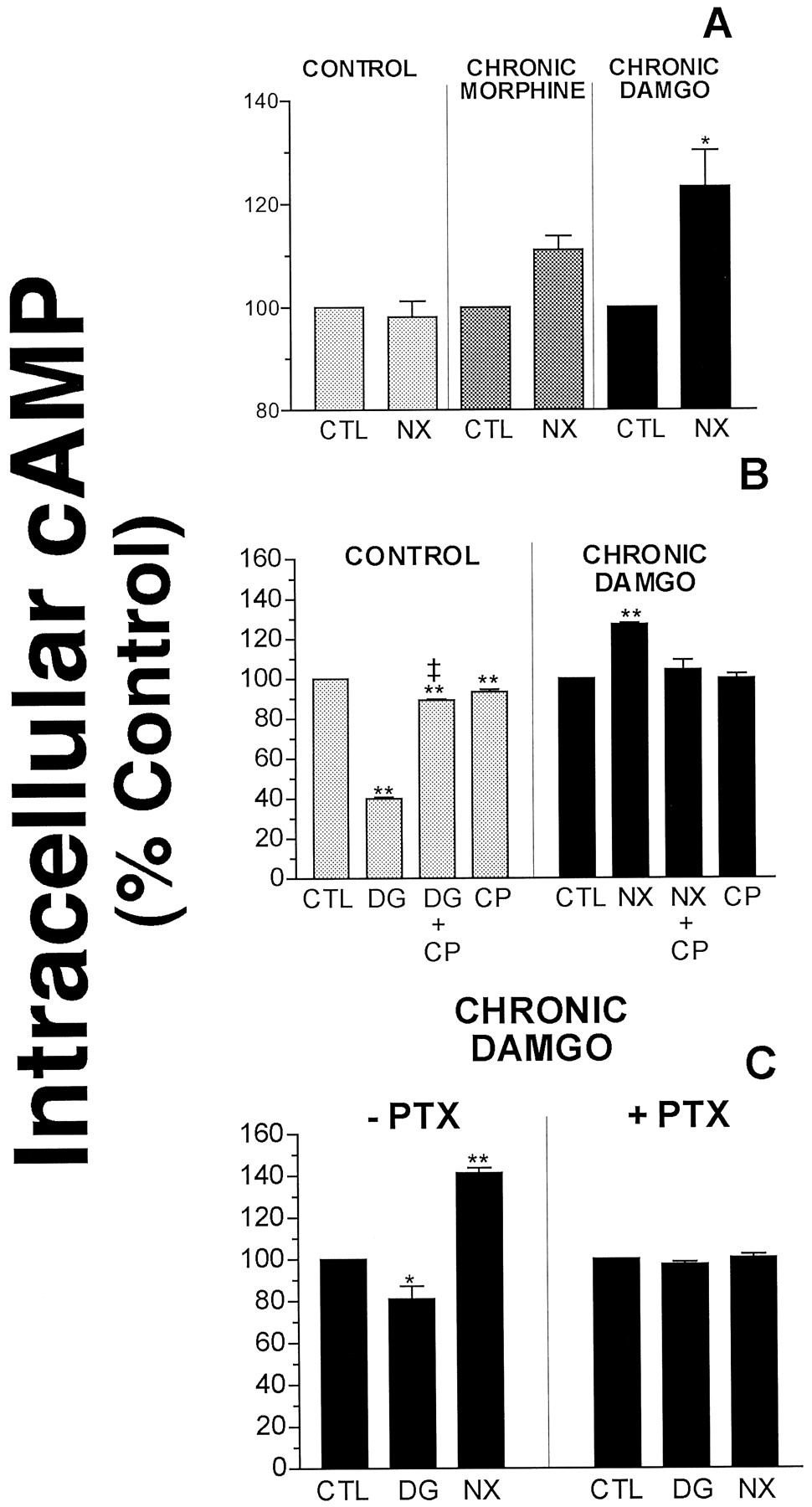
It has been well established that challenge of cells chronically exposed to opioid agonists with the antagonist naloxone results in a “rebound” or increase in cAMP production above basal levels (Yu et al., 1990; Nestler et al., 1993). Although the mechanisms responsible for this observation are the subject of much research, cAMP rebound has been proposed to represent a cellular model of withdrawal (Sharma et al., 1975). It was determined whether this adaptive process occurred in GH3MOR cells. As presented in Fig. 2A, 10 μM naloxone significantly enhanced forskolin-stimulated cAMP accumulation in chronic DAMGO-pretreated cells, but not in control or morphine pretreated cells (p < 0.05). Naloxone potentiated the forskolin response by 11.1 ± 2.7% and 23.3 ± 6.9% in morphine- and DAMGO-treated cells, respectively. As demonstrated previously for desensitization and down-regulation, this observation indicated that naloxone produced a rebound in cAMP levels in cells chronically treated with morphine and DAMGO in direct proportion to agonist efficacy. To confirm that the acute inhibition of adenylyl cyclase activity by DAMGO and the effect of naloxone potentiation of the forskolin response was mediated specifically by μ-opioid receptors, the ability of the μ-opioid antagonist CTAP to attenuate these responses was examined (Fig. 2B). CTAP (1 μM) produced no effect on cAMP levels when administered alone in control cells or in cells chronically exposed to DAMGO. However, concurrent addition of CTAP blocked the effect of naloxone potentiation of forskolin-stimulated cAMP accumulation in DAMGO-treated cells and also reversed the acute inhibition of forskolin-stimulated cAMP accumulation by DAMGO in control cells. This is in agreement with a previous study in which CTAP demonstrated similar neutral antagonist activity in SHSY5Y cells chronically treated with morphine (Wang et al., 1994).

