Article Text

Abstract
The incidence of QT prolongation and torsades de pointes is on the rise due to the use of cardiovascular and non-cardiovascular drugs. Robust efforts have been made and are still ongoing to understand the underlying mechanisms that can enhance or prevent the development of drug-induced proarrhythmia. A caveat in the use of antiarrhythmic drugs is the ability to obtain safe action potential prolongation therapeutic effects, through IKr blockade. This remains as yet completely unachievable, as blockers of the potassium channel have not provided complete safe measures. Because of this, efforts at understanding the mechanisms of proarrhythmia have continued. PI3K/Akt signalling pathway appears to possess some potential advantage in this regard because cardiomyocytes intracellular dialysis with phosphatidylinositol (3,4,5)-trisphosphate (PIP3) normalises ion channel alterations and eliminates proarrhythmic features. However, there is a conundrum. Increased activities of PIP3 signalling can enhance cell proliferation and survival, and reduced activities of PIP3 signalling can lead to proarrhythmia. PI3K inhibitors used in cancer treatment have been found to cause proarrhythmia, and represent a potential avenue for the research and evaluation of potential effectiveness of a battery of antiarrhythmic and cancer drugs that are either currently in use or in development. Despite this knowledge, limited information is available on PI3K/Akt signalling and arrhythmogenesis. This highlights the need to search for new ways to improve testing of antiarrhythmic drugs and increase our understanding in PI3K/Akt signalling and arrhythmogenesis.
- arrhythmias
- ventricular fibrillation
- cardiac remodelling
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Torsades de pointes (TdP) is a distinct electrocardiographic type of polymorphic ventricular tachycardia that is rare. It is marked by QT interval prolongation and systematic changes in the QRS amplitude or rather twisting of QRS complexes around isoelectric line, meaning that the morphology of QRS complex varies from one beat to another. QT prolongation and TdP may be as a result of bradycardia, hypokalaemia, and heart diseases such as congestive heart failure, left ventricular hypertrophy and myocarditis. Its aetiology may be described as congenital, acquired or drug-induced. Secondary causes of drug-induced TdP include concomitant IKr blockade, metabolic inhibitors, undetected human ether-a-go-go-related gene (hERG), female gender, atrial fibrillation, congenital heart failure, left ventricular hypertrophy, hypokalaemia and bradycardia. The primary cause of drug-induced TdP is known to be IKr blockade. TdP high-risk drugs have their therapeutic effects linked to IKr blockade, and include dofetilide, ibutilide, quinidine, disopyramide and sotalol. Those drug poising at low risk of causing TdP have their therapeutic effects, perhaps independent of IKr blockade, and include antibiotics, antihistamines, psychotropics and antiviral agents. The management of TdP is different from ventricular tachycardia. Particularly, the use of class 1A antiarrhythmic drugs in TdP, which tend to prolong QT interval, can be catastrophic. Proarrhythmia can be defined as a predisposition to new arrhythmia following the administration of antiarrhythmic agents or pharmacological substances. Such substances when administered at a certain therapeutic dosage can cause new arrhythmia or maintain an existing one. The effect may be atrioventricular disturbance, ventricular or supraventricular arrhythmias. The incidence of QT prolongation and TdP following administration of some cardiovascular and non-cardiovascular drugs is on the rise.1 Nonetheless, the basic mechanisms of proarrhythmia are not known clearly, and several cardiovascular antiarrhythmic Ca2+ blockers, including lidoflazine and terodiline, were withdrawn from the market in the UK and Europe between 1980 and 1991. The risk of proarrhythmia poses a significant challenge in drug development and results in a worrying number of cardiovascular emergencies, and perhaps sudden cardiac deaths, through polymorphic and monomorphic ventricular tachycardia and ventricular fibrillation.
