

Abstract
Opioid-induced hyperalgesia is characterized by hypersensitivity to innocuous or noxious stimuli during sustained opiate administration. Microinjection of lidocaine into the rostral ventromedial medulla (RVM), or dorsolateral funiculus (DLF) lesion, abolishes opioid-induced hyperalgesia, suggesting the importance of descending pain facilitation mechanisms. Here, we investigate the possibility that cholecystokinin (CCK), a pronociceptive peptide, may drive such descending facilitation from the RVM during continuous opioid administration. In opioid-naive rats, CCK in the RVM produced acute tactile and thermal hypersensitivity that was antagonized by the CCK2 receptor antagonist L365,260 or by DLF lesion. CCK in the RVM also acutely displaced the spinal morphine antinociceptive dose-response curve to the right. Continuous systemic morphine elicited sustained tactile and thermal hypersensitivity within 3 d. Such hypersensitivity was reversed in a time-dependent manner by L365,260 in the RVM, and blockade of CCK2 receptors in the RVM also blocked the rightward displacement of the spinal morphine antinociceptive dose-response curve. Microdialysis studies in rats receiving continuous morphine showed an approximately fivefold increase in the basal levels of CCK in the RVM when compared with controls. These data suggest that activation of CCK2 receptors in the RVM promotes mechanical and thermal hypersensitivity and antinociceptive tolerance to morphine. Enhanced, endogenous CCK activity in the RVM during sustained morphine exposure may diminish spinal morphine antinociceptive potency by activating descending pain facilitatory mechanisms to exacerbate spinal nociceptive sensitivity. Prevention of opioid-dose escalation in chronic pain states by CCK receptor antagonism represents a potentially important strategy to limit unintended enhanced clinical pain and analgesic tolerance.
- cholecystokinin
- CCK2 receptor
- opioid hyperalgesia
- antinociceptive tolerance
- morphine
- rostral ventromedial medulla
- microdialysis
- L365,260
Introduction
Preclinical studies have shown that acute or prolonged opioid administration can elicit abnormal pain states (Woolf, 1981; Yaksh and Harty, 1988; Trujillo and Akil, 1991; Celerier et al., 2001). Continuous morphine administration by subcutaneous pellets or osmotic minipump results in increased sensitivity to normally non-noxious probing of the paws with von Frey filaments and to noxious thermal stimuli (Vanderah et al., 2000, 2001a,b). Opioid-induced mechanical and thermal hypersensitivity can be reversed by lidocaine administration in the rostral ventromedial medulla (RVM) or by lesion of the dorsolateral funiculus (DLF) (Vanderah et al., 2001b), suggesting that descending pain facilitation from the RVM to the spinal cord is necessary for the manifestation of chronic opioid-induced sensitivity to noxious and non-noxious stimuli. The same opioid administration protocol that elicits tactile and thermal hypersensitivity also results in a rightward displacement of the spinal or systemic opioid dose-response curve (i.e., opioid antinociceptive tolerance) (Laulin et al., 1999; Vanderah et al., 2000, 2001b). Importantly, decreased spinal morphine antinociceptive potency (i.e., antinociceptive tolerance) is also prevented by lidocaine in the RVM or DLF lesion (Vanderah et al., 2001b). These findings provide neuroanatomical evidence that descending pain facilitation from the RVM may underlie the expression of morphine antinociceptive tolerance. The mechanisms by which such pain facilitation occurs during sustained morphine exposure are unknown. Here, the role of cholecystokinin (CCK) activity in the RVM in opioid-induced mechanical and thermal hypersensitivity and antinociceptive tolerance was explored.
CCK has a similar distribution with opioid peptides (Stengaard-Pedersen and Larsson, 1981) and receptors (Ghilardi et al., 1992) in the CNS, and the excitatory actions of CCK often counteract opioid antinociception. CCK may act as an endogenous “anti-opioid” (Faris et al., 1983). Thus, morphine tolerance coincides with an upregulation of CCK mRNA and increased concentration of CCK-like immunoreactivity (CCK-LI) in tissue extracts of the brainstem, the hypothalamus, and the spinal cord (Zhou et al., 1992, 1993; Ding and Bayer, 1993). Furthermore, studies have shown that morphine elicits CCK release in the spinal cord and frontal cortex (de Araujo Lucas et al., 1998; Becker et al., 1999; Gustafsson et al., 2001). Moreover, CCK antiserum or receptor antagonists prevent antinociceptive tolerance to morphine (Ding et al., 1986; Dourish et al., 1990; Hoffmann and Wiesenfeld-Hallin, 1994). CCK immunoreactivity is predominantly associated with fibers within the RVM, including the raphe nuclei and the medullary reticular formation (Hokfelt et al., 1988; Baber et al., 1989). Recently, CCK-8 in the RVM was shown to produce an enhanced nociceptive response to colon distension, and RVM administration of a CCK2 antagonist enhanced morphine antinociception in this model (Friedrich and Gebhart, 2003). Collectively, these findings led us to hypothesize that CCK in the RVM may engage descending pain facilitatory pathways to enhance spinal nociceptive transmission and attenuate morphine antinociception.
Materials and Methods
Animals. Male Sprague Dawley rats (Harlan Sprague Dawley, Indianapolis, IN), 250-350 gm at the time of testing, were maintained in a climate-controlled room on a 12 hr light/dark cycle (lights on at 7:00 A.M.), and food and water were available ad libitum. All of the testing was performed in accordance with the policies and recommendations of the International Association for the Study of Pain and the National Institutes of Health guidelines for the handling and use of laboratory animals and received approval from the Institutional Animal Care and Use Committee of the University of Arizona.