Effect of CTAP and PTX on acute DAMGO inhibition of cAMP levels and naloxone-induced cAMP rebound in GH3MOR cells chronically pretreated with DAMGO. A, naloxone (10 μM) produced a significant elevation in cAMP above basal levels in GH3MOR cells chronically pretreated with DAMGO (10 μM), but not morphine (10 μM) for 24 h. B, the μ-opioid antagonist CTAP reversed both acute DAMGO inhibition of cAMP levels and naloxone-induced cAMP rebound in DAMGO pretreated GH3MOR cells. C, pertussis toxin blocked both acute DAMGO inhibition of cAMP levels in control cells and naloxone-induced cAMP rebound in DAMGO pretreated GH3MOR cells. Each bar on all graphs represents the mean ± S.E.M. of three or four independent experiments conducted in triplicate. CTL, control; NX, naloxone; DG, DAMGO; CP, CTAP; PTX, pertussis toxin. ∗p < 0.05; significantly different from control (i.e., no opioid) (Dunnett's test). ∗∗p < 0.01; significantly different from control (i.e., no opioid) (Dunnett's test). ‡p < 0.01; significantly different from acute DAMGO inhibition of adenylyl cyclase activity (unpaired Student'st test).
Acute μ-Opioid Receptor Inhibition of Adenylyl Cyclase Activity and Naloxone Precipitated cAMP Rebound Are Mediated by PTX-Sensitive G Proteins.
Opioid receptors are known to produce inhibition of adenylyl cyclase activity by coupling to Gi/Goα proteins (Law and Loh, 1999). Therefore, the role of G proteins in naloxone-induced elevation of cAMP level and acute inhibition by DAMGO was next determined in GH3MOR cells treated with PTX (Fig. 2C). Treatment of GH3MOR cells concurrently with DAMGO and PTX (100 ng/ml, 24 h) abolished the ability of naloxone to potentiate forskolin-stimulated cAMP accumulation and acute inhibition by DAMGO. These results indicated that the potentiation effect on cAMP levels of naloxone after chronic opioid exposure and acute inhibition by DAMGO were mediated via functional coupling to Gi/Goα proteins.
Prolonged Exposure of GH3MOR Cells to Opioid Agonists Converts the Opioid Antagonists Naloxone and β-CNA to Inverse Agonists as Measured by [35S]GTPγS Binding.
Agonists for GPCRs stimulate the binding of the hydrolysis resistant GTP analog, [35S]GTPγS, to G protein α subunits and this can be used as a measure of receptor activation. Interestingly, for many GPCRs some antagonists have been shown to not only attenuate the effect of agonists, but also to produce effects that are opposite those observed by agonists when given alone. These findings suggest that a population of receptors must exist in a constitutively active state that can be stabilized by ligands that posses negative intrinsic activity (i.e., inverse agonists) (Costa et al., 1992). This is reflected by the demonstration that inverse agonists decrease [35S]GTPγS binding to G proteins (Szekeres and Traynor, 1997; Neilan et al., 1999). It has also been reported that chronic treatment with morphine increases the proportion of μ-opioid receptors in a constitutively active state (Wang et al., 2000). Therefore, we compared the effect of naloxone and β-CNA on [35S]GTPγS binding to membranes prepared from control and morphine- and DAMGO-pretreated cells. NaCl was replaced by KCl in the binding buffer for all of these experiments because this modification has been shown to increase constitutive activity of GPCRs and thus maximize the observation of potential inverse activity of test ligands (Costa et al., 1992). To establish the validity of our assay, we first examined the well-known ability of the agonist DAMGO to stimulate [35S]GTPγS binding in membranes prepared from control cells (Fig.3A). As expected, a maximal concentration of DAMGO (1 μM) increased [35S]GTPγS binding from 138 ± 3.3 to 315 ± 10.1 fmol/mg of protein in control membranes (i.e., a 128% increase). In contrast, the same concentration of DAMGO only elevated [35S]GTPγS binding from 161 ± 4.8 to 209 ± 18.6 fmol/mg of protein in membranes prepared from cells chronically exposed to DAMGO (i.e., a 29.8% increase). Importantly, chronic treatment of GH3MOR cells with DAMGO resulted in a small but significant (p < 0.01), increase in the level of basal [35S]GTPγS binding (i.e., 138 versus 161 fmol/mg) (Fig. 3B). These results suggest that the activation of G proteins by the μ-opioid agonist DAMGO is dramatically reduced in membranes prepared from GH3MOR cells chronically pretreated with DAMGO (i.e., desensitization). In addition, prolonged exposure to DAMGO significantly increases the basal activation of G proteins in GH3MOR cells, possibly because of constitutive activation of μ-opioid receptors.

