

Abstract
Amyotrophic lateral sclerosis (ALS) is a rapidly evolving and fatal adult-onset neurological disease characterized by progressive degeneration of motoneurons. Our previous study showed that glycinergic innervation of spinal motoneurons is deficient in an ALS mouse model expressing a mutant form of human superoxide dismutase-1 with a Gly93→Ala substitution (G93A–SOD1). In this study, we have examined, using whole-cell patch-clamp recordings, glycine receptor (GlyR)-mediated currents in spinal motoneurons from these transgenic mice. We developed a dissociated spinal cord culture model using embryonic transgenic mice expressing enhanced green fluorescent protein (eGFP) driven by the Hb9 promoter. Motoneurons were identified as Hb9–eGFP-expressing (Hb9–eGFP+) neurons with a characteristic morphology. To examine GlyRs in ALS motoneurons, we bred G93A–SOD1 mice to Hb9–eGFP mice and compared glycine-evoked currents in cultured Hb9–eGFP+ motoneurons prepared from G93A–SOD1 embryos and from their nontransgenic littermates. Glycine-evoked current density was significantly smaller in the G93A–SOD1 motoneurons compared with control. Furthermore, the averaged current densities of spontaneous glycinergic miniature IPSCs (mIPSCs) were significantly smaller in the G93A–SOD1 motoneurons than in control motoneurons. No significant differences in GABA-induced currents and GABAergic mIPSCs were observed between G93A–SOD1 and control motoneurons. Quantitative single-cell reverse transcription-PCR found lower GlyRα1 subunit mRNA expression in G93A–SOD1 motoneurons, indicating that the reduction of GlyR current may result from the downregulation of GlyR mRNA expression in motoneurons. Immunocytochemistry demonstrated a decrease of surface postsynaptic GlyR on G93A–SOD1 motoneurons. Our study suggests that selective alterations in GlyR function contribute to inhibitory insufficiency in motoneurons early in the disease process of ALS.
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by progressive loss of motoneurons (Rowland and Shneider, 2001). Approximately 10% of ALS cases are familial ALS (fALS) with inheritance patterns, and 90% are sporadic ALS with no known genetic component. Autosomal dominant mutations in the Cu/Zn superoxide dismutase-1 (SOD1) gene occur in ∼20% of fALS cases (Rosen et al., 1993). Transgenic mice expressing human mutated SOD1 genes develop fatal motoneuron disease resembling ALS in humans (Gurney et al., 1994).
The mechanisms causing motoneuron death in ALS are still not understood. Evidence supports the contributions of glutamate receptor-mediated excitotoxicity in human ALS and animal models of ALS (Plaitakis, 1990; Rothstein et al., 1992; Heath and Shaw, 2002). Excessive synaptic excitation might mediate some hyperexcitability (Pieri et al., 2003a,b; van Zundert et al., 2008); however, insufficient synaptic inhibition could be important too. Spinal cord slice cultures from mutant SOD1 transgenic mice showed an imbalance between excitatory and inhibitory innervation (Avossa et al., 2006). Glycine and GABA are the two main inhibitory neurotransmitters in the CNS that activate different ionotropic receptors permeable to chloride ions. Spinal cord motoneurons express glycine receptors (GlyRs) and GABAA receptors (GABAARs) that modulate motoneuron excitability (Rekling et al., 2000). We found that transgenic ALS mice develop an age-related loss of glycinergic innervation of motoneurons, whereas GABAergic innervation is mostly spared (Chang and Martin, 2009). In serum or autopsy tissues of ALS patients, abnormal glycine and GABA levels were observed (Malessa et al., 1991; Niebroj-Dobosz and Janik, 1999) and glycine binding sites have been reported to be reduced in anterior horn (Hayashi et al., 1981; Whitehouse et al., 1983). However, the relationships between changes in inhibitory synaptic function and mechanisms of motoneuron degeneration in ALS are still unknown in part because of the paucity of studies that analyze motoneuron physiology in cell models of ALS.
Motoneuron physiology can be studied in primary cultures of spinal cord neurons, but limitations arise from the difficulty of identifying motoneurons in mixed spinal cord cultures. Motoneurons in fixed spinal cord cultures can be identified using antibodies to nonphosphorylated neurofilament SMI-32 and acetylcholine-synthesizing enzyme choline acetyltransferase (ChAT) (Carriedo et al., 1995; Richards et al., 1995). However, no specific criteria for motoneurons maintained in live cultures are available. In this study, we used transgenic mice expressing enhanced green fluorescent protein (eGFP) driven by the Hb9 promotor (Wichterle et al., 2002) to identify a population of motoneurons in mixed spinal cord cultures. Hb9 is a homeodomain transcription factor that is expressed by embryonic motoneurons and functions during development to consolidate motoneuron identity (Arber et al., 1999; Thaler et al., 1999), but its specificity as an exclusive motoneuron marker is debatable (Wilson et al., 2005). We showed that Hb9–eGFP labels a subset of living neurons that had morphological characteristics of motoneurons in dissociated spinal cord cultures. We then examined functionality of GlyRs and GABAARs in spinal motoneurons from Hb9–eGFP/mutant SOD1 double-transgenic mice.
Materials and Methods
Transgenic mice.
Transgenic mice expressing a human mutant SOD1 gene encoding the glycine/alanine substitution at codon 93 (G93A) driven by the human SOD1 promoter (Gurney et al., 1994) and B6.Cg-Tg (Hlxb9–gfp)1Tmj/j transgenic mice expressing eGFP driven by the mouse Hlxb9 (Hb9) promoter (Wichterle et al., 2002) were originally obtained from The Jackson Laboratory and then housed in our animal facilities. The G93A–SOD1 transgenic mice with a high copy number of mutant allele (∼20 copies) and a rapid disease onset were used. The mice first show signs of spasticity at ∼10 weeks of age and then unilateral or bilateral hindlimb paresis at ∼11–12 weeks of age; the disease then progresses to end stage when the mice are quadriplegic at ∼16 weeks of age (Gurney et al., 1994; Martin et al., 2007). The institutional Animal Care and Use Committee approved the animal protocols. Every effort was made to minimize the number of animals used and their suffering.
Tissue preparation and fluorescence immunocytochemistry.
Male and female Hb9–eGFP mice were used for histological confirmation of eGFP expression in spinal motoneurons. Hb9–eGFP-expressing (Hb9–eGFP+) neurons were studied in spinal cord sections in embryonic, early postnatal, and adult mice. Embryos were removed from dams by cesarean section and immersion fixed in 4% paraformaldehyde. Postnatal mice were overdosed with sodium pentobarbital (50 mg/kg) and subsequently perfused transcardially with ice-cold 100 mm PBS, pH 7.4, followed by 4% paraformaldehyde. All mice were perfused under identical conditions by the same individual. Spinal cords were postfixed in paraformaldehyde for 3 h and then were cryoprotected in 20% glycerol overnight. Transverse serial sections (40-μm-thick) through lumbar spinal cord were cut on a sliding microtome (American Optical) and stored individually in 96-well plates in cyroprotectant buffer (1% polyvinylpyrrolidone, 40% ethylene glycol, and 0.1 m potassium acetate, pH 6.5) at −20°C until used for immunocytochemistry.
Free-floating sections were rinsed in Tris-buffered saline (TBS), pH 7.4, permeabilized with 0.4% Triton X-100, blocked with 10% donkey serum, and then incubated in a mixture of primary antibodies diluted in TBS containing 2% donkey serum and 0.1% Triton X-100 for 48 h at 4°C. The primary antibodies used in different combinations were as follows: mouse anti-glutamic acid decarboxylase 67 (GAD67) (monoclonal, 1:10,000; Millipore Corporation); guinea pig anti-glycine transporter-2 (GlyT2) (polyclonal, 1:10,000; Millipore Corporation); goat anti-ChAT (polyclonal, 1:200; Millipore Corporation); and mouse anti-calbindin (monoclonal, 1:200; Sigma). These antibodies were used to visualize GABAergic terminals, glycinergic terminals, motoneurons, and subsets of interneurons including Renshaw cells, respectively (Chang and Martin, 2009). After incubation in primary antibodies, the sections were washed extensively with TBS and then were incubated for 2 h at room temperature in a mixture of species-specific secondary antibodies (all raised in donkey) conjugated to Alexa Fluor 488, Alexa Fluor 594, and/or Alexa Fluor 647 (Invitrogen). Sections were washed again and mounted using Prolong anti-fade medium (Invitrogen).
