Article Text

Abstract
A 50 year old woman with a previous diagnosis of epilepsy presented to the emergency department with a generalised seizure. Her admission ECG showed QT prolongation secondary to bradycardia and a subsequent seizure in the department demonstrated that these events were secondary to cerebral hypoperfusion during episodes of torsades de pointes. This case illustrates how long QT syndrome can masquerade convincingly as epilepsy, delaying treatment and exposing the patient to a high risk of sudden cardiac death. Careful ECG analysis is recommended for all patients presenting with seizures.
- Arrhythmia
- ECG
- Long QT Syndrome
- Torsades de pointes
Statistics from Altmetric.com
A 50 year old woman presented to the emergency department with a seizure. Over the past 18 months she had experienced five similar episodes. On each occasion she collapsed with loss of consciousness, and then subsequently developed rigid flexor posturing with loss of urinary continence. Typically she would regain consciousness within several minutes with no residual neurological deficit. A diagnosis of epilepsy was made by her general practitioner on the basis of this history and a normal computed tomogram (CT) of the head, and she was treated with sodium valproate. She and her family were otherwise fit and well.
On admission the only abnormality on physical examination was bradycardia of 40 beats per minute. Routine admission blood tests including serum potassium and magnesium were normal and a chest x ray was unremarkable.
Her admission electrocardiogram (ECG) was markedly abnormal (fig 1A). It showed complete heart block with a ventricular rate of 40 beats per minute. The escape rhythm showed a partial right bundle branch block pattern. Most significantly, the corrected QT interval was highly prolonged at 0.64 seconds with broad, tall T waves.

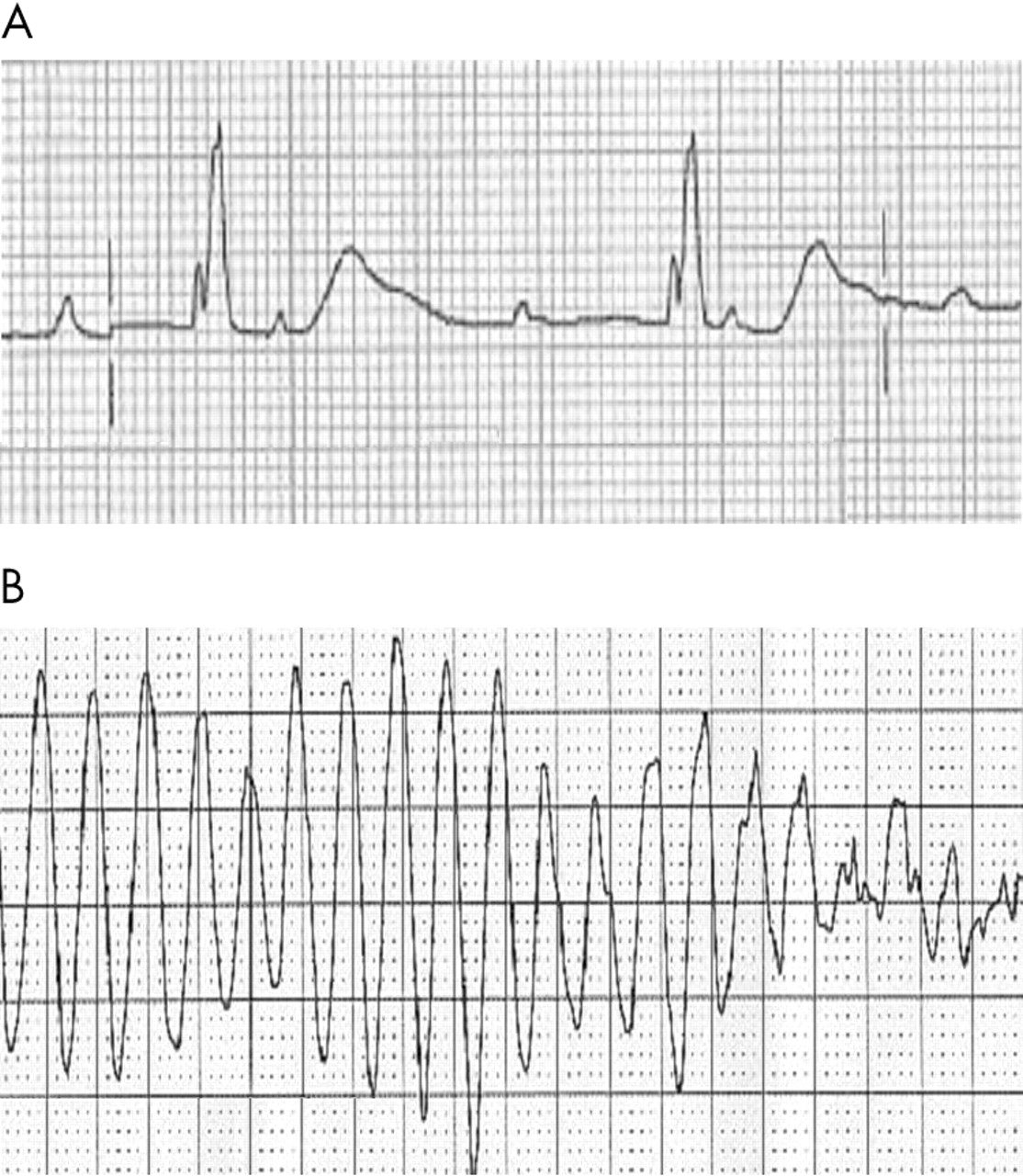{kind=link}
(A) Admission ECG (lead V3) and (B) torsades de pointes.
The patient was admitted to the coronary care unit for continuous monitoring and temporary pacing. While the procedure was being explained to the patient, she developed a VT arrest which was terminated by DC cardioversion. She lost consciousness at the onset of the arrest and subsequently developed a seizure characterised by rigid flexor posturing and loss of urinary continence. Her rhythm during the episode shows the classic features of torsades de pointes, with a short–long–short initiation sequence and a polymorphic ventricular tachycardia (fig 1B). Her seizures were secondary to cerebral hypoxia during episodes of torsades.