The stimulation of [35S]GTPγS binding by DAMGO to membranes prepared from control or DAMGO pretreated GH3MOR cells. A, a maximal concentration of DAMGO (1 μM) produced an increase in [35S]GTPγS binding of 128 ± 3.2% in control membranes not exposed to chronic DAMGO. In contrast, the same concentration of DAMGO only elevated [35S]GTPγS binding by 29.8 ± 8.9% increase in membranes prepared from cells chronically pretreated with DAMGO. B, importantly, chronic treatment of GH3MOR cells with DAMGO resulted in a small but significant (p < 0.01) increase in the level of basal [35S]GTPγS binding from 138 ± 3.3 fmol/mg of protein in control membranes, to 161 ± 4.8 fmol/mg of protein after prolonged DAMGO exposure. Each bar on the graph represents the mean ± S.E.M. of four independent experiments conducted in triplicate. CTL, control; DG, DAMGO. ∗∗p < 0.01; significantly different from corresponding control (i.e., no opioid) (unpaired Student'st test).
As presented in Fig. 4A, naloxone showed slight (14.0 ± 1.6%) but significant stimulation of [35S]GTPγS binding to membranes prepared from control cells (p < 0.01). Although this is indicative of weak partial agonist activity, the maximal amount of stimulation produced was only 11% of that demonstrated by the full agonist DAMGO. Furthermore, naloxone (10 μM) showed no inhibition of adenylyl cyclase in control cells, a characteristic of agonists (Fig. 2A). In marked contrast to the observation in control cells, naloxone produced significant, dose-dependent inhibition of [35S]GTPγS binding in membranes prepared from cells chronically treated with opioids (p < 0.01). Specifically, [35S]GTPγS binding was reduced maximally by 10.4 ± 1.4% (IC50 = 16.1 nM) and 21.7 ± 1.4% (IC50 = 63.8 nM) in morphine- and DAMGO-pretreated conditions, respectively. In addition, naloxone produced significantly greater maximal inhibition in GH3MOR cells chronically pretreated with the full agonist DAMGO relative to the partial agonist morphine (p < 0.01).
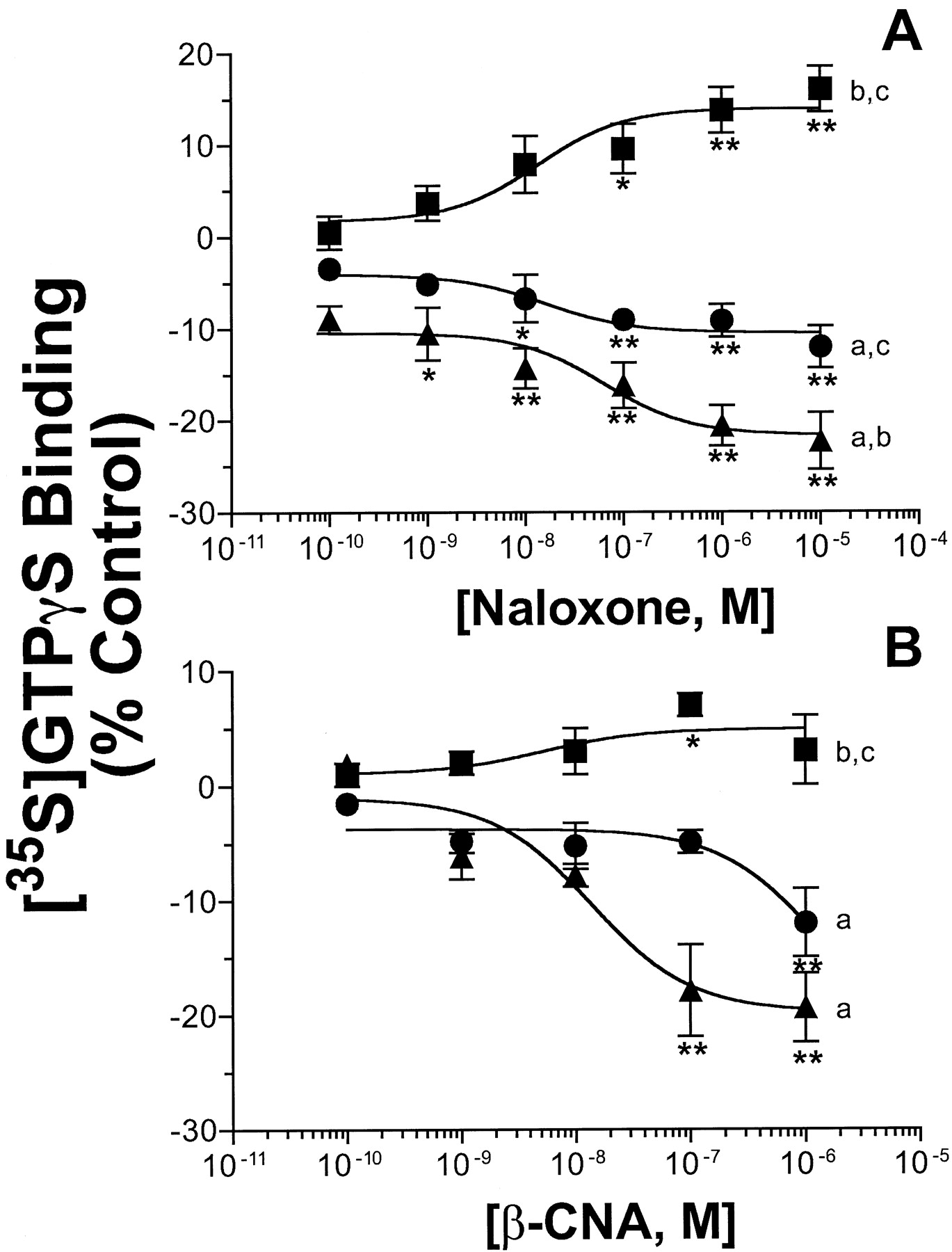
Effect of naloxone and β-CNA on [35S]GTPγS binding to membranes prepared from control and morphine- or DAMGO-pretreated GH3MOR cells. GH3MOR cells were treated with no opioid (▪), 10 μM morphine (●), or 10 μM DAMGO (▴) for 48 h. Membranes were prepared from extensively washed pretreated cells as described underExperimental Procedures. [35S]GTPγS binding to membranes