Cell culture.
To obtain embryos for spinal cord culture, G93A–SOD1 mice were mated with Hb9–eGFP mice. Breeder pairs were screened for the presence of the transgenes by PCR on tail DNA. On embryonic day 12 (E12) to E14, female mice with potential double-transgenic G93A–SOD1/Hb9–eGFP embryos and single-transgenic Hb9–eGFP or G93A–SOD1 embryos were anesthetized with isoflurane, and all embryos were harvested by caesarean section. Hb9–eGFP expression in embryos was confirmed under a fluorescence microscope. Primary cultures were obtained from total spinal cords of male and female Hb9–eGFP+ embryos. Each spinal cord was cultured individually, and each embryo was genotyped by PCR for the human SOD1 gene using genomic DNA isolated from the body. The spinal cord was dissected and incubated for 30 min in 0. 025% trypsin and then dissociated by gentle trituration. The resulting mixed cultures were plated on poly-d-lysine/laminin-coated glass coverslips (12 mm in diameter; BD Transduction Laboratories) and maintained in DMEM supplemented with 5% fetal bovine serum and 5% horse serum (Invitrogen). At 4–6 h after plating, the medium was replaced with Neurobasal medium supplemented with B-27 (Invitrogen). On the second day after plating, uridine and 5-fluoro-2-deoxyuridine were added to the culture medium to inhibit the proliferation of non-neuronal cells. The motoneurons were used for experiments between 12 and 16 d in vitro (DIV). At this age in culture, the motoneurons are fully mature with an inhibitory action of the neurotransmitter glycine or GABA (Jackson et al., 1982; Nicola et al., 1992).
In experiments using chimeric embryonic cultures from mixtures of mutant SOD1/Hb9–eGFP and non-mutant SOD1/Hb9–eGFP transgenic mouse spinal cords, individual motoneurons were captured after acquiring recordings. Individual motoneurons were genotyped by single-cell PCR for the human SOD1 gene after whole-genome amplification using a GenomePlex single-cell whole-genome amplification kit (Sigma).
Electrophysiology.
The Hb9–eGFP+ neurons were identified under fluorescent microscope and then recorded under differential interference contrast (DIC) scope. Whole-cell patch-clamp recordings were made with glass pipettes pulled on a P-97 electrode puller (Sutter Instruments). The resistance of the pipettes was 2–4 MΩ when filled with an intracellular solution (in mm: 120 CsCl, 21 tetraethylammonium chloride, 2 MgCl2, 10 HEPES, 10 EGTA, 2 Mg-ATP, 0.3 NaGTP, and 10 phosphocreatine, pH 7.25 adjusted with CsOH). Cells were superfused at a rate of 2 ml/min with an external bath solution containing the following (in mm): 150 NaCl, 2.5 KCl, 10 HEPES, 10 d-glucose, 2 CaCl2, and 1 MgCl2, pH 7.3–7.4. Experiments were performed at room temperature (22–24°C) and in the presence of tetrodotoxin (TTX) (0.5 μm).
Currents from neurons were monitored with an Axopatch 200B amplifier (Molecular Devices) and acquired through Digidata 1440A (Molecular Devices) onto a computer using pClamp 10 software (Molecular Devices). Peak amplitudes were measured using Mini Analysis software (Synaptosoft). To assess the differences between motoneurons independently of cell size, the glycine or GABA current recorded for each motoneuron was normalized with respect to the whole-cell surface area derived from measurements of the membrane capacitance. At least four cultures were used for the measurements of each parameter given in Results, and four to six motoneurons were recorded from each culture. All recordings were made blinded of human SOD1 genotype.
Glycine or GABA was applied through a small tipped (∼1 μm) pipette that was moved under visual control to within ∼50 μm of the soma of the neuron under study. The glycine or GABA solution was pressure ejected (100 ms, 1–2 psi) directly onto the cell body and proximal dendrites using a Picospritzer (General Valve). The pipette solution contained 1 mm glycine or 100 μm GABA dissolved in extracellular solution as described above. Under physiological conditions, GABA or glycine is present at the postsynaptic receptor for durations in the millisecond range (Beato, 2008). Most Cl− current during IPSCs flows after removal of GABA or glycine from the synaptic cleft (Jones and Westbrook, 1995). The physiological situation of the postsynaptic site of the synapse was mimicked by experiments with application of short (100 ms) agonist pulses to GABAAR channels or GlyR channels.
For the recording of glycinergic miniature IPSCs (mIPSCs), 0.5 μm TTX, 5 μm 6-cyano-7-nitroquinoxaline-2, 3-dione (CNQX), 50 μm dl-2-amino-5-phosphonovalerate (APV), and 5 μm bicuculline were added to bath solution to block action potential, glutamatergic, and GABAergic activities. For the recording of GABAergic mIPSCs, 5 μm bicuculline was substituted with 0.5 μm strychnine to block glycinergic activity. Recordings were performed for enough time to observe at least 100 events. Currents were analyzed using Mini Analysis software. mIPSCs were detected with a threshold amplitude of 5 pA (Mini Analysis) and verified by eye. Peak amplitude, rise time from 10 to 90% of the peak amplitude, and decay time from peak to 37% of the peak amplitude were measured for each event. For each continuous recording, frequency histograms of peak amplitudes, rise times, and decay times were computed, and the mean values were used to characterize the synaptic activity. The statistical difference between these graphs was determined by the Kolmogorov–Smirnov test (Mini Analysis).
All chemicals used for electrophysiological recordings were purchased from Sigma. All drugs and solutions were made fresh from drug stock solutions.
Quantitative single-cell reverse transcription-PCR.
Single-cell reverse transcription (RT) was performed on spinal cord Hb9 motoneurons and Hb9 interneurons maintained in dissociated cultures. The cytoplasm of individual Hb9 neurons was aspirated into sterile RNase-free patch pipettes, in some instances after electrophysiological recordings. The recording intracellular solution was prepared with RNase-free water and sterilized, and RNaseOUT RNase inhibitor (0.1 U/μl; Invitrogen) was added. The contents of the pipette (∼2 μl) were immediately expelled into the bottom of a chilled PCR tube containing 11 μl of RT reaction mixture (1 μl 5% NP-40, 300 ng of random primer, 1 μl of 10 mm dNTPs; Invitrogen). The tubes were kept frozen in an ethanol/dry ice bath, and RT was performed at the end of the day. Cells were sonicated at 4°C (ice water) for 5 min to promote lysis. Mixtures were heated at 65°C for 5 min and incubated on ice for at least 1 min; then 4 μl of 5× first-strand buffer, 1 μl of 0.1 m DTT, 40 U of RNaseOUT RNase inhibitor, and 200 U of Superscript III reverse transcriptase (Invitrogen) were added. The RT reaction (final volume, 20 μl) proceeded for 5 min at 25°C, then 1 h at 50°C, followed by inactivation at 70°C for 15 min. Finally, 1 μl of Escherichia coli RNase H was added at 37°C for 20 min to remove RNA complementary to the cDNA.
