

Abstract
Benztropine and its analogs are tropane ring–containing dopamine uptake inhibitors that produce behavioral effects markedly different from cocaine and other dopamine transporter blockers. We investigated the benztropine binding site on dopamine transporters by covalently attaching a benztropine-based photoaffinity ligand, [125I]N-[n-butyl-4-(4‴-azido-3‴-iodophenyl)]-4′,4"-difluoro-3α-(diphenylmethoxy)tropane ([125I]GA II 34), to the protein, followed by proteolytic and immunological peptide mapping. The maps were compared with those obtained for dopamine transporters photoaffinity labeled with a GBR 12935 analog, [125I]1-[2-(diphenylmethoxy)ethyl]-4-[2-(4-azido-3-iodophenyl)ethyl]piperazine ([125I]DEEP), and a cocaine analog, [125I]3β-(p-chlorophenyl)tropane-2β-carboxylic acid, 4′-azido-3′-iodophenylethyl ester ([125I]RTI 82), which have been shown previously to interact with different regions of the primary sequence of the protein. [125I]GA II 34 became incorporated in a membrane-bound, 14 kDa fragment predicted to contain transmembrane domains 1 and 2. This is the same region of the protein that binds [125I]DEEP, whereas the binding site for [125I]RTI 82 occurs closer to the C terminal in a domain containing transmembrane helices 4–7. Thus, although benztropine and cocaine both contain tropane rings, their binding sites are distinct, suggesting that dopamine transport inhibition may occur by different mechanisms. These results support previously derived structure–activity relationships suggesting that benztropine and cocaine analogs bind to different domains on the dopamine transporter. These differing molecular interactions may lead to the distinctive behavioral profiles of these compounds in animal models of drug abuse and indicate promise for the development of benztropine-based molecules for cocaine substitution therapies.
- cocaine
- benztropine
- dopamine transporter
- photoaffinity label
- dopamine uptake
- proteolytic peptide mapping
- immunoprecipitation
One of the major structural contributors to high-affinity binding of cocaine and its analogs to the dopamine transporter (DAT) is the tropane ring (Carroll et al., 1992a). Benztropine, a dopamine uptake inhibitor equipotent to cocaine, also contains a tropane ring that is considered essential for its high-affinity binding. However, several studies have revealed that benztropine and its analogs display binding and behavioral profiles substantially different from those of cocaine-like compounds (Meltzer et al., 1994, 1996, 1997; Newman et al., 1994, 1995; Agoston et al., 1997a; Katz et al., 1997; Kline et al., 1997), leading to the suggestion that the two series of ligands interact at different sites on the protein (Newman et al., 1994, 1995). To investigate this hypothesis further, we have developed a benztropine-based photoaffinity ligand, which binds irreversibly to the DAT, to identify its binding regions and compare them with those of cocaine (Agoston et al., 1997b).
The characterization of molecular targets of drug action has been aided by the use of selective high-affinity probes and the application of molecular and immunological techniques. These approaches can be combined to identify discrete binding sites and elucidate molecular mechanisms underlying drug action. One of the primary central targets for cocaine action is the DAT where synaptic dopamine is cleared using the energy of electrochemical ion gradients to drive transport of extracellular dopamine into presynaptic terminals. When the DAT is blocked by cocaine, dopamine levels are increased, and the resulting stimulation of postsynaptic dopamine receptors is believed to be the primary neurochemical mechanism underlying the psychostimulant and reinforcing actions of the drug (Ritz et al., 1987; Kuhar et al., 1991). Although there is compelling evidence to support the pivotal role the dopamine transporter plays in the reinforcing actions of psychostimulant drugs, subtle uptake inhibitor binding properties, at the molecular level, may be requisite to effect neurochemical and behavioral endpoints.