Basic mechanisms
The basic mechanisms of proarrhythmia are being elucidated. Interaction of the drugs with ion channels causes concomitant drug-induced TdP and QT prolongation. TdP is a cyclic shift in QRS axis, with repetitive polymorphic ventricular tachycardia, and it is consequent to delayed repolarisation, a typical feature of proarrhythmia, characterised by elongation of action potential duration (APD). Triangulation of action potential may be as a result of the APD prolongation. However, it is not an obligate sequela of APD prolongation, and there is no conclusive evidence that APD prolongation inevitably causes proarrhythmia. Triangulation may be regarded as linearisation of repolarisation. Linearisation as an analogous measure of action potential triangulation is the time interval from 30% to 90% repolarisation of the action potential. Reduced repolarisation currents lead to the linearisation (figure 1). This causes abnormal repolarisation and leads to the APD prolongation (figure 1A), early afterdepolarisation (EADs), re-entry, conduction abnormality, TdP and prolonged QT interval. Triangulation and spatiotemporary dispersion of ventricular repolarisation2 promote the TdP and QT prolongation.2 3 Spatial-temporal dispersion is the local and short-term variability in cardiac repolarisation. Spatial dispersion of ventricular repolarisation may result from variability of cardiac APD within transmural, trans-septal and apical regions of the myocardium.4 This may create vulnerable window current across myocardial ventricular wall and cause formation of EADs and TdP. In prolonged QT interval, accompanied by the EADs and TdP, the action potential morphology may be described as triangular or rectangular depending on the extent of alteration of APD50 and APD90. The QT interval represents ventricular repolarisation and depolarisation that occur for about 300–400 ms. The interval is indirectly proportional to heart rate. At a slow heart rate, the interval elongates, while at fast heart rate the interval shortens. The corrected QT interval (QTc) estimates the QT interval at a heart rate of 60 beats per minute. This allows comparison of ventricular depolarisation and repolarisation (QT) values over time at varied heart rates, and enhances detection of patients with increased risk of arrhythmias such as TdP. QTc can be calculated by Framingham’s newer formula. From this formula, QTcFra=QT+0.154 (1-RR), where RR interval=60/heart rate.
At the ionic level, reports by Coppini et al 5 and Soliman et al 6 used patch-clamp electrophysiology to demonstrate that the basic mechanisms may be associated with increased late INa. Increasing depolarising currents, such as the late INa and ICa,L, prolong APD. Prolongation of cardiac repolarisation also occurs by reduction in delayed rectifier potassium current—IKr (figure 1A,C). These ionic changes together produce APD prolongation, EADs, re-entry and TdP. The association between TdP formation and these mechanisms or the ionic remodelling may be complex, but may be basically explained in part by decreases in