She subsequently underwent successful temporary pacing and was transferred to a tertiary cardiology centre where a DDD pacemaker was inserted which corrected the bradycardia and secondary QT prolongation. Further investigations to elucidate the cause of her complete heart block were negative (echocardiography, Borrelia serology, sarcoid and infiltrative disorder screen). She has since experienced no further syncopal episodes. Her daughters were contacted and their ECGs show no QT prolongation.
DISCUSSION
The long QT syndrome represents a variety of congenital and acquired disorders of ventricular repolarisation characterised by a prolonged corrected QT interval (Bazett’s formula: QTc = QT/√RR) on the electrocardiogram.1 It is associated with a life threatening polymorphic ventricular tachycardia known as torsades de pointes (twisting of points).2 The QT interval on the ECG reflects the length of ventricular action potential and an interval over 0.44 seconds should be considered prolonged.3,4 A common presentation is syncope or sudden cardiac death.3 Initial presentation with epileptic seizures is less well recognised, especially in adults. The commonest causes of acquired QT prolongation are drugs, hypokalaemia, and hypomagnesaemia. Bradycardia is a rare but recognised cause of QT prolongation. Treatment is directed at the underlying cause.
Our case illustrates how long QT syndrome can masquerade convincingly as epilepsy, delaying both diagnosis and treatment, thereby exposing the patient to a high risk of sudden cardiac death. It is therefore important that long QT syndrome should feature in the differential diagnosis of seizures, particularly if there are atypical features or if there is no response to antiepileptic medications.5,6
A thorough history is important in the identification of these patients. There is often a precipitating factor for the seizures, such as loud noises or an adrenergic surge, and typically consciousness is lost for a brief period of time before seizures begin, as in our patient.3,7 A family history of sudden unexplained death or deafness also suggests congenital long QT syndrome, and recent introduction of new drugs, in particular antiarrhythmics may be an acquired cause of QT prolongation. Careful analysis of the 12-lead ECG at the time of admission is important to identify these patients. The ECG changes may be subtle and prolongation of the QT interval can be easily overlooked.
Our case also demonstrates the need for caution when explaining potentially frightening diagnoses and procedures to patients with QT prolongation, since torsades de pointes can be precipitated by adrenergic surges, especially in patients with the congenital form of the disease.3,5,8 β Blockade may reduce this risk.3
Footnotes
-
Competing interests: none declared