RVM cannulation. Rats were anesthetized with ketamine-xylazine (100 mg/kg, i.p.) for stereotaxic placement of bilateral cannula in the RVM. The skull was exposed, and two 26 gauge guide cannulas separated by 1.2 mm (Plastics One, Roanoke, VA) were directed toward the lateral portions of the RVM using the atlas of Paxinos and Watson (1986) (anteroposterior, -11.0 mm from bregma; lateral, ±0.6 mm; dorsoventral, -7.5 mm from the dura mater). The guide cannulas were cemented in place and secured to the skull by small stainless steel machine screws. The animals were allowed to recover for 5 d after surgery before any pharmacological manipulations were made. Drug administrations into the RVM were performed by slowly expelling 0.5 μl of drug solution through a 33 gauge injection cannula inserted through the guide cannula and protruding an additional 1 mm into fresh brain tissue to prevent backflow of drug into the guide cannula. At the termination of the experiments, pontamine blue was injected into the site of RVM injections, and cannula placement was verified histologically. Data from animals with incorrectly placed cannula were not included within the data analysis. Data from animals with misplaced cannulas are included as off-site controls.
Intrathecal catheterization. While under halothane anesthesia, some groups of rats were implanted with intrathecal catheters (polyethylene 10, 7.8 cm) for drug administration at the level of the lumbar spinal cord, as described previously (Yaksh and Rudy, 1976). Animals were allowed to recover for 7 d.
Spinal DLF lesions. Spinal lesions at T8 were performed in halothane-anesthetized rats as described previously (Vanderah et al., 2001b). The spinal cord was exposed by laminectomy, and the DLF bilaterally was crushed with fine forceps. Sham spinal surgery was performed by exposing the vertebrae and performing a laminectomy but without damaging neuronal tissue. Animals were allowed to recover for 5 d. Hemostasis was confirmed, and the wound lesions were verified histologically at the termination of the experiments by fixing the spinal sections obtained from the lesion site in paraffin. Sections (40 μm thick) were mounted and stained with Luxor Fast Blue myelin stain to visualize intact and disrupted white matter. Only animals that had appropriately placed DLF lesions were included in the data analysis; however, animals with incomplete lesions and the corresponding paw-withdrawal latencies are reported in Results as controls.
Drug administration. Sustained, systemic morphine administration was accomplished by subcutaneous implantation of two 75 mg morphine free base pellets. Control groups received two placebo pellets containing excipient only. The pellets were a generous gift from the National Institute on Drug Abuse Drug Supply Program. Intrathecal morphine (Sigma, St. Louis, MO) was administered 30 min before the tail-flick assay through intrathecal catheter in a volume of 5 μl, followed by a 9 μl flush. Each animal was used only once to avoid possible acute tolerance. Saline was injected as a control for morphine administration. Sulfated CCK-8 [CCK-8(s)] (30 ng or 26.3 pmol in each side; American Peptide Company, Sunnyvale, CA), L365,260 (3S (-)[N ′-2,3-dehydro-1-methyl-2-oxo5-phenyl-1H-1,4-benzodiazepin-3-yl]-1H-indole-2-carboxamide) (2.5 ng in each side; Merck, Rahway, NJ), 3S(-)-N-(2,3-dihydro-1-methyl-2-oxo-5-phenyl-1H-1,4-benzodiazepine-3-yl)-1H-indole-2-carboxamide (L364,718) (2.5 or 25 ng in each side; Merck), or vehicle were administered bilaterally in 0.5 μl/side into the RVM. CCK-8(s) was dissolved in distilled water, whereas L365,260 and L364,718 were dissolved in propylene glycol-DMSO (80 and 20%, respectively); distilled water or propylene glycol-DMSO were used in vehicle administrations. In the tail-flick test, CCK-8(s), L365,260, L364,718, or vehicle were given into the RVM 5 min before intrathecal morphine administration. For antagonism of CCK in the RVM, L365,260 or L364,718 were injected 5 min before CCK into the RVM.
Tactile thresholds. The withdrawal threshold of the hindpaw in response to probing with a series of eight calibrated von Frey filaments (Stoelting, Wood Dale, IL) in logarithmically spaced increments ranging from 0.41 to 15 gm (4-150 N) was determined. Each filament was applied perpendicularly to the plantar surface of the left hindpaw of rats kept in suspended wire-mesh cages. Measurements were taken both before and after administration of drug or vehicle. Withdrawal threshold was determined by sequentially increasing and decreasing the stimulus strength (“up and down” method), analyzed using a Dixon nonparametric test (Chaplan et al., 1994), and expressed as the mean withdrawal threshold. Significant changes from baseline control values were detected by ANOVA, followed by Fisher's least significant difference test. These evaluations were all performed with the aid of the pharmacological statistics package FlashCalc (Dr. Michael Ossipov, University of Arizona, Tucson, AZ). Significance was set at p < 0.05.
Thermal hyperalgesia. The method of Hargreaves et al. (1988) was used to assess paw-withdrawal latency to a thermal nociceptive stimulus. Rats were allowed to acclimate within a Plexiglas enclosure on a clear glass plate maintained at 30°C for 30 min. A radiant heat source (i.e., high-intensity projector lamp) was directed onto the plantar surface of the left hindpaw. A motion detector halted both lamp and timer when the paw was withdrawn. The paw-withdrawal latency from the radiant heat source was determined both before and after drug or vehicle administration. Baseline latencies were established at ∼20 sec to allow a sufficient window for the detection of possible hyperalgesia. A maximal cutoff of 40 sec was used to prevent tissue damage. Significant changes from baseline control values were detected by ANOVA, followed by Fisher's least significant difference test. These evaluations were all performed with the aid of the pharmacological statistics package FlashCalc. Significance was set at p < 0.05.