in response to naloxone (A) or β-CNA (B) was determined with 0.1 nM [35S]GTPγS in the presence of a increasing concentrations of naloxone (10−10 to 10−5 M) or β-CNA (10−10 to 10−6 M). Nonspecific binding was defined by the inclusion of 10 μM GTPγS. Data obtained at each opioid concentration were calculated as binding relative to that observed in the absence of opioid (i.e., percentage of control). Each data point on both graphs represents the mean ± S.E.M. of at least four independent experiments conducted in triplicate. ∗p < 0.05; significantly different from control (i.e., no opioid) (Dunnett's test). ∗∗p < 0.01; significantly different from control (i.e., no opioid) (Dunnett's test). Significantly different from maximal inhibition determined for chronic DAMGO (a), chronic morphine (b), or control group (c), p < 0.01 (Tukey's test).
Similar to naloxone, a second μ-opioid selective antagonist β-CNA also dose-dependently (IC50 = 13.9 nM) inhibited [35S]GTPγS binding to a maximal level of 19.8 ± 3.1% in membranes prepared from DAMGO pretreated cells (Fig. 4B). Interestingly, β-CNA only produced significant inhibition (12.0 ± 3%) at the highest concentration tested (1 μM) in GH3MOR cells chronically exposed to morphine (p < 0.01). No higher concentrations of β-CNA could be tested because nonspecific inhibition of [35S]GTPγS binding occurred at 10 μM and above in wild-type GH3 cells not expressing μ-opioid receptors (data not shown). Because β-CNA is known to alkylate μ-opioid receptors (Portoghese et al., 1979), higher concentrations might directly alkylate nonspecific targets including G proteins, interfering with [35S]GTPγS binding in a manner unrelated to receptor/G protein coupling. Finally, maximal concentrations of naloxone (10 μM) or β-CNA (1 μM) did not produce any significant reduction in [35S]GTPγS binding in membranes prepared from GH3MOR cells treated with 10 μM DAMGO for only 30 min (data not shown).
To confirm that the reduction in [35S]GTPγS binding after chronic DAMGO pretreatment by naloxone and β-CNA specifically involved μ-opioid receptors, reversal of this effect by the selective μ-opioid antagonist CTAP was examined (Fig.5). When tested alone, CTAP (1 μM) had no effect; however, it significantly reversed the ability of both naloxone (1 μM) and β-CNA (1 μM) to decrease [35S]GTPγS binding in GH3MOR cells chronically exposed to DAMGO (p < 0.05).