Real-time PCR quantification of GlyRα1 subunit (Glra1) mRNA in individual motoneurons or interneurons was performed using the TaqMan technique. As an endogenous control for normalization, PCR reaction was performed concurrently with amplification of a housekeeping gene β-actin (Calvo et al., 2008) in the same tube. GlyRα1 primer/probe (TaqMan Gene Expression Assay) and β-actin TaqMan Endogenous Control were purchased from Applied Biosystems. Real-time PCR reaction was performed on a Bio-Rad CFX96 Real-Time PCR Detection System. The reaction mixture (40 μl) consisted of 20 μl of 2× TaqMan Gene Expression Master Mix (Applied Biosystems), 2 μl of GlyRα1 TaqMan Gene Expression Assay (20×), 2 μl of β-actin TaqMan Endogenous Control, and 10 μl of sample neuron cDNA. PCR amplification was performed at 50°C for 2 min, heated to 95°C for 10 min, and then followed by 50 cycles of 15 s at 95°C and 1 min at 60°C.
Primer/probes for GlyRα1 and β-actin were tested for efficiency by generating standard curves using 10-fold dilutions of mouse total spinal cord mRNA transcripts. Total RNA was extracted from mouse spinal cord using Trizol Reagent (Invitrogen). The RNA concentration was determined using UV spectrometry, providing the basis for serial dilution. RT was performed on 0.5 μg of total RNA using random primers and Superscript III (Invitrogen) in a 20 μl reaction mix (1:1). Standard curves were derived from plotting cycle threshold (Ct) against concentration (picograms per microliters).
Because the GlyRα1 and β-actin genes have similar and nearly 100% amplification efficiencies (see Results), the ΔΔCt method (Livak and Schmittgen, 2001) was used for studying the relative difference in expression levels of the GlyRα1 gene in the different motoneurons. Real-time RT-PCR signal for GlyRα1 was first normalized to β-actin using the equation ΔCt = Ct,GlyR − Ct,β-actin. Samples were then normalized again by a “calibrator” using the equation ΔΔCt = ΔCt,Sample − ΔCt,Calibrator, to enable comparisons across reaction plates. We used 0.5 pg of total spinal cord RNA as the calibrator, and the calibrator was subjected to the same RT-PCR as the single-motoneuron samples on each plate. This ΔΔCt was changed to a linear value by the conversion 2−ΔΔCt and then compared between each sample.
Immunocytochemistry.
Spinal cord cultures grown on glass coverslips were fixed with 4% paraformaldehyde/PBS for 20 min (5 min for anti-GlyR and anti-gephyrin antibody stainings), washed with PBS, permeabilized with 0.2% Triton X-100, blocked with 10% donkey serum, and then incubated in a mixture of primary antibodies diluted in PBS containing 2% donkey serum and 0.05% Triton X-100 overnight at 4°C. The primary antibodies used in different combinations were as follows: mouse anti-SMI-32 (monoclonal, 1:1000; Covance); mouse anti-GAD67 (monoclonal, 1:10,000; Millipore Corporation); guinea pig anti-GlyT2 (polyclonal, 1:10,000; Millipore Corporation); goat anti-ChAT (polyclonal, 1:200; Millipore Corporation); rabbit anti-GlyR (polyclonal, 1:50; Millipore Corporation); mouse anti-gephyrin (monoclonal, 1:50; BD Transduction Laboratories); and mouse anti-synaptophysin (monoclonal, 1:100; Dako). After four washes with PBS, coverslips were incubated for 2 h at room temperature in a mixture of species-specific secondary antibodies (all raised in donkey) conjugated to Alexa Fluor 594 and Alexa Fluor 647 (Invitrogen). Coverslips were washed again and mounted using anti-fade mounting solution (Vectashield; Vector Laboratories).
Image acquisition and quantification.
Labeled cells were imaged using a 100× oil-immersion objective (1.3 numerical aperture lens) mounted on a Carl Zeiss LSM 510 confocal microscope. All specimens were imaged under identical conditions and analyzed using identical parameters. Four to six embryonic cultures for each of the groups were performed. On each coverslip, six to eight motoneurons were quantified.
Measurements of GlyR cluster numbers were performed using NIH Image J software. For each Hb9–eGFP+ motoneuron, z-series optical sections through the cell were collected, and maximum intensity projection was derived from these sections. Surface clusters are defined as aggregates surrounding soma or proximal neurites. Threshold values of 0.1 μm2 and 50% of maximal intensity were chosen for defining clusters. For the determination of the number of clusters on proximal dendrites, Hb9–eGFP+ motoneurons were displayed in the center of the digitized field, and all neurites within the field were analyzed. Intracellular labeling was excluded from analysis by manual delineation of the surface neuronal region. Standardized cluster number (receptor density) was determined by dividing the number of clusters on the surface of a motoneuron soma or proximal dendrite by its perimeter, which was measured using NIH Image J software.
Colocalization analysis was performed as described previously (Chang and Martin, 2009). Briefly, images were imported into NIH Image J software in which regions of interest (ROIs) were outlined, and regions outside ROIs were cleared using “segmenting assistant” plug-in. Each channel was then subjected to background subtraction and analyzed by “colocalization threshold” plug-in (zero–zero pixels was excluded in threshold calculation). The thresholds for each channel were then calculated, and pixels below this value were ignored for colocalization quantification. A pixel was defined as having colocalization when the intensities of both labels were above their respective thresholds. Colocalization of GlyR with GlyT2, gephyrin, or synaptophysin immunoreactivity was given as the colocalization coefficients M1 and M2. These coefficients are proportional to the number of colocalizing pixels in either channel (channel 1 or channel 2) of the composite image, relative to the total number of pixels above threshold in that channel.
Results are presented as mean ± SEM. Statistical significance was determined by Student's t test. The level of significance was set of p < 0.05.
Results
The identity of Hb9–eGFP-expressing neurons in mouse spinal cord sections
In spinal cord sections of E12–E14 embryos and early postnatal Hb9–eGFP mice, numerous cells throughout ventral spinal cord express eGFP (Fig. 1A, E13, P1). Only large Hb9–eGFP cells in ventral spinal cord express the motoneuron marker ChAT but not small cells (Fig. 1B, E13, P1). Approximately 90% of ChAT-positive neurons in developing ventral horn are Hb9–eGFP positive.

Hb9–eGFP-positive neurons in spinal cord sections of embryonic, early postnatal, and adult Hb9–eGFP transgenic mice. A, In E13, postnatal day 1 (P1), and adult Hb9–eGFP transgenic mice, low-power fluorescent micrographs illustrate the distribution of eGFP+ neurons in the spinal cord. B, Higher-magnification images show large-sized, but not small-sized (arrow), Hb9–eGFP+ neurons colabeled (yellow) with the motoneuron marker ChAT (red). C, A representative confocal image showing an Hb9–eGFP+ motoneuron in a ventral lumbar spinal cord section from an adult Hb9–eGFP mouse contacted with GlyT2 (red) and GAD (blue) boutons. D, E, In medial lamina VIII, intermediomedical cell column, and lamina X of adult Hb9–eGFP mice, subsets of small Hb9–eGFP+ interneurons (arrows) are neither calbindin+ (Cal; D, red) nor ChAT+ (E, red). CC, Central canal; VH, ventral horn. Scale bars: A, D, E, 100 μm; B, C, 20 μm.
In adult mouse spinal cord sections, eGFP expression is mostly restricted to ventral spinal cord in laminae VIII and IX (Fig. 1A, Adult). A few Hb9–eGFP+ cells were also observed near the central canal in the intermediomedial cell column and lamina X (Fig. 1A, Adult). The majority (∼96%) of Hb9–eGFP+ cells in ventral horn are large, ChAT-positive cells (Fig. 1B, Adult) that are surrounded by GlyT2- and GAD-positive boutons (Fig. 1C). Approximately half (∼55%) of the large ChAT-positive neurons in adult mouse spinal cord ventral horn are Hb9–eGFP positive (Fig. 1B, Adult). In medial lamina VIII, intermediomedial cell column, and lamina X, subsets of small Hb9–eGFP+ neurons were also observed (Fig. 1A, Adult). They were neither calbindin positive nor ChAT positive (Fig. 1D,E).