Many dopamine uptake blockers, including highly selective and potent analogs of cocaine, GBR 12909, benztropine, mazindol, and methylphenidate, have been characterized extensively and are being evaluated preclinically as potential treatments for cocaine abuse (for review, see Carroll et al., 1997). Despite the large amount of pharmacological and behavioral data that has been collected on these structurally diverse compounds, the molecular mechanism by which they bind to the protein and block transport is not known. For example, it is currently unclear whether or not these compounds bind at single or multiple sites or whether they interfere with the same or different aspects of substrate translocation (Berger et al., 1990; Meiergerd and Schenk, 1994; Pristupa et al., 1994; Reith et al., 1996). Although some studies have indicated that many of these compounds access a common binding domain (Froimowitz, 1993; Froimowitz et al., 1997), more extensive structure–activity relationships (SAR) have identified molecular requirements indicating that some ligands may be accessing different domains (Meltzer et al., 1994, 1996, 1997; Newman et al., 1994, 1995; Agoston et al., 1997a). Elucidating the nature of these ligand–protein interactions is essential for understanding the molecular basis of DAT action and for the development of potential cocaine pharmacotherapies.
Herein, we report the discovery of a novel photoaffinity ligand, based on benztropine, and the identification of its recognition site on the dopamine transporter via the use of proteolytic and immunological peptide mapping. Furthermore, despite its tropane ring pharmacophore, this probe does not covalently bond to the same transmembrane domain region and thus may not be accessing the same binding site as cocaine. This study confirms the divergent SAR in these classes of compounds and may be relevant to their distinctive pharmacological profiles in animal models of cocaine abuse.
MATERIALS AND METHODS
Synthesis of [125I]GA II 34. The radiosynthesis of [125I]N-[n-butyl-4(4‴-azido-3‴-iodophenyl)]-4′,4"-difluoro-3α-(diphenylmethoxy)tropane ([125I]GA II 34) has been communicated in brief (Agoston et al., 1997b) and is reported here in full. Detailed methods for the preparation of [125I]1-[2-(diphenylmethoxy)ethyl]-4-[2-(4-azido-3-iodophenyl)ethyl]piperazine ([125I]DEEP) and [125I]3β-(p-chlorophenyl)tropane-2β-carboxylic acid, 4′-azido-3′-iodophenylethyl ester ([125I]RTI 82) have been reported previously (Grigoriadis et al., 1989; Wilson et al., 1989; Lever et al., 1993). To prepare [125I]GA II 34, we treated a solution ofN-[n-butyl-4-(4′-aminophenyl)]-4′,4"-difluoro-3α-(diphenylmethoxy)tropane (50 μl; 3.0 mm) in a 2:1 mixture of aqueous sodium acetate buffer (0.3 m), pH 4.0, and methanol at ambient temperature with no carrier-added Na125I (20 μl; 2.05 mCi; 1.0 nmol) followed by Chloramine-T trihydrate (15 μl; 3.5 mm). After 30 min, the mixture was chilled at −4°C and treated with acetic acid (50 μl; 3.0 m) followed by sodium nitrite (25 μl; 0.5 m). After 15 min, sodium azide (25 μl; 0.5 m) was added, and the mixture was allowed to warm to ambient temperature. After 10 min, the reaction was quenched with sodium metabisulfite (10 μl; 50 mm) and taken up in a syringe along with a rinse of the reaction vessel with 0.2 ml of the mobile phase to be used for HPLC purification. This ternary solvent consisted of methanol (30%), acetonitrile (30%), and an aqueous solution (40%) of triethylamine (2.1% v/v) and acetic acid (2.8% v/v). The HPLC system was equipped with a UV absorbance detector (254 nm), a flow-through radioactivity detector, and a Waters C-18 Nova-Pak column (radial compression module; 8 × 100 mm; 6 μm). At a flow rate of 4 ml/min, [125I]GA II 34 (retention time, 20.1 min; capacity factor, 29.4) was resolved from both nonradioactive and radioactive side products. The radioligand (1.56 mCi; 76.1%) was collected in a 10 ml volume, diluted to 50 ml with distilled water, and passed through an activated solid phase extraction cartridge (Waters Sep-Pak Light t-C-18) that was flushed with water (2.5 ml) to remove salts and then with argon. Elution of the cartridge with ethanol (1.5 ml) containing 1% (v/v) Tris buffer (5 mm), pH 7.4, gave a concentrated solution of [125I]GA II 34 that had ≥99.8% radiochemical purity by HPLC. The specific radioactivity was calculated as 2075 mCi/μmol using HPLC to determine the mass of carrier in a sample having known radioactivity. The UV absorbance peak height of the carrier was related to the equation for a linear (r2 = 0.97) six-point standard curve established by HPLC with the nonradioactive compound over a concentration range (35–700 pmol) chosen to bracket the region of interest.