Tail-flick test. The hot-water tail-flick test was performed by placing the distal third of the tail in a water bath maintained at 52°C. The latency until tail withdrawal from the bath was determined and compared among the treatments. A 10 sec cutoff was used to avoid tissue damage. Tolerance to the antinociceptive effect of morphine was indicated by a significant reduction in tail-flick latency after challenge with an A90 (dose of morphine that results in a 90% antinociceptive effect in the 52°C warm-water tail-flick test) dose of 10 μg/5 μl, intrathecally. Data were converted to percentage of antinociception by the following formula: (response latency - baseline latency)/(cutoff - baseline latency) - 100 to generate dose-response curves. Linear regression analysis of the log dose-response curves was used to calculate the A50 (dose of morphine that resulted in a 50% antinociceptive effect) values and the 95% confidence intervals (CIs). Relative potency was determined as a ratio of the A50 values. The significance of the relative potency and the confidence intervals were determined by applying the t test at p = 0.05. When differences within a treatment were measured, the significant differences among means within treatments were determined by ANOVA, followed by Fisher's least significant difference test. These evaluations were all performed with the aid of the pharmacological statistics package FlashCalc.
In vivo microdialysis. Three hours before the experiment, rats were placed in a recording chamber with access to food and water ad libitum. After acclimation, a microdialysis probe [CMA/12 (CMA Microdialysis, North Chelmsford, MA); polycarbonate membrane; outer diameter of 0.5 mm; length of 2 mm; molecular cutoff of 20 kDa] was inserted into the guide cannula such that the probe membrane extended 2 mm into the RVM. Microdialysis of the RVM was performed in freely moving, awake animals. At a flow rate of 3.5 μl/min, the microdialysis probe was continuously perfused with an artificial CSF (in mm: 138 NaCl, 5 KCl, 1 MgCl2, 1 CaCl2, 11 NaHCO3, and 1 NaH2PO4, pH 7.4) containing 0.2% bovine serum albumin, 0.2% glucose, and 0.03% of the peptidase inhibitor Bacitracin. To allow the dialysis to reach a steady state after insertion, the probe was perfused for a 1 hr washout period before collection of the first sample. In all experiments, nine dialysate fractions of 100 μl (30 min of perfusion each) were collected at 8°C and then immediately frozen at -20°C until their CCK-LI content could be measured using the radioimmunoassay (RIA) procedure described below. Thirty minute fraction collections began after the washout period to establish baseline levels of CCK-LI in the RVM. In some experiments, the ability of morphine to acutely release CCK was evaluated by collecting two baseline samples and then administering morphine (100 μm) into the RVM by reverse microdialysis. The same technique and a similar dose of morphine induces a significant release of CCK in the dorsal horn (Gustafsson et al., 1999). Microdialysate was then collected for 30 min fractions over the entire 2.5 hr period. At no point during the microdialysis did the animals exhibit any behavioral signs of discomfort, abnormal behavior, or motor hyperactivity.
CCK RIA. CCK-LI was determined using a modified RIA procedure with improved sensitivity developed especially for the determination of neuropeptide concentrations in microdialysates (Brodin et al., 1983; You et al., 1994; Gustafsson et al., 1999). An increased sensitivity of the RIA was achieved by minimizing the total incubation volume and limiting the amount of radioligand (∼1000 cpm/assay tube), as well as a sequential incubation procedure. This modified protocol yielded an eightfold increase in the sensitivity of the assay, as indicated by a shift in the average IC50 value, which was reduced from 105.6 ± 10.1 (n = 6) to 12.9 ± 1.5 pm (n = 5; mean ± SEM). The microdialysates and standard samples (100 μl) were preincubated for 24 hr at 4°C with the C-terminal-directed CCK/gastrin antiserum (2609/10, respectively, in 0.1 m barbital buffer containing 0.2% BSA) (Rehfeld, 1978). After addition of the radioligand (∼1000 cpm/assay tube of 125I-gastrin-I; specific activity, 70 MBq/nmol), all samples were incubated at 4°C for another 72 hr. Free radioligand was separated from antibody-bound radioligand in all RIA samples, except those used for determination of the total radioactivity, by the addition of 250 μl sheep anti-rabbit antibody-coated Sepharose suspension. After 30 min of incubation and centrifugation at 2600 × g for 10 min, the supernatant was discarded by aspiration, and the radioactivity in the pellets containing the bound fraction was measured in a gamma counter.
Results
CCK in the RVM elicits tactile and thermal hypersensitivity
Administration of CCK-8(s) by bilateral microinjection in the RVM resulted in a time-related decrease in the paw-withdrawal threshold to non-noxious mechanical stimuli, as well as to noxious thermal stimuli (Fig. 1A,B). Baseline paw-withdrawal thresholds diminished from 14.0 ± 1.1 to 5.7 ± 1.7 gm within 30-40 min after CCK microinjection. Baseline paw-withdrawal latencies to noxious thermal stimuli were 20.7 ± 1.0 sec and were decreased to 15.4 ± 1.7 sec within 30-40 min after CCK microinjection. Mechanical and thermal thresholds returned to baseline paw-withdrawal latencies within 120 min after CCK administration. Administration of vehicle by bilateral microinjection in the RVM did not change either mechanical or thermal thresholds compared with baselines. Pretreatment with L365,260 in the RVM, a CCK2 receptor antagonist, prevented CCK-induced mechanical and thermal hypersensitivity (Fig. 1A,B). Pretreatment with L364,718 in the RVM, a CCK1 receptor antagonist (Ebenezer, 2002; Alttoa and Harro, 2004), at the dose of 2.5 ng, as well as a 10-fold higher dose (25 ng), did not antagonize CCK-induced mechanical and thermal hypersensitivity. Paw-withdrawal thresholds in animals receiving L364,718 (25 ng bilaterally) in the RVM 5 min before CCK were 5.8 ± 2.3 gm (Fig. 1A). Similarly, response thresholds to noxious thermal stimuli in animals receiving L364,718 (25 ng bilaterally) in the RVM 5 min before CCK were 13.7 ± 1.9 sec (Fig. 1B). These values did not differ from post-CCK baselines. L365,260 in the RVM alone did not alter thresholds to either non-noxious tactile (15.0 ± 0.0 gm) or noxious thermal (19.0 ± 1.5 sec) stimuli. Likewise, L364,718 in the RVM alone did not alter thresholds to either non-noxious tactile (15.0 ± 0.0 gm) or noxious thermal (20.2 ± 2.1 sec) stimuli. In addition, control studies with the administration of vehicle for L365,260 and L364,718 were performed. Vehicle administration into the RVM 5 min before CCK administration into the RVM resulted in no significant differences (6.1 ± 2.1 gm and 12.9 ± 2.0 sec for mechanical and thermal testing, respectively) from animals receiving CCK alone. Bilateral RVM cannula placement for all of the injections are displayed in Figure 7A.