Reversal of naloxone and β-CNA inhibition of [35S]GTPγS binding to membranes prepared from DAMGO pretreated cells by CTAP. Membranes were prepared from extensively washed GH3MOR cells pretreated for 48 h with 10 μM DAMGO as described under Experimental Procedures. [35S]GTPγS binding to membranes in response to 1 μM naloxone or β-CNA was determined with 0.1 nM [35S]GTPγS. The inhibition of [35S]GTPγS binding produced by both naloxone and β-CNA was reversed by the presence of CTAP (1 μM). Each bar of on all graphs represents the mean ± S.E.M. of three or four independent experiments conducted in triplicate. CTL, control; NX, naloxone; CP, CTAP. ∗p < 0.05; significantly different from control (i.e., no opioid) (Dunnett's test).
In conclusion, results presented for naloxone and β-CNA concerning [35S]GTPγS binding suggest that chronic exposure of GH3MOR cells to opioid agonists converts μ-opioid antagonists into inverse agonists. Because inverse agonism can only be observed for ligands acting at constitutively active receptors, this implies that chronic opioid agonist pretreatment results in a conversion to, or enhancement of, μ-opioid receptors in a constitutively active state. Finally, as observed previously for desensitization, down-regulation, and cAMP rebound, the transformation from antagonist to inverse agonist occurs in direct proportion to the efficacy of the agonist used for chronic pretreatment.
β-CNA Displays Receptor Binding Characteristics of an Inverse Agonist Only in Membranes Prepared from GH3MOR Cells Chronically Treated with Opioid Agonists.
A two-state receptor model has been proposed to account for inverse agonism at GPCRs in which receptors exist in an equilibrium between inactive (R) and active (R*) states. Agonists stabilize the R* state, inverse agonists stabilize the R state and antagonists have equal preferences for both states (Costa et al., 1992). Therefore, conditions known to uncouple GPCRs from G proteins, such as the addition of guanine nucleotides and sodium chloride (Childers and Snyder, 1980), serve to decrease the binding of agonists, have no effect on the binding of antagonists, and enhance the binding of inverse agonists to GPCRs (Neilan et al., 1999). The previous functional studies using [35S]GTPγS binding suggested that β-CNA acted as an inverse agonist in membranes prepared from cells chronically exposed to opioids. Therefore, the ability of this compound to compete with the binding of [3H]DAMGO to μ-opioid receptors was determined in the presence or absence of the GTP analog GppNHp and NaCl in control GH3MOR cells and in cells pretreated with morphine or DAMGO (Fig.6; Table2). β-CNA binds to μ-opioid receptors in both a reversible and irreversible manner (Portoghese et al., 1979). Therefore, the IC50 values for β-CNA obtained from competition binding studies are presented rather than their conversion to Ki values, because this calculation assumes freely reversible binding (Cheng and Prusoff, 1973). Competition binding between 3 nM [3H]DAMGO and increasing concentrations of β-CNA in the presence or absence of 100 mM NaCl and 25 μM GppNHp was presumed to reflect binding to the low- or high-affinity state of the receptor, respectively (Table 2). The concentration of [3H]DAMGO used in these competition binding experiments was saturating (approximately five times the K d value of the drug); this relatively high concentration restricts the maximal observed competition by β-CNA. In GH3MOR cells not exposed to opioids, the amount of β-CNA required to produce half-maximal inhibition of [3H]DAMGO binding to the high- (IC50 = 3.83 ± 0.84 nM) and low- (IC50 = 10.20 ± 4.02 nM) affinity states of the receptor was not significantly different (Fig. 6A). This is in agreement with the [35S]GTPγS binding results for β-CNA in control membranes and suggests that under these conditions, β-CNA displays receptor binding properties of a neutral antagonist. In contrast, in membranes prepared from cells pretreated with morphine, more than 3-fold less β-CNA was needed to reduce [3H]DAMGO binding to the low- (IC50 = 1.57 ± 0.35 nM) versus the high- (IC50 = 5.03 ± 0.89 nM) affinity state of the μ-opioid receptor (Fig. 6B; p < 0.05). This enhancement in the affinity of β-CNA for the low- (IC50 = 0.33 ± 0.09 nM) relative to the high- (IC50 = 4.30 ± 0.61 nM) affinity state of μ-opioid receptors was even greater in membranes prepared from GH3MOR cells chronically treated with DAMGO. This is reflected by a 13-fold leftward shift in the competition curve of β-CNA for [3H]DAMGO binding in the presence of GppNHp/NaCl in membranes prepared from chronically treated cells (Fig. 6C, p < 0.01). The IC50 for the competition of β-CNA with [3H]DAMGO to the high-affinity state of μ-opioid receptors (i.e., in the absence of GppNHp/NaCl), was not significantly altered by any of the pretreatment conditions. Importantly, in membranes prepared from cells chronically exposed to DAMGO, the addition of GppNHp/NaCl to the binding buffer produced no shift in the competition curve by the neutral antagonist CTAP (data not shown). These observations support our previous studies examining [35S]GTPγS binding and indicate that after chronic exposure of GH3MOR cells to opioids, the μ-opioid antagonist β-CNA displays receptor binding properties of an inverse agonist.