net outward potassium current (figure 1C). It is known that IKr inhibition by the drugs are proarrhythmic. Inherited channelopathies such as the long QT syndromes are also proarrhythmic because IKr could be reduced. Reduced IKr slows ventricular repolarisation (figure 1A) and prolongs the QT interval (figure 1B). This may result in suitable substrate and trigger EADs. If these EADs reach a threshold, they can give rise to premature ventricular beat and cause TdP by either a re-entrant mechanism with suitable substrate or by recurrent ectopy. The effects are diverse, and include decreased conduction velocity and increased refractoriness, as well as potentials to cause heterogeneous action potentialprolongation. The maintenance of trigger depends on the formation of EADs and delay after depolarization (DAD), while the maintenance of re-entry depends on substrate properties, with adequate balance between determinants of membrane excitability and refractory. EAD formation depends on action potential prolongation, while DAD formation depends on the degree by which ryanodine receptors release Ca2+ in response to transmembrane Ca2+ entry.
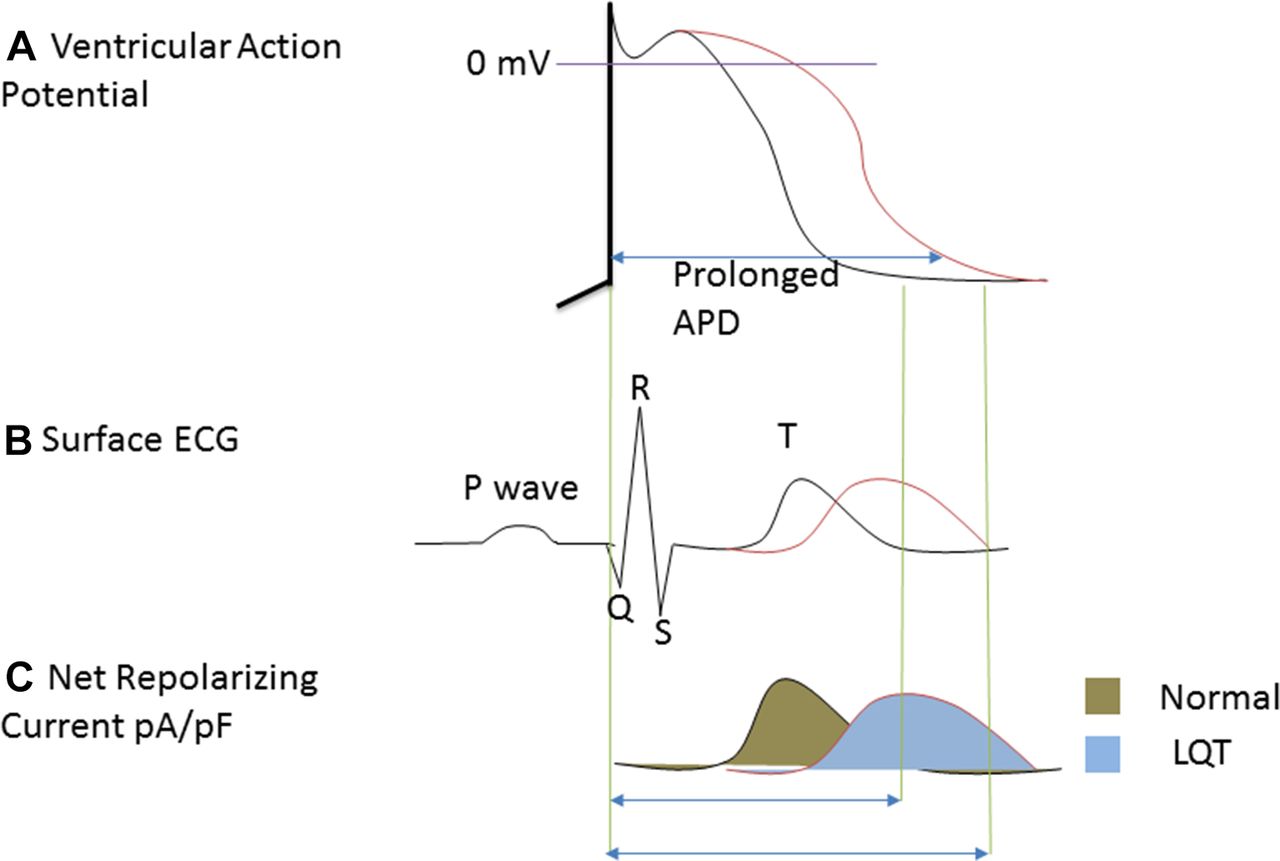
(A) Ventricular action potential depicting prolonged action potential duration (APD) at single-cell level. (B) Surface ECG showing prolonged QT interval at organ level as a result of prolonged APD. (C) Net reduction in current density of IKr.
IKr blockade
The aim in the use of antiarrhythmic drugs is to eliminate the underlying mechanisms discussed, which cause drug-induced proarrhythmia. Obtaining this goal is not usually the case because proarrhythmic APD prolongation may occur following administration of the antiarrhythmic drugs, which interact with ion channels and worsen or even cause arrhythmia, by facilitating re-entry and producing triggers. Non-cardiovascular drugs or commonly used drugs, including antibiotics, can as well cause proarrhythmia by blocking the IKr and elongating APD. In designing the antiarrhythmic drugs, a goal has been to obtain a therapeutic effect of APD prolongation, through blockade of the K+ channel, without initiating TdP arrhythmias.7 An approach in achieving this is by the inhibition of an augmenting ICa, together with hERG channel that carries the IKr. This is since APD prolongation can also be attributed to increased transmembrane inward currents such as INa and ICa, and repolarisation is governed by the activities of inward and outward ionic currents. Thus, integrating blockers of IKr with blockers of ICa protected against repolarisation collapse and preserved APD.8 However, IKr blockers can increase inward currents, such as late INa at the plateau phase of AP. This makes the goal completely unachievable, and the approach is therefore not a complete measure. However, it raised the expectation that blockers of late INa plus IKr together might also eliminate proarrhythmia, and it is clear that non-specific drugs such as amiodarone and verapamil appear to have less proarrhythmic effects than highly specific IKr blockers like dofetilide.