Photoaffinity labeling. DATs were photoaffinity labeled with [125I]GA II 34, [125I]DEEP, or [125I]RTI 82 as described previously (Vaughan, 1995; Vaughan and Kuhar, 1996; Agoston et al., 1997b). Male Sprague Dawley rats (150–250 gm) were decapitated, and the striata were rapidly removed and weighed. Tissue was homogenized in 10 ml of ice-cold sucrose-phosphate (SP) buffer (10 mmNa2HPO4, pH 7.4, containing 0.32m sucrose), using a Brinkman Polytron homogenizer. The homogenate was centrifuged at 20,000 × g for 12 min, the supernatant was discarded, and the homogenization and centrifugation were repeated. The resulting membranes were suspended in SP at 5–10 mg/ml original wet weight followed by addition of 5 nm [125I]DEEP or [125I]RTI 82 or 40 nm[125I]GA II 34. Samples were incubated for 1 hr at 0°C and irradiated with UV light for 45 sec, and the membranes were washed twice by centrifugation. For subsequent gel purification, membranes were solubilized with SDS-PAGE sample buffer (62.5 mm Tris, pH 6.8, 0.5% SDS, 10% glycerol, and 10 mm dithiothreitol) at 20 mg/ml original wet weight. For subsequent in situ proteolysis, the unsolubilized membranes were suspended in 50 mm Tris-HCl, pH 8.0, at 50 mg/ml original wet weight.
Proteolytic peptide mapping. Peptide mapping of photoaffinity-labeled DAT was done with two different procedures, (1) proteolysis after gel purification and (2) in situproteolysis of membrane suspensions. For comparison of DATs labeled with different photoaffinity ligands, samples were simultaneously prepared and analyzed exactly in parallel. For gel purification, solubilized photolabeled membranes were electrophoresed on 8% gels, and DATs were identified by autoradiography. The 80 kDa region of the gel containing the DAT was excised, and the protein was passively eluted from the gel with 0.1 m ammonium bicarbonate, pH 8.0, containing 0.1% SDS. Aliquots were treated with 2–2000 μg/ml trypsin for 1 hr, followed by addition of sample buffer and electrophoresis on 14% SDS-PAGE gels. For in situproteolysis, photolabeled membrane suspensions were treated with trypsin, followed by separation of membranes and supernatants and immunoprecipitation of the membrane samples. This permits determination of the proximity of label incorporation to transmembrane helices, because retention of a proteolyzed fragment in the membranes denotes the presence of integral membrane structure.
Immunoprecipitation. Immunoprecipitation of photolabeled DAT fragments was performed using antisera generated against the following epitopes of the DAT primary sequence: antibody 15, amino acids (aa) 6–30; antibody 16, aa 42–59; antibody 5, aa 225–236; and antibody 18, aa 580–608. Epitopes 15 and 16 are in the N-terminal tail, epitope 5 is at the C-terminal side of the large extracellular loop between transmembrane domains (TMDs) 3 and 4, and epitope 18 is in the C-terminal tail. These antibodies have been well characterized by immunoprecipitation and immunohistochemistry in a variety of studies and shown to be highly specific for the DAT (Vaughan, 1995; Nirenberg et al., 1996; Vaughan and Kuhar, 1996). For immunoprecipitation, photolabeled striatal membranes, with or without trypsin treatment, were solubilized with SDS-PAGE sample buffer and diluted to 0.1% SDS with 50 mm Tris-HCl, pH 8.0, plus 0.1% Triton X-100–containing antiserum diluted 1:10–1:100. Samples were incubated at 4°C for 1 hr followed by the addition of 20 μl of protein Sepharose CL4B (Pharmacia, Piscataway, NJ) for an additional hour. Immune complexes were washed twice with the Tris–Triton buffer, and samples were eluted from the beads with SDS-PAGE sample buffer. The samples were electrophoresed on 14% polyacrylamide gels followed by autoradiography using Kodak BioMax film for 1–5 d or by computer image analysis using a Molecular Dynamics PhosphorImager (Sunnyvale, CA). For peptide-blocking experiments, diluted antiserum 16 was preincubated with peptide 16 or peptide 12 (aa 541–550) at 50 μg/ml (Vaughan, 1995) before addition of sample. Amersham Rainbow molecular mass markers (Arlington Heights, IL) were standards on all gels.