Competition of β-CNA for [3H]DAMGO binding in the presence or absence of GppNHp/NaCl in membranes prepared from either control or morphine- or DAMGO-pretreated GH3MOR cells. GH3MOR cells were treated with no opioid (A), 10 μM morphine (B), or 10 μM DAMGO (C) for 48 h. Membranes were prepared from extensively washed pretreated cells as described underExperimental Procedures. Competition binding was determined using 3 nM [3H]DAMGO and increasing concentrations of β-CNA (10-12 to 10-6 M) in the presence [open symbols (■, ○, ▵)] or absence [closed symbols (▪, ●, ▴)] of 25 μM GppNHp and 100 mM NaCl. The IC50 values are reported in Table 2. Each data point on both graphs represents the mean ± S.E.M. of three or four independent experiments conducted in triplicate.
Competitive inhibition of [3H]DAMGO binding by β-CNA to GH3MOR cell membranes in the presence or absence of GppNHp/NaCl. The displacement of [3H]DAMGO binding by β-CNA to membranes prepared from control and morphine or DAMGO-pretreated GH3MOR cells was assessed. Cells were treated with or without 10 μM morphine or DAMGO for 48 h. Membranes were prepared from extensively washed cells as described under Experimental Procedures. Competition binding was determined with 3 nM [3H] DAMGO and various concentrations of β-CNA in the presence or absence of 25 μM GppNHp and 100mM NaCl. Results are the mean ± S.E.M. from three or four experiments preformed in triplicate.
Discussion
This study tested the hypothesis that prolonged exposure to morphine would produce greater constitutive activation of μ-opioid receptors than exposure to the full agonist DAMGO. Initially, it was determined that GH3MOR cells were an appropriate cellular model to investigate the adaptive changes that occur in response to chronic opioid administration. First, morphine and DAMGO acted as partial and full agonists, respectively, to inhibit adenylyl cyclase activity in GH3MOR cells. Next, chronic treatment with either morphine or DAMGO resulted in: 1) a decrease in the ability of DAMGO to acutely inhibit forskolin-stimulated cAMP accumulation (i.e., desensitization), 2) a reduction in the density of μ-opioid receptors in the plasma membrane (i.e., down-regulation), and 3) an increase of cAMP levels above baseline in response to a challenge with the μ-opioid antagonist naloxone (i.e., cAMP rebound). Importantly, the degree of desensitization, down-regulation, and cAMP rebound were all directly correlated to the efficacy of the agonist used for chronic treatment. These observations are similar to those reported in other recent studies (Yabaluri and Medzihradsky, 1997; Zaki et al., 2000).
Many GPCRs exhibit constitutive activity, activating G proteins in the absence of agonists (Lefkowitz et al., 1993). Hence, membranes prepared from cells that contain constitutively active receptors demonstrate higher basal binding of the hydrolysis resistant GTP analog [35S]GTPγS to G protein α subunits. Inverse agonists can reduce constitutive activity and thus decrease basal [35S]GTPγS binding when given alone (Milligan et al., 1995). Therefore, the inverse activity of a ligand can be observed only in membranes containing constitutively active receptors. The inverse agonist ICI-174,864 demonstrated that δ-opioid receptors can exist in a constitutively active state (Chiu et al., 1996;Merkouris et al., 1997; Szekeres and Traynor, 1997; Neilan et al., 1999). However, with the exception of a few initial reports (Wang et al., 1999; Burford et al., 2000), the evidence for the existence of constitutive activity of μ-opioid receptors is lacking. It has also been reported that chronic treatment with morphine increases the proportion of μ-opioid receptors in a constitutively active state (Wang et al., 2000). Therefore, we compared the effect of two μ-opioid receptor antagonists, naloxone and β-CNA, on [35S]GTPγS binding to membranes prepared from GH3MOR cells chronically treated with either no opioid, morphine, or DAMGO. Prolonged exposure to DAMGO significantly increased basal [35S]GTPγS binding, suggesting that constitutively active μ-opioid receptors were present and spontaneously activated G proteins in the absence of agonist. This was confirmed by the finding that chronic treatment of GH3MOR cells with either DAMGO or morphine converted the μ-opioid receptor antagonists naloxone and β-CNA into inverse agonists. Both ligands produced a concentration-dependent reduction of basal [35S]GTPγS binding. Furthermore, in cells chronically exposed to DAMGO, the inverse agonism was reversed by the neutral μ-opioid antagonist CTAP. This indicates that the inverse agonism of both ligands was mediated specifically through action at μ-opioid receptors. Additionally, because a maximal concentration of CTAP produced no effect on [35S]GTPγS binding when administered alone, it is unlikely that