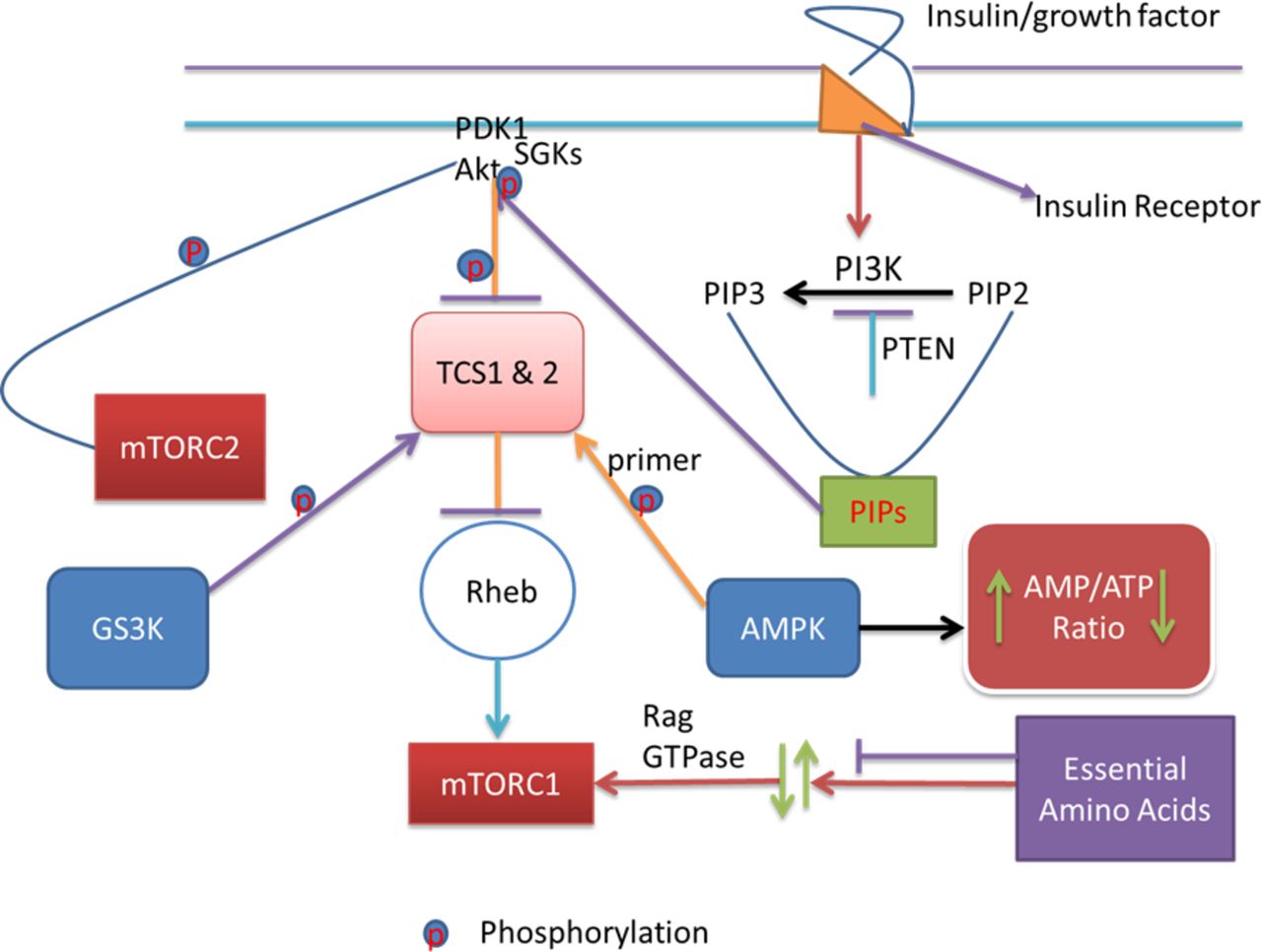
QT prolongation, TdP and drug-induced long QT syndrome (diLQTS) caused by using the antiarrhythmic agents/IKr blockers are the major causes of drug relabelling and withdrawals.1 9 Targeting proarrhythmia whether supraventricular, ventricular or both is a major challenge in cardiovascular interventions. Functional roles of PI3K/Akt pathway (figure 2) have not been clearly elucidated in arrhythmogenesis. The functional role of the pathway (figure 2) is contrasting to cardiovascular and cancer biologists. The pathway regulates persistent cell growth, proliferation and survival through phosphatidylinositol (3,4,5)-trisphosphate (PIP3) activation of downstream signalling effectors such as Akt. Akt activation stimulates downstream anabolic signalling required for the cell growth, proliferation and survival. Whereas this function may be detrimental in cancer management, it may be advantageous in cardiovascular interventions because persistent cell proliferation and survival is a feature the cardiovascular biologists have always searched for to improve in other to alleviate cardiac cellular damage. Conversely, inhibiting the function of the pathway through reduction in PIP3 signalling may be advantageous in cancer management, where cell proliferation and growth malignancy are the disease mechanisms. In this line, tyrosine kinase inhibitors (ibrutinib and nilotinib) highlight the protective effects of PI3K/Akt signalling pathway (figure 2) in cancer treatment, but possess a potential threat for the occurrence of atrial fibrillation and diLQTS.10 11

{kind=link}
{kind=link}
The pathway signalling starts with the binding of insulin or growth factors to insulin receptors. This binding in turn activates PI3K. Activated PI3K converts PIP2 to PIP3. These PIPs then mop up PDK1 and Akt to the cell membrane. When PDK1 and Akt are taken to the cell membrane, Akt gets activated and phosphorylated. Phosphorylation and activation of Akt phosphorylate and inactivate TSC1 and TSC2. The consequence of this in turn is the activation of Rheb and mTOR1, part of the two functional complexes of mTOR. At this point, AMPK, an energy sensor that detects changes in the intracellular ATP/AMP ratio, comes in and directly phosphorylate TSC2. This phosphorylation is very significant as it conditions TSC2 for further phosphorylation by GSK3. Further phosphorylation of TSC2 by GSK3 inhibits mTOR1. Amino acid availability activates mTORC1 via Rag GTPases. The PI3K downstream effectors including Akt, PIP3, mTOR, GSK3 and PDK1 regulate cell growth, proliferation and survival. Akt, PIP3, GSK3 and PDK1 downstream of PI3K effect the ion channels. The tyrosine kinase inhibitors that block the ion channels and/or enhance cancer management act on some of these effectors and effect their signalling, thereby causing cardiovascular toxicity and APD elongation, through interactions with the ion channels in the heart, conversely. APD, action potential duration; GSK3, glycogen synthase kinase 3; PDK1, phosphoinositide-dependent kinase 1; PIP3, phosphatidylinositol (3,4,5)-trisphosphate; TSC, tuberous sclerosis.
Modulation of PI3K/Akt pathway