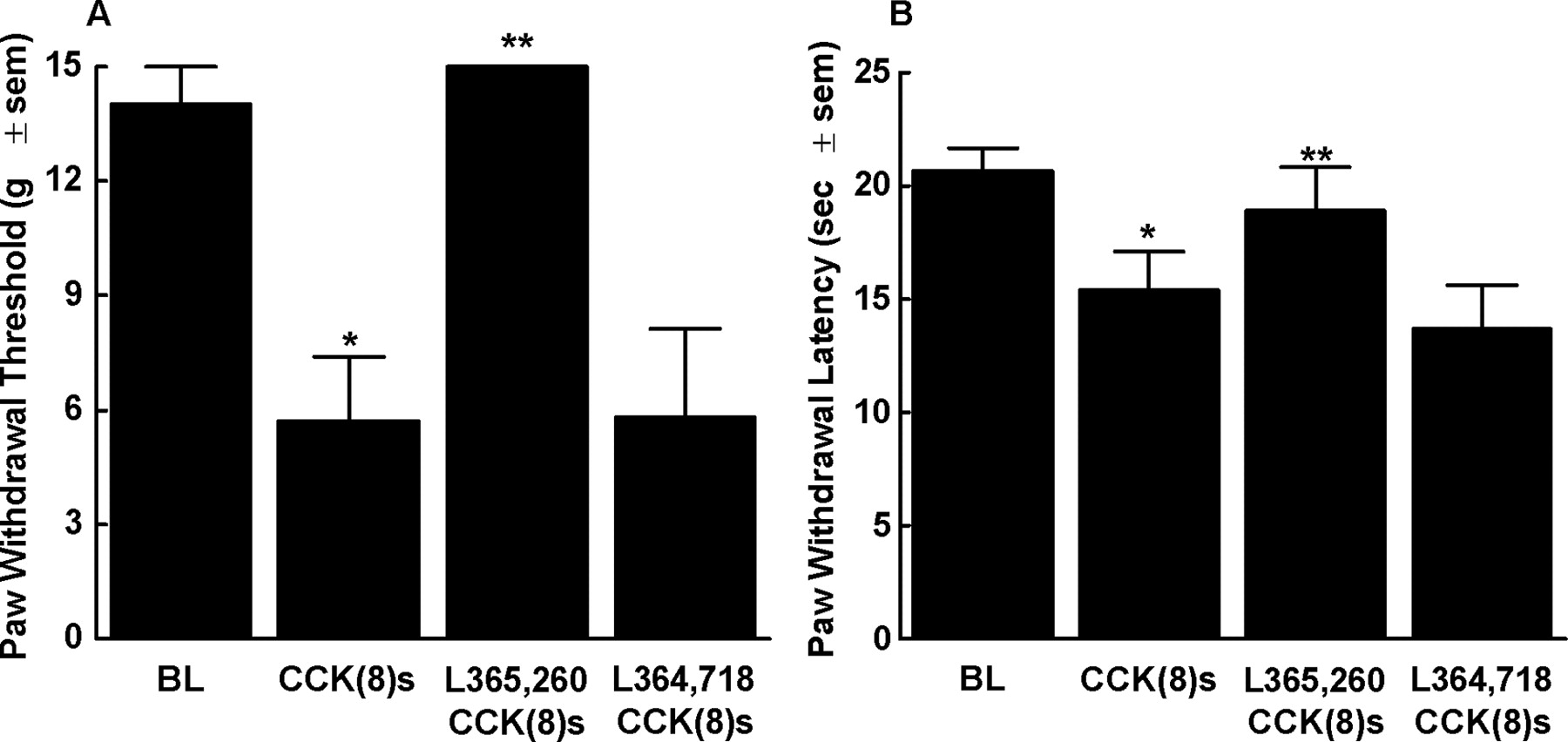
Male Sprague Dawley rats received CCK-8(s) (30 ng/0.5 μl) bilaterally into the RVM and were tested for mechanical (A) or thermal (B) hypersensitivity using von Frey filaments or radiant heat, respectively. The bilateral RVM administration of CCK-8(s) resulted in significant mechanical (A; n = 8) and thermal (B; n = 7) hypersensitivity (*p < 0.05) that was significantly blocked by the preadministration of the CCK2 receptor antagonist L365,260 (2.5 ng/0.5 μl, -5 min) administered bilaterally into the RVM (**p < 0.05; n = 8) but not by the CCK1 antagonist L364,718 (25 ng/0.5 μl, -5 min; n = 6). The preadministration of vehicle (0.5 μl, -5 min) had no effect on CCK-8(s)-induced mechanical and thermal hypersensitivity (n = 6). BL, Baseline.

Schematic representation of cannula and probe placement for injection and microdialysis in the RVM. Bilateral cannula placement (A) was histologically determined and marked as correct (filled circle) when the cannula tip fell within 1 mm rostral and caudal; 0.5 mm lateral from our designated RVM area of 11.5-10.0 mm anteroposterior; 0-1.5 mm mediolateral; 8.25-10.5 mm dorsoventral. Data from animals with cannulas that projected beyond this area (filled square) were not included. Similarly, the location of the microdialysis probes (B) were histologically confirmed and marked as correct (black bar) when the cannula tip fell with in the above stated range. Data from animals with probes located beyond this area (gray bar) were not included. This figure was adapted from the atlas of Paxinos and Watson (1986), and the abbreviations used on the figure can be found in that atlas.