Identification of motoneurons in Hb9–eGFP mouse spinal cord cultures
In dissociated spinal cord cultures obtained from E12–E14 embryos, Hb9–eGFP+ cells are divided into three groups according to their somal sizes at DIV 12–16: large-sized (diameter > 28 μm), medium-sized (diameter of 10–28 μm), and small-sized (diameter < 10 μm) cells. Cells in cultures were double stained with the motoneuron markers SMI-32 and ChAT. Of the large-sized Hb9–eGFP cells, 98.2 ± 2.1% (n = 68) were double labeled with SMI-32 and ChAT immunoreactivities. These SMI-32/ChAT-positive large Hb9–eGFP cells had characteristic motoneuron morphology with a fusiform, triangular, or polygonal soma, a prominent dendritic arborization, at least one broad dendrite whose diameter rivaled that of the soma and emanates from the pole of the cell, frequently branching into thinner secondary processes, and generally a single long axon-like neurite, often extending over several millimeters (Fig. 2Ai,Aii). The percentage of medium-sized Hb9–eGFP cells that were SMI-32/ChAT positive was 42.7 ± 3.2% (n = 224), and these cells had characteristic morphologies that mimic the large-sized Hb9–eGFP cells (Fig. 2Aiii). Of the small-sized Hb9–eGFP cells, 96.9 ± 1.2% (n = 98) were SMI-32 negative (SMI-32−)/ChAT−, with an oval or round soma and bipolar or multiple thin dendrites emerging from the soma (Fig. 2Aii, arrow). A total of 3.1 ± 1.2% (n = 98) small-sized Hb9–eGFP cells were SMI-32+ and/or ChAT+, which are likely to be ChAT+ interneurons or Hb9–eGFP+ interneurons (data not shown). Therefore, numerous living motoneurons in the dissociated spinal cord cultures were identified reliably by their Hb9–eGFP expression, size, and characteristic morphology. The surfaces of eGFP+ putative motoneurons were decorated with neurotransmitter markers GlyT2- and GAD-positive boutons in the dissociated spinal cord cultures (Fig. 2B), demonstrating glycinergic and GABAergic presynaptic innervations.

Identification of motoneurons in Hb9–eGFP mouse spinal cord cultures. A, Hb9–eGFP+ neurons in DIV 12–16 dissociated spinal cord cultures colabeled (see Overlay) with SMI-32 (red) and ChAT (blue). Large eGFP+ cells (Ai, Aii) that are double labeled with SMI-32 and ChAT immunoreactivities, as well as small eGFP+ cells (Aii, arrow) that are mostly SMI-32−/ChAT− are shown. Medium-sized eGFP+ cells (Aiii) with characteristic morphologies that mimic the large Hb9 motoneurons are also SMI-32+/ChAT+. B, Hb9–eGFP+ spinal motoneurons are contacted with presynaptic glycinergic GlyT2 (red) and GABAergic GAD (blue) boutons. Scale bars, 20 μm.
Glycine-induced currents are decreased in G93A–SOD1 motoneurons
Using whole-cell patch-clamp recording technique, we examined the electrophysiological properties of GlyRs in cultured Hb9–eGFP motoneurons (Fig. 3A,B). Experiments were performed in the presence of the voltage-dependent sodium channel blocker TTX (0.5 μm), the GABAAR antagonist bicuculline (5 μm), and non-NMDA and NMDA ionotropic glutamate receptor antagonists CNQX (5 μm) and APV (50 μm), respectively. Bath application of 5 μm bicuculline was reported to block the majority of GABAAR-mediated responses but with little effect on GlyR-mediated responses (O'Brien and Berger, 1999). Pressure ejection was used to apply 100 ms pulses (1–2 psi) of 1 mm glycine onto Hb9–eGFP+ neurons. In 61 of 64 Hb9–eGFP motoneurons, the glycine pulses produced large inward currents when the membrane potential was held at −65 mV (mean current density of 118.1 ± 8.2 pA/pF) (Fig. 3B). Glycine-induced small inward currents (<20 pA/pF) were seen in 3 of 64 cells. In small-sized Hb9–eGFP neurons, the peak amplitudes of glycine-evoked currents were highly variable, with the current densities ranging from ∼50 to ∼150 pA/pF (n = 15). Only Hb9–eGFP motoneurons with large inward currents were analyzed in the following experiments comparing responses in control and G93A–SOD1 motoneurons. The inward current was completely blocked by bath application of the GlyR antagonist strychnine (1 μm) (Fig. 3B), indicating that the glycine-induced responses are mediated by GlyR activation.

Glycine-induced currents are decreased in G93A–SOD1 motoneurons. A, An Hb9–eGFP+ motoneuron is identified under fluorescent microscope and recorded under DIC optics, along with a patch electrode (right) and a drug application pipette (left) containing 1 mm glycine. Scale bar, 20 μm. B, A 100 ms pulse (2–3 psi) of glycine (1 mm) evokes an inhibitory current that is blocked by strychnine (Stry; 1 μm). C, Representative recordings of glycine-evoked currents in control and G93A–SOD1 motoneurons. D, Glycine current densities in G93A–SOD1 motoneurons (n = 17) are significantly smaller than in controls (n = 20). Data represent the mean ± SEM (**p < 0.01, Student's t test). E, Examples of traces of glycine-evoked currents at the holding potentials indicated next to each trace and their I–V plots in control and G93A–SOD1 motoneurons. The peak amplitude of glycine currents at different holding potentials in G93A–SOD1 motoneurons are smaller than in control motoneurons, whereas no difference is observed in the reversal potential of glycine currents between G93A–SOD1 and control motoneurons.
To examine GlyRs in ALS mouse motoneurons, we bred G93A–SOD1 mice to Hb9–eGFP mice and recorded glycine-evoked currents in cultured Hb9–eGFP motoneurons prepared from G93A–SOD1 embryos and from their non-mutant SOD1 transgenic littermates. In each group, Hb9–eGFP motoneurons with characteristic morphologies were randomly selected and recorded. At equivalent days in culture, G93A–SOD1 motoneurons were slightly larger in size (membrane capacitance of 43.5 ± 2.4, n = 20) than control motoneurons (membrane capacitance of 39.8 ± 1.8, n = 17), but no significant difference was detected. Pressure ejection of glycine at −65 mV produced an inward current markedly smaller in the G93A–SOD1 motoneurons than in the control motoneurons (Fig. 3C,D). The decrease of glycine-evoked current density was significant as evidenced by the ∼35% reduction in G93A–SOD1 motoneurons compared with control motoneurons (control, 121.2 ± 9.5 pA/pF, n = 20; G93A–SOD1, 78.6 ± 4.9 pA/pF, n = 17). No significant difference was observed in the rise and decay time of the glycine-evoked currents between control and G93A–SOD1 motoneurons (data not shown).
Current–voltage (I–V) relationships for glycine-evoked currents were also examined in control and G93A–SOD1 motoneurons (Fig. 3E). Measurements of the glycine-evoked currents at holding potentials ranging from −70 to +50 mV produced an approximately linear current–voltage relationship. G93A–SOD1 motoneurons had decreased peak amplitude of glycine currents at all holding potentials, but no significant difference was observed in the reversal potential of glycine-evoked currents (Fig. 3E).
Glycinergic miniature postsynaptic currents are altered in G93A–SOD1 motoneurons
The data so far demonstrate that glycine-induced currents are decreased in ALS mouse motoneurons. However, GlyRs could differ in functional properties and numbers at the synapse compared with nonsynaptic receptors. To directly examine synaptic GlyRs in spinal cord cultures, we performed an analysis of glycinergic mIPSCs. mIPSCs represent postsynaptic responses to the spontaneous release of single quanta of neurotransmitter and can therefore be used to detect changes in synaptic receptor properties without the complicating presynaptic variables of action potential propagation and evoked calcium influx into the presynaptic terminal. We recorded glycinergic mIPSCs in Hb9–eGFP motoneurons voltage clamped at −65 mV in the presence of TTX (0.5 μm), CNQX (5 μm), APV (50 μm), and bicuculline (5 μm). Under this condition, fast rising and fast decaying spontaneous synaptic currents were detected in most Hb9–eGFP motoneurons (55 of 62) (Fig. 4A), reflecting in our motoneuron culture model the local release of glycine from presynaptic terminals as determined subsequently by immunofluorescence studies using antibodies against GlyT2 and synaptophysin (see Fig. 8A). The peak amplitude of these mIPSCs ranged from −5 to −620 pA and occurred at a frequency of 0.21 ± 0.03 Hz. Perfusion of the GlyR antagonist strychnine (1 μm) abolished the responses (Fig. 4A), confirming that these events are mediated by GlyRs.