the reduction in [35S]GTPγS binding produced by naloxone and β-CNA after chronic opioid administration was simply caused by an antagonism of the stimulation of [35S]GTPγS binding produced by residual morphine or DAMGO used for pretreatment. This conclusion is further supported by the observation that maximal concentrations of naloxone or β-CNA produced no decrease in [35S]GTPγS binding after only a brief 30-min exposure of GH3MOR cells to 10 μM DAMGO. CTAP also demonstrated neutral antagonist activity in a previous study in which in SHSY5Y cells were chronically treated with morphine (Wang et al., 1994). Importantly, the maximal decrease in [35S]GTPγS binding in response to either inverse agonist was greater after exposure to the full agonist DAMGO, relative to the partial agonist morphine. This supports the initial hypothesis that chronic treatment with opioids converts μ-opioid receptors to a constitutively active state. However, it was quite unanticipated that this transition occurred in direct proportion to the efficacy of the opioid agonist used for pretreatment.
Conditions known to uncouple GPCRs from G proteins, such as the addition of guanine nucleotides and NaCl (Childers and Snyder, 1980), decrease the binding of agonists, have no effect on the binding of antagonists and enhance the binding of inverse agonists to GPCRs (Neilan et al., 1999). For example, the δ-opioid receptor inverse agonist ICI-174,864 exhibited a 7-fold increase in its affinity for δ-receptors in the presence of NaCl and GppNHp in transfected C6 glioma cells (Neilan et al., 1999). Similarly, in the present study, the addition of NaCl and GppNHp produced over a 3- and 13-fold leftward shift in the competition curve of β-CNA for [3H]DAMGO binding only in membranes prepared from cells chronically treated with morphine or DAMGO, respectively. In contrast, these conditions produced no shift in the competition curve by the neutral antagonist CTAP in cells exposed to chronic DAMGO. The leftward shift in the β-CNA competition curve by the inclusion of GppNHp and NaCl was significantly greater in membranes prepared from cells pretreated with the full agonist DAMGO, relative to the partial agonist morphine. Collectively, the results obtained from experiments examining both receptor binding and [35S]GTPγS binding indicate that the μ-opioid antagonist β-CNA displays properties of an inverse agonist after chronic exposure of GH3MOR cells to opioids. Because inverse agonism can be observed only if constitutively active receptors are present, this supports the original hypothesis that chronic opioid treatment increases the proportion of μ-opioid receptors in a constitutively active state. Unexpectedly, this conversion is greater after prolonged exposure to full, compared with partial, opioid agonists.
Although the mechanisms underlying these observations are not known, they are likely to involve differences between the adaptation of μ-opioid receptors to chronic exposure to agonists with different relative efficacies, such as morphine and DAMGO. For example, both acute and chronic administration of morphine and DAMGO produce distinct μ-opioid receptor complexes that are differentially sensitive to phosphorylation by protein kinases (Chakrabarti et al., 1998). Therefore, chronic morphine or DAMGO exposure could result in uniquely phosphorylated forms of the receptor, each producing differential levels of constitutive activity. In addition, full μ-opioid agonists produce greater amounts of receptor phosphorylation than partial agonists (Yu et al., 1997; Ferguson et al., 1998). This suggests a direct correlation may exist between the extent of μ-opioid receptor phosphorylation and the production of constitutive activity.
Constitutive activation of GPCRs is most readily observed in transfected cells containing high levels of receptors (Lefkowitz et al., 1993). However, GH3MOR cells express a relatively low, physiological density of μ-opioid receptors (i.e., 0.39 pmol/mg), similar to endogenous amounts reported in several brain regions such as the striatum (i.e., 0.30 pmol/mg) (Sim et al., 1996). Furthermore, constitutive activation of μ-opioid receptors was observed only after chronic opioid pretreatment, which produced a reduction in the number of receptors. In fact, greater constitutive activity occurred after chronic DAMGO than morphine pretreatment. This was interesting, because prolonged exposure to DAMGO resulted in a 2-fold larger reduction of μ-opioid receptor density than morphine. The fact that more constitutive activity was observed in membranes containing fewer available receptors provides further evidence that prolonged exposure to the full agonist DAMGO converts a greater proportion of the remaining μ-opioid receptors to a constitutively active state, relative to the partial agonist morphine.