Lesion of the DLF prevents CCK in the RVM-induced tactile and thermal hypersensitivity
Bilateral DLF, or sham DLF, lesions at the T8 level were performed 5 d before CCK in the RVM administration. Correct placement of the DLF lesions were confirmed post hoc, and only data from animals with correctly placed lesions were used in the behavioral analysis. Baseline paw-withdrawal thresholds to light mechanical stimuli were 15 ± 0.0 gm (cutoff for non-noxious mechanical thresholds) in sham DLF and DLF-lesioned rats, indicating that the DLF lesion alone did not significantly alter sensory thresholds in opioid naive rats. Rats that underwent a sham DLF lesion resulted in a decrease in mechanical threshold from baseline readings of 15.0 ± 0.0 to 5.4 ± 1.3 gm after CCK in the RVM (Fig. 2A). Thresholds returned to baseline levels within 120 min after CCK administration. In contrast, animals that received DLF lesions did not significantly express mechanical hypersensitivity after administration of CCK into the RVM with tactile thresholds of 10.5 ± 1.9 gm after CCK (Fig. 2A). Baseline paw-withdrawal thresholds to noxious thermal stimuli were 22.5 ± 2.1 and 21.1 ± 1.7 sec in sham DLF and DLF-lesioned rats, respectively, indicating that the DLF lesion alone did not significantly alter sensory thresholds. CCK in the RVM-induced thermal hypersensitivity in rats that underwent a sham DLF lesion resulted in a decrease from paw response baseline readings of 22.5 ± 2.1 to 14.1 ± 0.4 sec after CCK (Fig. 2B). Thresholds returned to baseline paw-withdrawal latencies within 120 min after CCK administration. In contrast, CCK in the RVM did not produce significant thermal hypersensitivity in animals that received DLF lesions. In these animals, thermal paw-withdrawal thresholds were 19.1 ± 3.3 sec after CCK (Fig. 2B), which was not significantly different from the pre-CCK paw-withdrawal baseline thresholds of 21.1 ± 1.7 sec. The histological placement of DLF lesions after testing was verified in all animals. Only one animal from the group that received CCK in the RVM had an incomplete lesion. Hence, the mechanical and thermal thresholds of 3.7 gm and 10.8 sec, respectively, for this animal were not included in the data analysis.

Male Sprague Dawley rats received bilateral lesions of the DLF or sham lesions at the T8 spinal level. Five days later, rats received CCK-8(s) (30 ng/0.5 μl) bilaterally into the RVM and were tested for mechanical (A) or thermal (B) hypersensitivity using von Frey filaments or radiant heat, respectively. CCK-8(s)-induced mechanical and thermal hypersensitivity (*p < 0.05) were significantly blocked in animals that underwent a bilateral DLF lesion compared with the sham DLF-lesioned animals (**p < 0.05). n = 6-8 animals in all studies. BL, Baseline.
CCK in the RVM decreases spinal morphine antinociceptive potency
Spinal morphine produced dose-related antinociception in the tail-flick test that was modulated by RVM administration of CCK. CCK or vehicle was microinjected into the RVM 5 min before spinal morphine. The morphine A50 (and 95% CI) value (constructed at 30 min after morphine) in animals receiving microinjection of vehicle into the RVM was 5.3 μg (4.1-7.0) (Fig. 3). In contrast, the morphine A50 (and 95% CI) (constructed 30 min after morphine) in animals receiving an RVM microinjection of CCK was 30.8 μg (21.8-43.5), which represents a sixfold rightward increase in the A50 value for spinal morphine. This decrease in spinal morphine antinociceptive potency was prevented by the administration of L365,260, a CCK2 receptor antagonist, 5 min before the injection of CCK into the RVM, i.e., 10 min before spinal morphine. In these animals, the spinal morphine antinociceptive A50 value (and 95% CI) calculated at 30 min after morphine was 5.2 μg (3.5-7.5), which was not significantly different from the control group that received vehicle into the RVM (Fig. 3). Injection of L365,260 alone into the RVM did not alter the antinociceptive potency of spinal morphine; the A50 value in this group was 4.4 μg (2.7-6.9).

Spinal morphine antinociceptive dose-response curves in male Sprague Dawley rats using the hot-water (52°C) tail-flick test. The preadministration of vehicle bilaterally into the RVM resulted in a spinal morphine dose-response curve with an A50 of 5.3 μg (circles). The preadministration of CCK-8(s) (30 ng/0.5 μl, -5 min) bilaterally into the RVM resulted in a significant rightward shift of the spinal morphine dose-response curve with an A50 of 30.8 μg (squares). The CCK-8(s) in the RVM-induced rightward shift of the spinal morphine dose-response curve was completely blocked by the RVM administration of the CCK2 antagonist L365,260 (2.5 ng/0.5 μl) 5 min before CCK-8(s) in the RVM with an A50 of 5.2 μg (triangles). Vehicle in the RVM before CCK-8(s) in the RVM had no effect on the rightward shift of the spinal morphine dose-response curve (data not shown), and L365,260 in the RVM (2.5 ng/0.5 μl) 5 min before vehicle in the RVM had no effect on the spinal morphine dose-response curve (upside-down triangles). All experiments included six to eight animals per dose tested. i.th., Intrathecal.