Glycinergic mIPSCs are altered in G93A–SOD1 motoneurons. A, Glycinergic mIPSCs are detected in Hb9–eGFP motoneurons in the presence of TTX (0.5 μm), CNQX (5 μm), APV (50 μm), and bicuculline (Bic; 5 μm). Strychnine (Stry; 1 μm) blocks all the events that remain after application of the inhibitor mixture. B, C, Glycinergic mIPSCs in control and G93A–SOD1 motoneurons. Representative traces of mIPSCs, averaged mIPSC events (B), and the amplitude distributions/cumulative fraction histograms of mIPSCs (C) are shown for a given recording. Note the increase in the number of low-amplitude events in the G93A–SOD1 motoneurons (for group averages, see Results). Holding potential, −65 mV.
The average amplitude of glycinergic mIPSCs were significantly smaller in the G93A–SOD1 motoneurons than in the control (Fig. 4B,C), with the mean current densities of mIPSCs for control and G93A–SOD1 motoneurons of 2.48 ± 0.25 pA/pF (n = 20) and 1.79 ± 0.19 pA/pF (n = 17), respectively. Cumulative amplitude histograms for mIPSCs showed a shift toward smaller amplitudes for G93A–SOD1 motoneurons (Fig. 4C). The percentage of small-amplitude mIPSCs (<20 pA) in G93A–SOD1 motoneurons (22.7 ± 2.3%, n = 17) was significantly higher than in the control motoneurons (14.3 ± 1.6%, n = 20). The decay phase of mIPSC events was slightly faster in G93A–SOD1 than in the control motoneurons (Fig. 4B), with decay times of 21.33 ± 2.18 ms for G93A–SOD1 and 25.51 ± 2.59 ms for control, respectively, but no significant difference was detected. The frequency of mIPSC events in G93A–SOD1 motoneurons (0.13 ± 0.02 Hz, n = 17) was lower than in the control motoneurons (0.21 ± 0.03 Hz, n = 20), but no significant difference was detected (p = 0.09) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). No significant differences in the rise time of mIPSC events were observed between control and G93A–SOD1 motoneurons (data not shown).
GlyR currents are rescued in G93A–SOD1 motoneurons from chimeric cultures
We also designed motoneuron recording experiments using spinal cord chimeric cultures composed of mixtures of normal (wild-type) cells and cells that express G93A–SOD1, e.g., a culture prepared from mixtures of Hb9–eGFP/G93A–SOD1 embryonic spinal cords and their Hb9–eGFP/non-mutant SOD1 transgenic littermate spinal cords. Individual motoneurons were harvested after recordings were made and were genotyped by single-cell PCR for the human SOD1 gene after whole-genome amplification (Fig. 5A, top). Surprisingly, no significant alternations were observed in glycine-induced current densities between G93A–SOD1/Hb9–eGFP motoneurons (127.2 ± 7.0 pA/pF, n = 8) and control motoneurons (130.7 ± 11.9 pA/pF, n = 7) (Fig. 5B). The amplitude of glycinergic mIPSCs was not different between G93A–SOD1 motoneurons and control motoneurons (Fig. 5C), with the mean current densities of mIPSCs for G93A–SOD1 and control motoneurons were 2.24 ± 0.21 pA/pF (n = 8) and 2.39 ± 0.26 pA/pF (n = 7), respectively. This indicates a neural network involvement in the ultimate decline of motoneuron GlyR physiology and that wild-type neurons can compensate for network failure.

Glycine receptor-mediated currents are rescued in G93A–SOD1 motoneurons in chimeric culture. A, Top, Single-cell genotyping of a non-mutant SOD1/Hb9–eGFP motoneuron and a G93A–SOD1/Hb9–eGFP motoneuron from the same culture. Bottom, Glycine-evoked currents in these two motoneurons. B, No significant differences in glycine-induced current densities are observed between G93A–SOD1 (n = 8) and control motoneurons (n = 7). Data represent the mean ± SEM (Student's t test). C, Representative traces and averaged glycinergic mIPSCs events in a control motoneuron and a G93A–SOD1 motoneuron.
GABAA receptor currents are not affected in G93A–SOD1 motoneurons
GABAAR-mediated currents were also examined in Hb9–eGFP motoneurons. In the presence of TTX (0.5 μm), CNQX (5 μm), APV (50 μm), and strychnine (0.5 μm), brief pulses of GABA (100 μm, 100 ms, 1–2 psi) evoked inward currents in 30 of 37 Hb9–eGFP motoneurons (mean current density of 49.8 ± 6.8 pA/pF). The current was reversibly abolished by bath application of bicuculline (50 μm), a competitive antagonist of GABAARs, thus indicating that the current was mediated by GABAAR activation (Fig. 6A). The peak amplitude of GABA-induced currents were slightly higher in G93A–SOD1 motoneurons than in control motoneurons, but no significant differences were observed (mean current densities for control and G93A–SOD1 were 49.8 ± 6.8 pA/pF, n = 30 and 51.4 ± 4.6 pA/pF, n = 24, respectively) (Fig. 6B,C). No significant differences were observed in the decay time of GABA-induced currents in G93A–SOD1 and control motoneurons (decay times for control and G93A–SOD1 were 3.94 ± 0.29 s, n = 30, and 4.63 ± 0.43 s, n = 24, respectively).