The incidence of QT prolongation and TdP may be as a result of inhibition of PIP3 signalling, reduction in IKr and increase in late INa. Anticancer drugs that inhibit IKr appear to reduce the PI3K-Akt/PIP3 signalling and cause proarrhythmia.11 Chronic exposure to nilotinib prolonged APD, by decreasing IKr, increasing late INa and by decreasing PI3K.12 Such exposure may cause interaction of the drugs with ion channels in a manner that might affect channel biophysics and result in enhanced channel recovery from activation, reduced channel deactivation and a positive shift in channel availability. Cardiac metabolic-induced arrhythmia appears to have similar effects. Hyperglycaemic-induced QT prolongation reduced PIP3 signalling, increased late INa and caused APD prolongation in diabetes.13 14 These observations recapitulate the challenges in the use of the channel blockers and predict drug relabelling and withdrawals. It is underlining for advancement in the observations that blockers of late INa plus IKr might eliminate the proarrhythmic features. It is therefore apparent that mechanisms of proarrhythmia demand better understanding and should be elucidated beyond the available evidence.
With the premise in mind that blockers of IKr, similar to tyrosine kinase inhibitors, may be inhibiting PIP3 signalling, thereby increasing late INa and initiating proarrhythmias, Roden’s group recently examined the effects of acute and chronic exposure to dofetilide on late INa in mouse cardiomyocytes, human-induced pluripotent stem cell-derived cardiomyocytes and Chinese hamster ovary (CHO) cells.15 The mouse cardiomyocytes lack IKr, but have the late INa. The human-induced pluripotent stem cell-derived cardiomyocyte has both IKr and the late INa. The CHO cells have transfected SCN5A carrying the human cardiac sodium current, but lack the IKr. These manipulations allowed the authors to examine the effects of the channel blockers when the late INa was either present or absent. They observed that prolonged APD resulted in early and delayed afterdepolarisations, following chronic exposure of dofetilide to the human and mouse cardiomyocytes. This observation, although surprising in the mouse which lacks IKr, indicates that other membrane excitable mechanisms, like an alteration in late INa, might be involved. They then measured the late INa in the CHO, human and mouse cardiomyocytes and found increased late INa with no changes in the expression of SCN5A that carries the current. They attributed the increase in late INa to the activation and inactivation kinetics of the Nav1.5, instead of alterations in the Nav1.5 expressions. They also observed that non-cardiovascular drugs like haloperidol, thioridazine and erythromycin and other IKr blockers except moxifloxacin and verapamil increased the late INa. The increased late INa following chronic exposure to 1 μmol/L of dofetilide, but not to 100 μmol/L of moxifloxacin, was evidently reversed to the levels of controls by PIP3 intracellular dialysis.15 Consistently, ATX II, a peptide toxin that binds to voltage-gated sodium channel, increased the late INa in a PIP3-independent fashion. The result indicates that the kinase inhibitors and blockers of IKr act by decreasing PIP3 signalling to produce proarrhythmic features. Thus, normalising PIP3 signalling may be a potential approach to addressing proarrhythmia. Whether this would completely address alterations in other ion channels including calcium ion channels implicated in proarrhythmia is not completely clear. It also remains to be elucidated whether IKr blockers enhance the kinase activity.