Morphine-induced hypersensitivity is blocked by microinjection of L365,260
Sustained morphine resulted in behavioral signs of mechanical hypersensitivity. Paw-withdrawal thresholds diminished from 14.7 ± 0.3 to 4.7 ± 0.8 gm after 7 d of continuous morphine exposure. RVM microinjection of L365,260 significantly reversed morphine-induced tactile thresholds to 13.1 ± 1.9 gm (Fig. 4A). This blockade of morphine-induced mechanical hypersensitivity was time related, and the mechanical threshold returned to pre-L365,260 value by 90 min after microinjection. Similarly, sustained morphine resulted in thermal hypersensitivity. The paw-withdrawal thresholds diminished from a pre-morphine baseline of 19.6 ± 0.7 to 14.5 ± 0.7 sec on day 7 after morphine implantation. Morphine-induced thermal hypersensitivity was reversed by injection of L365,260 into the RVM, after which the paw-withdrawal latency was 19.7 ± 1.9 sec (Fig. 4B). The effect of L365,260 was time related, and the latency of paw withdrawal returned to baseline levels within 90 min after the injection of the antagonist. Thermal and mechanical paw-withdrawal latencies-thresholds in animals implanted with placebo pellets were not different from baseline paw-withdrawal latencies-thresholds on day 7. Likewise, L365,260 administered into the RVM resulted in no difference in paw-withdrawal latencies-thresholds in placebo-pelleted animals compared with baseline thresholds (data not shown). The vehicle for L365,260 when administered into the RVM in morphine or placebo-pelleted rats had no significant effect on the paw-withdrawal thresholds or paw-withdrawal latencies.

Male Sprague Dawley rats received two 75 mg morphine pellets (MP) (sustained release from 7 to 10 d) or placebo pellets (PP) subcutaneously and were tested on day 7 after pellet implantation for morphine-induced mechanical (A) and thermal (B) hypersensitivity. On day 7, animals implanted with morphine pellets resulted in decreased mechanical (A) and thermal (B) thresholds compared with the placebo-pelleted animals (*p < 0.05). Bilateral RVM administration of the CCK2 antagonist L365,260 (2.5 ng/0.5 μl) on day 7 after pellet implantation resulted in a full reversal of morphine-induced mechanical (A) and thermal (B) hypersensitivity (**p < 0.05), yet no change was seen in placebo-pelleted animals. Vehicle in the RVM had no effect on morphine-induced hypersensitivities (data not shown). All experiments included six to eight animals. BL, Baseline.
Spinal morphine antinociceptive tolerance is blocked by L365,260 in the RVM
The antinociceptive dose-response curve for spinal morphine was generated 7 d after subcutaneous placebo or morphine pellets. Animals received an RVM microinjection of either L365,260 or vehicle 5 min before spinal morphine administration, and antinociceptive activity was evaluated an additional 30-40 min later. In animals with placebo pellets and receiving vehicle in the RVM, the A50 value for spinal morphine was 2.6 μg (1.9-3.6) (Fig. 5). In animals implanted with morphine pellets and receiving vehicle in the RVM, the A50 for spinal morphine was 11.9 μg (9.0-15.7). The difference in the A50 values represents a 4.6-fold rightward shift, indicative of antinociceptive tolerance in the morphine-pelleted rats (Fig. 5). RVM injection of L365,260 5 min before spinal morphine in placebo-pelleted animals did not significantly alter spinal morphine antinociceptive potency; the A50 (and 95% CI) in this group was 2.9 μg (2.1-4.0). In contrast, L365,260 microinjection into the RVM of the morphine-pelleted animals prevented the expected rightward shift in morphine antinociceptive potency. In these animals, the morphine A50 was 3.1 μg (2.1-4.7), which was not significantly different from that in the RVM vehicle-placebo-pelleted group.

Spinal morphine antinociceptive dose-response curves in male Sprague Dawley rats subcutaneously implanted with either two 75 mg morphine pellets (MP) or two placebo pellets (PP) using the hot-water (52°C) tail-flick test. Tail-flick latencies were recorded on day 7 after pellet implantation. The spinal morphine antinociceptive dose-response curve in placebo-pelleted rats with vehicle in the RVM resulted in an A50 of 2.6 μg (circles). However, the spinal antinociceptive morphine dose-response curve was significantly shifted to the right in morphine-pelleted rats-RVM vehicle with an A50 of 11.9 μg (squares). Bilateral RVM administration of the CCK2 antagonist L365,260 had no effect on the spinal morphine dose-response curve in placebo-pelleted animals (A50 of 2.9 μg) (triangles) but completely blocked the rightward shift of the spinal morphine dose-response curve in the morphine-pelleted animals with an A50 of 3.1 μg (upside-down triangles). All experiments included six to eight animals per dose of morphine tested. i.th., Intrathecal.
Increased extracellular levels of CCK-LI in the RVM in animals exposed to sustained morphine
The basal levels of CCK-LI in the RVM in morphine- or placebo-pelleted animals were measured using in vivo microdialysis. Microdialysis probe placement was histologically verified in every animal, and only data with proper RVM probe placements were included. The basal extracellular levels of CCK-LI were higher in morphine-pelleted animals when compared with placebo-pelleted animals measured on both day 2 and day 7. Microdialysis was performed over a 2.5 hr period in 30 min fractions. Placebo-pelleted animals showed an average baseline CCK-LI of 1.3 ± 0.1 pm [area under the curve (AUC), 5.1 ± 0.7; AUC over the entire five 30 min time points collected] over the 2.5 hr analysis period. Morphine-pelleted rats showed an average baseline of CCK-LI of 3.2 ± 0.3 pm (AUC, 13.2 ± 0.8) on day 2 after pellet implantation (Fig. 6A,C). Rats with morphine pellets resulted in an ∼2.5-fold (2.7-fold AUC) increase in basal extracellular CCK-LI levels when compared with placebo controls at day 2 after pellet implantation (Fig. 6C). Similarly, morphine-pelleted rats showed significantly higher basal levels of CCK-LI on day 7 after morphine; the basal CCK-LI levels were 5.9 ± 0.7 pm (AUC, 24.2 ± 1.6) versus placebo (1.3 ± 0.2 pm; AUC, 5.4 ± 0.6) pelleted animals (Fig. 6B,C), indicating an ∼4.6-fold AUC increase in basal extracellular CCK-LI levels when compared with placebo controls (Fig. 6C). To determine whether morphine had a local effect on CCK-LI release, we directly perfused a high concentration of morphine (100 μm) via the microdialysis probe into the RVM over the 2.5 hr period and collected microdialysate in 30 min intervals in naive animals. Over the entire 2.5 hr microdialysis period, the extracellular CCK-LI remained unchanged compared with artificial CSF-perfused animals; the former showed an average of basal extracellular CCK-LI of 1.5 ± 0.1 pm. Analysis from two animals in which the probe was misplaced (confirmed histologically) (1 mm rostral and caudal; 0.5 mm lateral from our designated RVM area of 11.5-10.0 mm anteroposterior, 0-1.5 mm mediolateral, 8.25-10.5 mm dorsoventral) resulted in an undetectable basal level of CCK in two animals, suggesting that either no CCK exists immediately outside the RVM or the amount of basal level of CCK is below our detection level of 0.78 pg/100 μl. Probe misplacement 1 mm posterior from the RVM (one animal with morphine pellets and two animals with placebo pellets) resulted in detectable levels of CCK and followed the trend of being higher in morphine-pelleted animals compared with the placebo-pelleted animals; however, these animals were not used in the final results because of the misplacement of the probe. RVM probe placements for all of the microdialysis experiments are displayed in Figure 7B.