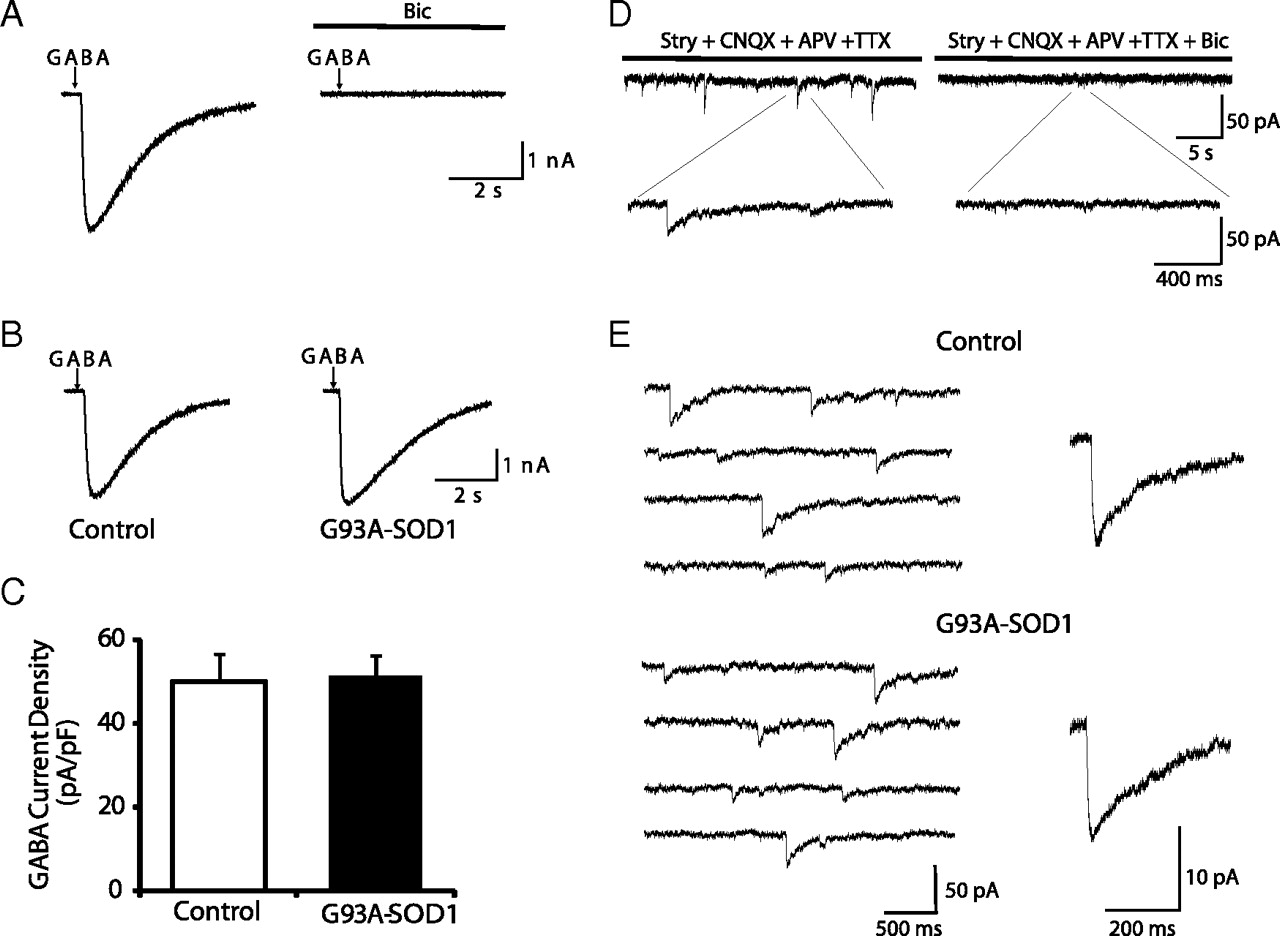
GABAA receptor-mediated currents are not affected in G93A–SOD1 motoneurons. A, A 100 ms pulse (2–3 psi) of GABA (100 μm) evokes an inhibitory current that is blocked by bicuculline (Bic; 50 μm). B, Sample recordings of GABA-evoked currents in control and G93A–SOD1 motoneurons. C, No significant differences in GABA-induced current densities are observed between control (n = 30) and G93A–SOD1 (n = 24) motoneurons. Data represent the mean ± SEM (Student's t test). D, GABAergic mIPSCs are detected in Hb9–eGFP motoneurons in the presence of TTX (0.5 μm), CNQX (5 μm), APV (50 μm), and strychnine (Stry; 0.5 μm). Bicuculline (Bic; 50 μm) blocks all the events that remain after application of the inhibitor mixture. E, Representative traces and averaged GABAergic mIPSCs events in a control motoneuron and a G93A–SOD1 motoneuron.
GABAergic mIPSCs were detected in 29 of 37 Hb9–eGFP motoneurons in the presence of an antagonist mixture containing TTX (0.5 μm), CNQX (5 μm), APV (50 μm), and strychnine (0.5 μm). The mean current densities of GABAergic mIPSCs was smaller (0.228 ± 0.031 pA/pF, n = 29) and the decay time phase was slower (59.02 ± 7.21 ms, n = 29) than that of glycinergic mIPSCs. These mIPSCs were blocked by bicuculline (50 μm), suggesting that they were mediated by activation of GABAARs (Fig. 6D). No significant differences existed in the peak amplitude of the mIPSCs between control and G93A–SOD1 motoneurons (mean current densities for control and G93A–SOD1 were 0.228 ± 0.031 pA/pF, n = 29 and 0.231 ± 0.041 pA/pF, n = 24, respectively) (Fig. 6E). No significant differences in the frequency, rise times, and decay times of GABAergic mIPSC events were observed between control and G93A–SOD1 motoneurons (data not shown).
GlyR mRNA expression is decreased in G93A–SOD1 motoneurons
To determine the molecular mechanisms underlying the GlyR current reduction in G93A–SOD1 motoneurons, we used quantitative single-cell real-time RT-PCR to examine GlyR mRNA expression in G93A–SOD1 and control Hb9 motoneurons. GlyRs isolated from mammalian spinal cord are pentameric membrane proteins composed of α and β subunits (Pfeiffer et al., 1982; Prior et al., 1992). GlyRs exhibit subtype heterogeneity as a result of four isoforms of α subunit (α1–α4) (Kuhse et al., 1995; Lynch, 2009). GlyRs in immature neurons contain the abundantly expressed α2 subunit, but GlyRs in mature neurons contain predominantly the α1 subunit within 2 weeks after birth (Malosio et al., 1991). The α3 subunit is mainly expressed in the cerebellum (Malosio et al., 1991), and the α4 subunit is a murine gene that is not expressed in human (Matzenbach et al., 1994). Because the majority of glycinergic neurotransmission in adults is mediated by α1β GlyRs and because we evaluated motoneurons that were morphologically well differentiated, only the α1 subunit (Glra1) was analyzed in this study. The housekeeping gene β-actin was used as an endogenous control (Calvo et al., 2008). The standard curves for the GlyRα1 and β-actin show approximately the same slope (Fig. 7A), indicating similar amplification efficiency (Halford, 1999; Medhurst et al., 2000). GlyRα1 mRNA was detected in 45 of 47 (96%) individual Hb9–eGFP motoneurons. Figure 7B shows amplification plots of GlyRα1 transcripts obtained from two individual Hb9–eGFP motoneurons (one from a control and one from a G93A–SOD1 culture). Ct values were obtained for GlyRα1 and β-actin (indicated by the bold line). Ct values for β-actin were similar in the two motoneurons (Fig. 7B). In contrast, GlyRα1 transcript amount in the G93A–SOD1 motoneuron was lower than that of the control motoneuron, because a higher Ct number was necessary to reach the threshold (Fig. 7B). The data were normalized to β-actin levels, and statistic analysis of the ΔCt = Ct,GlyR − Ct,β-actin revealed a significant difference between control motoneurons (ΔCt = 3.06 ± 0.16; n = 15) and G93A–SOD1 motoneurons (ΔCt = 4.27 ± 0.15; n = 28). The calibration of ΔCt values gives the relative amount of GlyRα1 transcripts in the respective motoneuron groups (Fig. 7C). G93A–SOD1 motoneurons had lower GlyRα1 mRNA levels compared with control motoneurons (Fig. 7C), suggesting that downregulation of GlyRα1 gene expression may underlie insufficient functioning of G93A–SOD1 motoneurons. To determine whether the reduction in GlyRα1 mRNA was motoneuron specific, a subset of small bipolar Hb9 interneurons (Fig. 7D, inset) were also examined. No significant change was observed in GlyRα1 mRNA expression in small bipolar Hb9 interneurons between control and G93A–SOD1 cultures (Fig. 7D), indicating that the decrease in GlyRα1 mRNA level is specific to motoneurons.