No inverse activity of either naloxone or β-CNA was observed in naive GH3MOR membranes, indicating a lack of constitutive activity of μ-opioid receptors. Although another recent report is in agreement with our observations (Neilan et al., 1999), two other studies showed that β-CNA acted as an inverse agonist in opioid naive human embryonic kidney 293 cells transfected with μ-opioid receptors (Wang et al., 1999; Burford et al., 2000). There are several possible explanations for the differences between these studies. First, the μ-opioid receptor density in studies where constitutive activity was observed was much higher than in GH3MOR cells (∼4.0 versus 0.39 pmol/mg). Second, β-CNA and naloxone may possess only partial inverse agonist activity and thus lack the efficacy to detect μ-opioid receptor constitutive activity in GH3MOR cells. Third, unknown factors have been suggested to suppress basal δ-opioid receptor activity in some cell lines (Chiu et al., 1996). Calmodulin might represent one such factor that reduces constitutive activity of μ-opioid receptors (Wang et al., 2000). Therefore, GH3MOR cells might contain higher levels of calmodulin or other unknown suppressive factors than cell lines in which μ-opioid receptor constitutive activity has been observed. Lastly, the assays used here might lack the sensitivity needed to detect low levels of inverse activity.
The potentiation of forskolin-stimulated cAMP accumulation by naloxone after chronic DAMGO treatment was observed in GH3MOR cells; this adaptive process is thought to represent a cellular model of withdrawal (Sharma et al., 1975; Yu et al., 1990; Nestler et al., 1993). Understanding the biochemical basis of cAMP rebound might lend insight into the mechanisms of opioid tolerance and dependence. The enhancement of adenylyl cyclase activity after chronic opioid withdrawal requires Gsαbut also involves stimulation of specific isoforms of adenylyl cyclase by Gβ γ subunits released form inhibitory G proteins (Avidor-Reiss et al., 1996; Ammer and Schulz, 1998). Additionally, constitutive activation of μ-opioid receptors after chronic opioid treatment might also contribute to cAMP rebound (Wang et al., 1994). If increased constitutive activation of μ-opioid receptors occurs in response to chronic opioid treatment, enhanced spontaneous coupling of receptors to Gi/Goα proteins and an augmented suppression of adenylyl cyclase might occur. Naloxone, as an inverse agonist, could relieve the constitutive suppression of the enzyme leading to a rebound of cAMP levels.
Whistler et al. (1999) recently suggested that the relative activity versus endocytosis value for individual opioids was predictive of the potential to produce tolerance and/or dependence. Therefore, drugs such as morphine that demonstrate relatively good efficacy but have a very poor ability to produce desensitization and/or endocytosis have a high relative activity versus endocytosis value and may readily promote physiological tolerance. In light of this interesting new hypothesis, two observations provided by the present study should be considered. First, rather surprisingly, morphine produced approximately half as much down-regulation of μ-opioid receptors as the full agonist DAMGO (33.5 versus 68.7%). This relatively pronounced effect suggests that the ability of morphine (and other potential candidate drugs) to promote endocytosis may vary significantly from tissue to tissue. Second, the apparent conversion of μ-opioid receptors to a constitutively active state upon chronic opioid exposure may also contribute to the development of dependence (Wang et al., 1994). If this is true, it is interesting that the full agonist DAMGO produced greater constitutive activity than the highly addictive partial agonist morphine.
In summary, it has been demonstrated that prolonged exposure to either morphine or DAMGO converted two μ-opioid antagonists into inverse agonists, indicative of constitutive activation of μ-opioid receptors. Surprisingly, chronic treatment with the more efficacious agonist DAMGO produced greater increases in both measures of inverse agonist activity than did morphine. These observations lend novel insight into the mechanisms of opioid tolerance and dependence.
Acknowledgments
We thank Nancy A. Martin and Xiao-Ping Liao (University of Arkansas for Medical Sciences) for their helpful discussions and expert assistance with word processing and graphic presentations.
Footnotes
-
This work was supported in part by National Institute on Drug Abuse Grant DA10936 (P.L.P.).
Abbreviations
- DAMGO
- [d-Ala2,N-MePhe4,Gly-ol5]enkephalin
- CTAP
- d-Phe-Cys-Tyr-d-Trp-Arg-Thr-Pen-Thr-NH2
- GppNHp
- 5′-guanylylimidodiphosphate
- IBMX
- 3-isobutyl-1-methylxanthine
- PTX
- pertussis toxin
- β-CNA
- β-chlornaltrexamine
- GTPγS
- guanosine 5′-O-(3-thio)triphosphate
- GPCR
- G protein-coupled receptor
- MOR
- μ-opioid receptor
- Received January 9, 2001.
- Accepted March 1, 2001.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}