RVM in vivo microdialysis for basal extracellular CCK-LI peptide in male Sprague Dawley rats implanted with either morphine or placebo pellets. Animals underwent microdialysis on day 2 or day 7 after pellet implantation. Thirty minute fraction collections over a 2.5 hr period were collected with RVM basal extracellular CCK-LI levels (1.3 pm on average) in placebo-pelleted animals on day 2 (A) or day 7 (B), similar to what has been reported in the literature (de Araujo Lucas et al., 1998; Gustafsson et al., 1999; Becker et al., 2000). However, there was a significant increase in RVM basal extracellular CCK-LI in animals with morphine pellets on day 2 (A), as well as on day 7 (B), after pellet implantation. The area under the curve (C) basal CCK-LI in the RVM on days 2 or 7 after either placebo pellet (open bars) or morphine pellet (filled bars) implantation. On day 2, animals with morphine pellets demonstrated a 2.6-fold increase in basal CCK-LI extracellular levels compared with placebo-pelleted animals, and, on day 7, animals with morphine pellets demonstrated a 4.6-fold increase in basal CCK-LI extracellular levels compared with placebo-pelleted animals. In vivo microdialysis experiments only include animals with proper cannula placement (n = 7 for all groups).
Discussion
The present study used pharmacological and physiological approaches to uncover the role of CCK in the RVM in opioid-induced mechanical and thermal hyperalgesia and the possible relationship of CCK in the RVM to opioid antinociceptive tolerance. Our data demonstrate that pharmacological administration of CCK in the RVM of naive rats elicits tactile and thermal hypersensitivity of the hindpaws, which is mediated by CCK2 receptor activation. Such hyperalgesia was likely the result of activation of a descending pain facilitatory system because this effect was prevented, or significantly attenuated, by previous lesions of the DLF. Furthermore, CCK in the RVM-induced activation of descending pain facilitation in opiate-naive rats resulted in a rightward displacement of the spinal morphine antinociceptive dose-response curve, which is similar to that seen after prolonged opioid administration and is characteristic of antinociceptive tolerance. Consistent with these pharmacological observations, RVM administration of L365,260, a CCK2 receptor antagonist, reversed the tactile and thermal hypersensitivity resulting from continuous morphine delivery. Blockade of CCK2 receptors in the brainstem also prevented the rightward displacement of the spinal morphine dose-response in these morphine-exposed animals. Finally, baseline levels of CCK in the RVM were found to be significantly elevated after sustained morphine administration, indicating the presence of enhanced CCK tone in these animals. Together, these findings point to a role of endogenous CCK activity in the RVM as a mediator of opioid-induced mechanical and thermal hypersensitivity, descending pain facilitation, and of spinal morphine antinociceptive potency (i.e., antinociceptive tolerance).
These studies have confirmed and extended our previous observations showing that pharmacological administration of CCK into the RVM, or into sites somewhat dorsal to the RVM (Kovelowski et al., 2000), induces thermal and mechanical hypersensitivity in naive rats. These effects were time dependent, were mediated via the CCK2 receptors, and were abolished by lesions of the DLF, suggesting activation of a descending pain facilitation mechanism. Descending modulation of pain from the RVM has been characterized previously as playing an important role in spinal nociception (Fields et al., 1983; Fields and Heinricher, 1985; Morgan and Fields, 1994; Friedrich and Gebhart, 2003) and is mediated by bulbospinal projections via the DLF (Zhou and Gebhart, 1992; Urban et al., 1996; Urban and Gebhart, 1997; Fields and Basbaum, 1999). CCK in the RVM inhibits the antinociceptive effect of systemic morphine by preventing morphine-induced increase in the firing of RVM “OFF” cells (Heinricher et al., 2001). A recent observation that microinjection of CCK into the RVM can also activate the firing of “ON” cell, suggests that CCK activity in the RVM may mediate descending pain facilitation (Heinricher and Neubert, 2004)). In addition, the antinociceptive effect of spinal morphine can be attenuated by systemic, spinal, or supraspinal administration of CCK (Faris et al., 1983; Li and Han, 1989; Stanfa and Dickenson, 1993). Consistent with these observations, our studies show that RVM administration of CCK displaced the spinal morphine dose-response curve to the right. The fact that this right shift in spinal morphine potency is coincident with hyperalgesia induced by CCK in the RVM and can be similarly blocked by L365,260 in the RVM suggests that CCK2 receptor activation in the RVM increased nociceptive drive to the spinal cord, which acutely diminishes spinal morphine antinociception. L365,260 given alone in the RVM, without the pharmacological action of exogenous CCK, did not alter the potency of spinal morphine, showing that the descending pronociceptive pathway is apparently not normally active and has little influence on normal spinal morphine antinociceptive efficacy. It is important to note that, unlike the present studies, previous findings showing that blockade of the CCK receptor enhanced the antinociceptive effects of opioids demonstrated these effects after administration of the opioid and CCK antagonist at the same anatomical region or by the use of systemic opiates (Dourish et al., 1990; Wiesenfeld-Hallin et al., 1990; Vanderah et al., 1994, 1996; Friedrich and Gebhart, 2003).