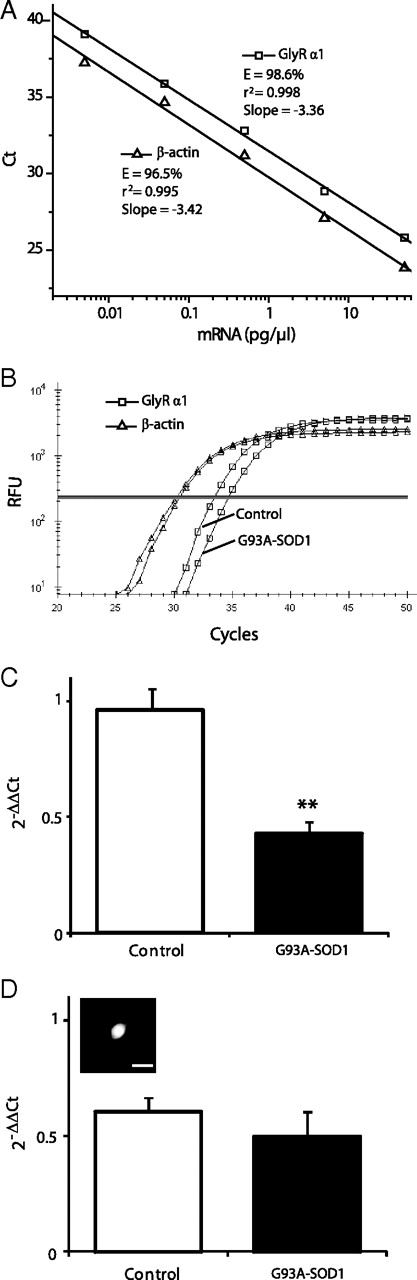
GlyR α1 subunit mRNA expression is decreased in G93A–SOD1 motoneurons. A, A validation experiment showing Ct values for GlyRα1 (squares) and β-actin (triangles) plotted against total spinal cord mRNA concentration. The amplification efficiencies (E) of the GlyRα1 and β-actin are similar. B, Representative real-time PCR amplification plots of GlyRα1 (squares) and β-actin (triangles) transcripts expression in a control and a G93A–SOD1 motoneuron. Ct for each transcript is shown as the intersection of the bold line with the RFU (relative fluorescence units) plot. C, Quantitative real-time RT-PCR analysis of GlyRα1 gene expression levels in control and G93A–SOD1 Hb9–eGFP motoneurons. Expression levels of GlyRα1 mRNA were calculated using the ΔΔCt method ΔΔCt = ΔCt,Sample − ΔCt,Calibrator, where the calibrator is 0.5 pg of total spinal cord mRNA, which was subjected to the same RT-PCR as the samples on each plate. Data are mean ± SEM; n = 15–28. **p < 0.01. D, Quantitative real-time RT-PCR analysis of GlyRα1 gene expression levels in small bipolar Hb9–eGFP interneurons in control and G93A–SOD1 cultures. Data are mean ± SEM; n = 6–8. The inset shows a representative fluorescent micrograph of a small bipolar Hb9–eGFP interneuron used for the single-cell RT-PCR. Scale bar, 10 μm.
Surface GlyRs are decreased in G93A–SOD1 motoneurons
To validate the mRNA expression data obtained from single-cell RT-PCR, we used immunocytochemistry to detect GlyRα1 subunit protein in cultured Hb9–eGFP motoneurons. The localization of GlyR was examined by confocal microscopy after labeling receptors with an anti-GlyR antibody and an antibody against gephyrin, the anchoring protein of GlyR (Triller et al., 1985; Betz et al., 1994). GlyR and gephyrin cluster postsynaptically at glycine-releasing nerve terminals (Triller et al., 1985, 1987). At DIV 14, GlyR and gephyrin staining was localized in clusters at the surface of the soma and dendrites of Hb9–eGFP motoneurons (Fig. 8Ai), and they were almost perfectly colocalized (colocalization coefficients M1 = 0.946 ± 0.012, n = 6; calculated as M1 = pixelsCh1, coloc/pixelsCh1, total, where Ch1 is GlyR). Most GlyR clusters on the surface of soma and dendrites of Hb9–eGFP motoneurons were associated with presynaptic GlyT2 boutons (colocalization coefficients M1 = 0.917 ± 0.019, n = 6; calculated as M1 = pixelsCh1, coloc/pixelsCh1, total, in which Ch1 is GlyR), suggesting a close association with glycinergic presynaptic terminals (Fig. 8Aii). Surface GlyR cluster labeling and gephyrin staining were also observed in close apposition to presynaptic synaptophysin (colocalization coefficients M1 = 0.911 ± 0.015, n = 6; calculated as M1 = pixelsCh1, coloc/pixelsCh1, total, in which Ch1 is GlyR; M1 = 0.922 ± 0.014, n = 6; calculated as M1 = pixelsCh1, coloc/pixelsCh1, total, in which Ch1 is gephyrin), a ubiquitous synaptic vesicle marker (Navone et al., 1986), demonstrating directly the existence of glycinergic synaptic connections in this cell culture model (Fig. 8Aiii,Aiv). Intracellular labeling of GlyRs was also detected within the soma, mostly around the nucleus (Fig. 8A,B). The intracellular GlyR immunoreactivity did not colocalize with gephyrin, GlyT2, or synaptophysin (Fig. 8Ai–Aiii), suggesting that these are extrasynaptic receptors. No attempts were made to study intracellular GlyRs. Surface GlyRs were detected in 47 of 56 Hb9–eGFP motoneurons, whereas the staining of GlyRs in small-sized Hb9–eGFP neurons varies: some have very few GlyR staining, whereas some have very intense GlyR staining (data not shown). These findings are consistent with the electrophysiological results.

Surface GlyR expression is decreased in G93A–SOD1 motoneurons. A, Confocal images show the localization of GlyRs (Ai–Aiii, red) in close apposition to postsynaptic scaffold protein gephyrin (Geph; Ai, blue), presynaptic GlyT2 (Aii, blue), and presynaptic synaptophysin (SYN; Aiii, blue) immunoreactivities on the surface of the soma and proximal dendrites of Hb9–eGFP motoneurons. Coincidence of Geph (red) and SYN (blue) immunoreactivities on Hb9–eGFP motoneurons is also shown (Aiv). B, Representative images showing the localization of GlyRs (red) on Hb9–eGFP motoneurons from control (Bi) and G93A–SOD1 (Bii) cultures. C, Quantitative analysis of GlyR densities on the surface of soma and proximal dendrites in control motoneurons (n = 47) and G93A–SOD1 motoneurons (n = 42). Data represent the mean ± SEM (*p < 0.05, Student's t test). Scale bar, 20 μm.
We examined the number of clusters on the surface of soma and proximal dendrites, which mainly represent the functional postsynaptic GlyRs. GlyR cluster number on the soma and proximal dendrites of G93A–SOD1/Hb9–eGFP motoneurons was significantly lower than control motoneurons (Fig. 8B,C) (control, 29.9 ± 1.6 for soma and 37.4 ± 1.1 for proximal dendrites, n = 47; G93A–SOD1, 24.5 ± 1.1 for soma and 31.7 ± 1.6 for proximal dendrites, n = 42, respectively). Receptor alternation patterns were similar in the soma and proximal dendrites (Fig. 8B,C).
Discussion
The major finding of this study is that GlyR-mediated currents were altered in cultured spinal motoneurons prepared from embryonic mice expressing human ALS-linked mutant SOD1. The abnormality was selective for GlyRs because GABAARs were not affected. The reduction of functional GlyRs could be attributable to decreased GlyRα1 mRNA expression and diminished surface postsynaptic GlyR proteins on G93A–SOD1 motoneurons.
Motoneuron cell culture as a model to study physiological consequences of genes causing ALS
It is necessary to determine consequences of ALS-causing mutant genes at the level of motoneuron synaptic function. We hypothesize that functional perturbations in motoneurons antedate structural and biochemical abnormalities that are studied most commonly. However, information on motoneuron physiology in models of ALS is sparse. The synaptic physiology of motoneurons in the context of genetic disease can be studied in slice and dissociated cell culture systems. We used a dissociated culture system because it offers advantages for studying motoneuron functions, such as better environment control and recording of responses in individual neurons. Cultured embryonic spinal cord neurons develop morphologically differentiated (Colin et al., 1996) and functional (Jackson et al., 1982) inhibitory synapses. However, motoneuron functional studies in culture have been limited because of the difficulty of identifying unequivocally motoneurons maintained in live spinal cord cultures.