Therefore, PI3K/Akt signalling pathway (figure 2) appears to offer some potential approach in addressing proarrhythmia. This makes it valid that understanding the signalling pathway better in arrhythmogenesis is critically important. Since the pathway regulates cell proliferation and survival through PIP3 activation of downstream signals, infusion of PIP3 at supraphysiological levels may cause abnormal cell growth that may be harmful to the myocardium. Apparently, while reduced activities of PIP3 signalling may cause proarrhythmia, increased activities of PIP3 signalling may enhance cell proliferation and survival and cause cancer development. Since PI3K produces PIP3 in the pathway, and PI3K inhibitors have been used in cancer treatment, reduced PIP3 production may be in part why PI3K inhibitors cause proarrhythmia.11 12 This requires extensive safety studies to be conducted to understand the interplay between the pathway and arrhythmogenic mechanisms.
Limited literature is available on this subject,14 and it is known that PI3K might regulate cardiac ion channels in arrhythmogenesis. With the focus on the molecular mechanisms evident, as so far discussed, it is known that the pathway might be very crucial in addressing proarrhythmia. This would enable identification of novel roles played by the kinase in the heart. What is now lacking is how to control, influence and direct the outcome of activation and target substrate phosphorylation of the pathway. It is therefore the aim of this review that an optimum strategy to target PI3K/Akt signalling pathway in cardiac protection is required. This underscores the importance of this work, calling for evaluation of the pathway in proarrhythmia. This work has succinctly summarised the advances made in our understanding of PI3K and proarrhythmia. An ability to dissect the roles of the pathway in proarrhythmia using in vivo genetic manipulations will confirm some of the predictions made by Ballou et al and increase the pace for serendipitous insights needed in this subject. PI3K/Akt signalling site-directed ion channel phosphorylation, together or rather with channels inhibitors, is recommended. In 2004, such sites were identified in neurons. Ser574A and ser574E phosphorylation on calcium channel (Cav)β2a that prevented and mimicked the effect of PI3K/Akt essentially and significantly promoted Cav channel plasma membrane translocation and expression.16 Enhanced or decreased membrane expression of ion channels has been a parsimonious explanation of the pathways downward or upward regulation of ion currents. Because membrane insertion of ion channels by PI3K controls membrane excitability, cardiac safety research should therefore importantly investigate in expanded details prolonged effects of new and candidate drugs on a number of cardiac excitable determinants. Expanding the roles of the pathway in membrane excitability will offer opportunities in appreciating the roles of the pathway in the mechanism of proarrhythmia, in developing it as a target to attenuate proarrhythmia and in screening for tyrosine kinase drug development in its current platform for drug discovery.
Summary
Clinical practice, clinical pharmacology, cardiac electrophysiology and genetics have raised awareness of incidence of QT prolongation and TdP. Biotechnology and pharmaceutical companies screen compounds for kv11.1 channel activity early during preclinical safety trials/evaluation, thus recognising the high risk for QT prolongation and TdP with potential pharmacological substances. Cellular and molecular mechanisms of QT prolongation are incompletely understood. QT interval is evaluated from the start of ventricular depolarisation (QRS complex) to the end of ventricular repolarisation (T wave), meaning that both repolarisation and conduction of the ventricle affect QT interval. Delayed ventricular repolarisation at phase 3 of APD has a major effect in causing QT prolongation. Drug interaction, upset of electrolyte imbalance, heterogenicity of the myocardium and genetic polymorphism can cause QT prolongation; however, a well-known mechanism of QT prolongation is the reduction in IKr current either by abnormal trafficking of Kv11.1 channel to cell surface or by blockade of the channel activity. This is the accepted view of the mechanism of diLQTS. However, as we learn from emerging evidence, and since diLQTS, particularly those caused by tyrosine kinase inhibitors, could be due to inhibition of PI3K signalling, a more current view should be that inhibition of the hERG only is not sufficient to detect risk for QT prolongation and TdP.
References
Footnotes
Contributors ME wrote the MS; SE organised and edited.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Additional data and information can be obtained by contacting the corresponding author.