The role of descending projections from the RVM to the spinal cord via the dorsolateral funiculus on the manifestation of morphine antinociceptive tolerance has been implicated in our previous studies (Vanderah et al., 2000, 2001a,b). Continuous exposure to opioids, either by osmotic minipump infusions of subcutaneous morphine or by morphine pellet implantation produced behavioral signs of abnormal pain and antinociceptive tolerance that were abolished by lesions of the dorsolateral funiculus or by lidocaine administration into the RVM. The present study substantiates these previous observations by demonstrating that activation of the CCK2 receptors in the RVM may underlie the pronociceptive descending input to the spinal cord, and blockade of these receptors is sufficient to simultaneously abolish opioid-induced hyperalgesia and antinociceptive tolerance. As noted above, because the CCK2-selective antagonist did not alter baseline sensory thresholds or spinal morphine potency in naive animals, it is likely that there is normally minimal endogenous CCK activity in the RVM. However, the evidence here shows that the CCK activity in the RVM must increase during sustained morphine administration because the basal level of CCK-LI rises substantially in the RVM and that CCK2 blockade in the RVM now significantly influences both sensory thresholds and spinal morphine potency. Furthermore, the significant upregulation of basal release of CCK-LI as well as the pharmacological action of L365,260 implicate a tonic activity of CCK in the RVM that would then result in a descending input to the spinal cord.
The source of this tonic CCK output in the RVM is at present unknown. Previous studies have demonstrated that morphine can enhance extracellular levels of CCK. Microdialysis studies in vivo demonstrated that systemic and spinal morphine increased CSF levels of CCK (de Araujo Lucas et al., 1998). Furthermore, systemic morphine resulted in an 89% increase in CCK levels in spinal cord perfusate (Zhou et al., 1993). Microdialysis studies also revealed a naloxone-sensitive, marked increase in extracellular CCK in the frontal cortex of conscious rats after systemic morphine (Becker et al., 1999). The CCK-LI release in the dorsal horn evoked by systemic administration of morphine was blocked by spinal naloxone (Gustafsson et al., 1999). However, our data show that direct, acute administration of morphine into the RVM did not elicit measurable CCK-LI release. These findings suggest that the release of CCK in the RVM is unlikely because of local release from RVM interneurons but may arise from excitatory input to the RVM. The latter remains to be elucidated.
Many studies have shown that CCK antagonists can block and reverse opioid-induced antinociceptive tolerance (Watkins et al., 1984; Dourish et al., 1990; Hoffmann and Wiesenfeld-Hallin, 1994; Tortorici et al., 2003). However, there is a major distinction between these previous observations and the ones made here. Whereas the anti-opioid effect of CCK observed previously concerns mainly the local mode of action of CCK (i.e., spinal CCK2 activation that antagonizes spinal morphine to attenuate afferent nociceptive input), the present findings concern an indirect, distant site of action of CCK on spinal morphine antinociception. The causal relationship between a CCK-responsive, pain facilitatory input to the spinal cord and morphine antinociceptive tolerance strongly supports the argument that the latter is an out-come of enhanced nociceptive transmission at the spinal level, and the enhanced CCK transmission in the RVM provides the first evidence for a supraspinal mechanism that may underlie spinal morphine antinociceptive tolerance. Furthermore, a loss of spinal morphine potency attributable to hyperactivity of the CCK-responsive neurons in the RVM may also underlie systemic opioid antinociceptive tolerance, because the potency of systemic morphine has been shown to depend on the synergy between spinal and supraspinal morphine activity, and such synergy is lost after sustained administration of opioids (Roerig et al., 1984).
One hypothesis that has been advanced to explain the phenomenon of opioid tolerance is the suggestion of an “upregulation of endogenous anti-opioids” (Rothman et al., 1993). Our current observations support this concept and have unraveled a specific mechanism by which enhanced pronociceptive tone in the RVM, driven at least in part from enhanced levels and activity of CCK, activates descending pain facilitation processes to produce a hyperalgesic state. We suggested previously that enhanced pain may be regarded as a “physiological antagonist” of antinociception and that manipulations that block enhanced pain can block the expression of opiate antinociceptive tolerance (for review, see Vanderah et al., 2001a). Accordingly, blockade of upregulated pronociceptive (i.e., anti-opioid) systems, such as those driven by RVM CCK that appear to be engaged by sustained morphine exposure, may provide significant therapeutic benefit. Recent studies have demonstrated that CCK antagonists can enhance opioid analgesia in humans (McCleane, 1998, 2000, 2003). Our studies suggest a mechanism by which this might occur and indicate that blockade of CCK receptors may prevent or reverse opioid-induced hyperalgesia and the expression of opioid analgesic tolerance, providing a potential strategy for improving the efficacy of opioids as therapy in chronic pain conditions.
Footnotes
These studies were supported by National Institutes of Health-National Institute on Drug Abuse Grant DA15205-01, and Swedish Medical Research Council Grant 6836, Magnus Bergvalls Stiftelse, the Swedish Society of Medicine, and the Karolinska Institute. We thank Annika Olsson for excellent technical assistance with the RIAs.
Correspondence should be addressed to Dr. Todd W. Vanderah, University of Arizona, Department of Pharmacology, 1501 N. Campbell, Tucson, AZ 85724. E-mail: vanderah{at}u.arizona.edu.
L. R. Gardell's present address: ACADIA Pharmaceuticals, 3911 Sorrento Valley Boulevard, San Diego, CA 92121.
Copyright © 2005 Society for Neuroscience 0270-6474/05/250409-08$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}