We used transgenic mice that express eGFP driven by the Hb9 promoter to identify motoneurons. Hb9–eGFP expression in motoneurons in vivo was confirmed in spinal cords of embryonic, newborn, and adult mice. A subset of ChAT+ neurons was Hb9–eGFP+ in embryonic and postnatal mouse spinal cord, and appreciable expression is maintained even in adult spinal motoneurons. The identification of Hb9–eGFP motoneurons in dissociated spinal cord culture was confirmed by SMI-32 and ChAT staining. Hb9–eGFP labeling reveals more exquisite morphological details than SMI-32 and ChAT immunostaining, and, more importantly, it labels living motoneurons necessary for electrophysiological studies. We used a dissociated total spinal cord culture that includes all neighboring cells of motoneurons and are capable of supporting the survival of cultured motoneurons and provide a more physiological environment than artificial substrates. In our cultures, motoneurons stay healthy for at least 3 weeks without attrition of morphology and physiological functions. Our approach is distinctly different from motoneuron enrichment techniques, including density gradients (Schnaar and Schaffner, 1981; Dohrmann et al., 1986; Martinou et al., 1989), retrograde labeling (Calof and Reichardt, 1984; Schaffner et al., 1987), and immunopanning (Camu and Henderson, 1992; Mettling et al., 1995), which generally give low yield of motoneurons or lack complete specificity for motoneurons.
In previous electrophysiological studies, putative motoneurons in mixed spinal cord culture were identified by morphology and diameter (>28 μm) (Pieri et al., 2003a; Carunchio et al., 2008). Because motoneuron number in total spinal cord culture is very low (<1%) and there are other cells that are large in size (>28 μm), this approach is insufficient for specific evaluation of motoneurons; thus, the number of motoneurons is likely to be overestimated and recordings could be from different populations of cells. Our culture model using genetic labeling of motoneurons solves these problems. However, we did find that Hb9–eGFP also labels a group of small spinal neurons in culture with less evident resemblance to motoneurons (SMI-32−/ChAT−). These cells could be glutamatergic excitatory interneurons (Wilson et al., 2005).
Expression of human mutant SOD1 alters GlyR in motoneurons
Studies have shown that primary spinal cord neuron cultures express functional GlyRs forming postsynaptic receptor aggregates (Jackson et al., 1982; Hamill et al., 1983; Nicola et al., 1992) and that adult GlyRα1 subunit was detected within the second week of culture (Hoch et al., 1992; St John and Stephens, 1993). Using immunocytochemistry and confocal microscopy, we found robust localization of the GlyRα1 on cultured motoneurons at times when recordings were made. Furthermore, we confirmed the postsynaptic localization of GlyRs and their associations with presynaptic glycinergic terminals by their colocalization with gephyrin, synaptophysin, and GlyT2.
We found that the current densities of glycine-evoked currents and glycinergic mIPSCs were significantly smaller in G93A–SOD1 motoneurons than in control. This could be caused by decreased expression of GlyRs. Indeed, using quantitative single-cell RT-PCR, we found decreased GlyRα1 mRNA expression in G93A–SOD1 motoneurons. By immunofluorescence, this finding was substantiated at the protein level by diminished expression of surface GlyRs on G93A–SOD1 motoneurons. Another possibility is increased removal of postsynaptic GlyRs from dendritic and somatic surfaces via endocytosis and decreased postsynaptic GlyRs aggregation. A change in single-channel conductance or channel open time or probability of opening may contribute to the decrease in glycine currents as well.
The analysis of glycinergic mIPSCs in G93A–SOD1 motoneurons demonstrated slightly faster decay kinetics than in control. The faster decay could be attributable to expression of synaptic GlyR channels with faster kinetic properties. Alternatively, the faster decay could be attributable to faster clearance of transmitter from the synaptic cleft. A comparison of responses induced by brief pulses of glycine onto motoneurons provides direct evidence for the kinetic properties of GlyRs. No apparent difference in the kinetics of glycine-evoked currents was detected in G93A–SOD1 motoneurons compared with control. Therefore, the kinetic change in mIPSCs could not be attributable to altered GlyR subunit composition. Additional studies on transmitter clearance kinetics are required. van Zundert et al. (2008) reported enhanced frequency of spontaneous inhibitory transmission in hypoglossal motoneurons and spinal interneurons in G93A–SOD1 mice, whereas mIPSCs were not significantly different. We did not find significant differences in the frequency and rise time of mIPSCs in G93A–SOD1 motoneurons, although there was a trend for decreased mIPSC frequency. The discrepancy between reduced cluster density and unaltered mIPSC frequency could have a presynaptic or postsynaptic basis. In G93A–SOD1 motoneurons, the normally low probability release sites could be changed to high probability release sites. Alternatively, because glycinergic silent synapses (Faber et al., 1991) and extrasynaptic receptors (Flint et al., 1998) have been identified, the normally silent synapses could become activated in G93A–SOD1 motoneurons, or G93A–SOD1 motoneurons could recruit extrasynaptic GlyRs that do not exhibit the typical receptor clustering and might not be detected by our immunofluorescence analysis. Physiological methods have a lower detection threshold than conventional morphological methods and enable the detection of extrasynaptic receptors (Triller and Choquet, 2005). In fact, we observed that G93A–SOD1 motoneurons had a significant increase in the number of small-amplitude mIPSCs. These would be incomplete compensation mechanisms whereby residual GlyRs (clusters and extrasynaptic receptors) in mutant motoneurons become more functional to keep the frequency unchanged but inadequate for maintaining average amplitude.
We found using a chimeric culture system that evoked and miniature GlyR currents in G93A–SOD1 motoneurons were rescued. This finding suggests that GlyR defects might be driven by factors extrinsic to motoneurons or that intrinsic defects in motoneurons can be compensated by extrinsic factors. Our spinal cord cultures are heterogeneous in cellular composition containing multiple neuronal types but fewer glial cells because proliferation is inhibited. These cultures develop a physiological network as evidenced by spontaneous synaptic activities of motoneurons that are contacted by numerous synapses. One possible explanation for the reduced GlyR functioning is that it is secondary to loss of innervations from glycinergic interneurons. We have shown in G93A–SOD1 mouse spinal cord that presynaptic glycinergic innervation of motoneurons is deficient, possibly as a result of an early degeneration of spinal interneurons (Chang and Martin, 2009). A similar process could be evolving in spinal cord cultures from G93A–SOD1 mice, but in chimeric cultures there is compensation by wild-type interneurons. Another possibility is that intrinsic insufficiency in motoneuron GlyRs can be rescued by other cells.
We did not find significant changes in the current density and decay kinetics of GABAARs in G93A–SOD1 motoneurons. Carunchio et al. (2008) also reported no differences in the current density of GABA (30–100 μm)-evoked responses in primary motoneurons cultured from G93A mice. Thus, of the two major ligand-gated chloride ion channels in spinal cord, the functional alteration appears to be selective for GlyR.
Relevance of our findings to disease mechanisms in ALS
Motoneuron degeneration from excessive excitation has been long suspected (Heath and Shaw, 2002). Primary cultured spinal cord neurons from G93A–SOD1 mice are reported to be hyperexcitable (Pieri et al., 2003b; Kuo et al., 2004). Work on brainstem slices of G93A–SOD1 mice revealed evidence for hypoglossal motoneuron hyperexcitability, as well as increased persistent Na+ current at postnatal day 4 (van Zundert et al., 2008), which has also been seen in dissociated spinal cord cultures (Kuo et al., 2005). In spinal cord acute slices from G85R–SOD1 mice, lumbar motoneurons show reduced input resistance and altered frequency–intensity relationships at the second postnatal week (Bories et al., 2007). Thus, emergent electrophysiological evidence indicates that the mechanisms of disease in ALS mouse models might be engaged long before adulthood and the disease could be much more protracted than generally appreciated. Our study lays a foundation for the novel idea that motoneurons in ALS might undergo chronic stress as a result of inhibitory insufficiency. This inhibitory insufficiency could be a motoneuron-autonomous process or secondary to glycinergic interneuron disease or dysfunction (Chang and Martin, 2009) and thus is a motoneuron non-autonomous process of degeneration involving Renshaw cells and neural networks.
Footnotes
This work was supported by National Institutes of Health Grants NS034100 and NS065895. We thank Yan Pan for technical assistance.
- Correspondence should be addressed to Dr. Lee J. Martin, Department of Pathology, Johns Hopkins University School of Medicine, 720 Rutland Avenue, 558 Ross Building, Baltimore, MD 21205. martinl{at}jhmi.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}