RESULTS
During our SAR study of a large series of 3- and N-substituted benztropine analogs, we identified a compound,N-(4"-phenyl-n-butyl)-3α-[bis(4′-fluorophenyl)methoxy]tropane, that demonstrated high affinity and selectivity for the DAT (Agoston et al., 1997a). This molecule provided an ideal template for addition of125I and an azido functionality to generate a radiolabeled-benztropine photoaffinity ligand. The synthesis and characterization of the photoaffinity compound GA II 34 has been described previously (Agoston et al., 1997b). When assessed by binding competition against [3H]WIN 35,428, GA II 34 displayed a Ki of 160 nmand, after photoactivation, bound to the DAT in a wash-resistant, irreversible manner. The structures of [125I]GA II 34, [125I]DEEP, and [125I]RTI 82 are shown in Figure 1.

Chemical structures of [125I]GA II 34, [125I]RTI 82, and [125I]DEEP.
Photoaffinity labeling of the DAT with [125I]GA II 34 is shown in Figure 2. Total photolabeled rat striatal membranes show many nonspecifically radiolabeled proteins similar to that seen when membranes are labeled with [125I]DEEP or [125I]RTI 82 (Vaughan and Kuhar, 1996). However, in contrast to [125I]DEEP- or [125I]RTI 82–labeled membranes, the membranes labeled with [125I]GA II 34 show no obvious protein at 80 kDa, possibly because of the 10-fold lower affinity of [125I]GA II 34 (Ki = 160 nm) relative to the other ligands (Ki = 15 nm) (Grigoriadis et al., 1989; Carroll et al., 1992b). The presence of [125I]GA II 34–labeled DATs in these membranes was demonstrated by immunoprecipitation with DAT antiserum 16 (Fig. 2), which shows that the extracted protein migrates at 80 kDa and is precipitated by immune but not preimmune serum. We have shown previously that [125I]GA II 34–labeled DATs comigrate with [125I]RTI 82–labeled DAT (Agoston et al., 1997b).

Photoaffinity labeling of the DAT with [125I]GA II 34. Striatal membranes were photoaffinity labeled with [125I]GA II 34, and aliquots of the solubilized membranes were electrophoresed directly (Total) or immunoprecipitated with preimmune or immune serum 16 as indicated. Molecular mass standards for all gels are shown on the left in kilodaltons.
Investigation of the binding site of [125I]GA II 34 was done with the proteolysis and immunoprecipitation strategies used to localize sites of interaction of [125I]DEEP and [125I]RTI 82. We first performed proteolysis of gel-purified [125I]GA II 34–labeled DATs and compared its peptide map pattern with that of gel-purified DATs labeled with [125I]DEEP and [125I]RTI 82 (Fig. 3). Gel purification before proteolysis provides a radioactively pure sample, so that all radioactive fragments generated originate from the DAT and can be visualized (Vaughan, 1995; Vaughan and Kuhar, 1996). Proteolysis of these samples showed that the mapping pattern of DATs labeled with [125I]GA II 34 is identical to that of DATs labeled with [125I]DEEP and clearly distinct from that of DATs labeled with [125I]RTI 82. DATs labeled with [125I]GA II 34 or [125I]DEEP each generated 45 kDa fragments at 20 μg/ml trypsin, 45 and 14 kDa fragments at 200 μg/ml trypsin, and 7 kDa fragments at 2000 μg/ml trypsin. In contrast, proteolysis of [125I]RTI 82–labeled DATs generated a 32 kDa fragment at 20 and 200 μg/ml trypsin and a fragment of <6.5 kDa at 2000 mg/ml trypsin. Minor nonspecific contaminants not routinely obtained are present in some samples. The proteolysis patterns for [125I]DEEP- and [125I]RTI 82–labeled DATs shown in Figure 3 are identical to those described previously (Vaughan, 1995).

Peptide maps of DATs labeled with [125I]GA II 34, [125I]DEEP, or [125I]RTI 82. DATs photoaffinity labeled with the indicated compound were gel purified to remove radiolabeled contaminants, and the samples were treated with the indicated concentrations of trypsin. The resulting samples were electrophoresed on 14% gels and were subjected to autoradiography.
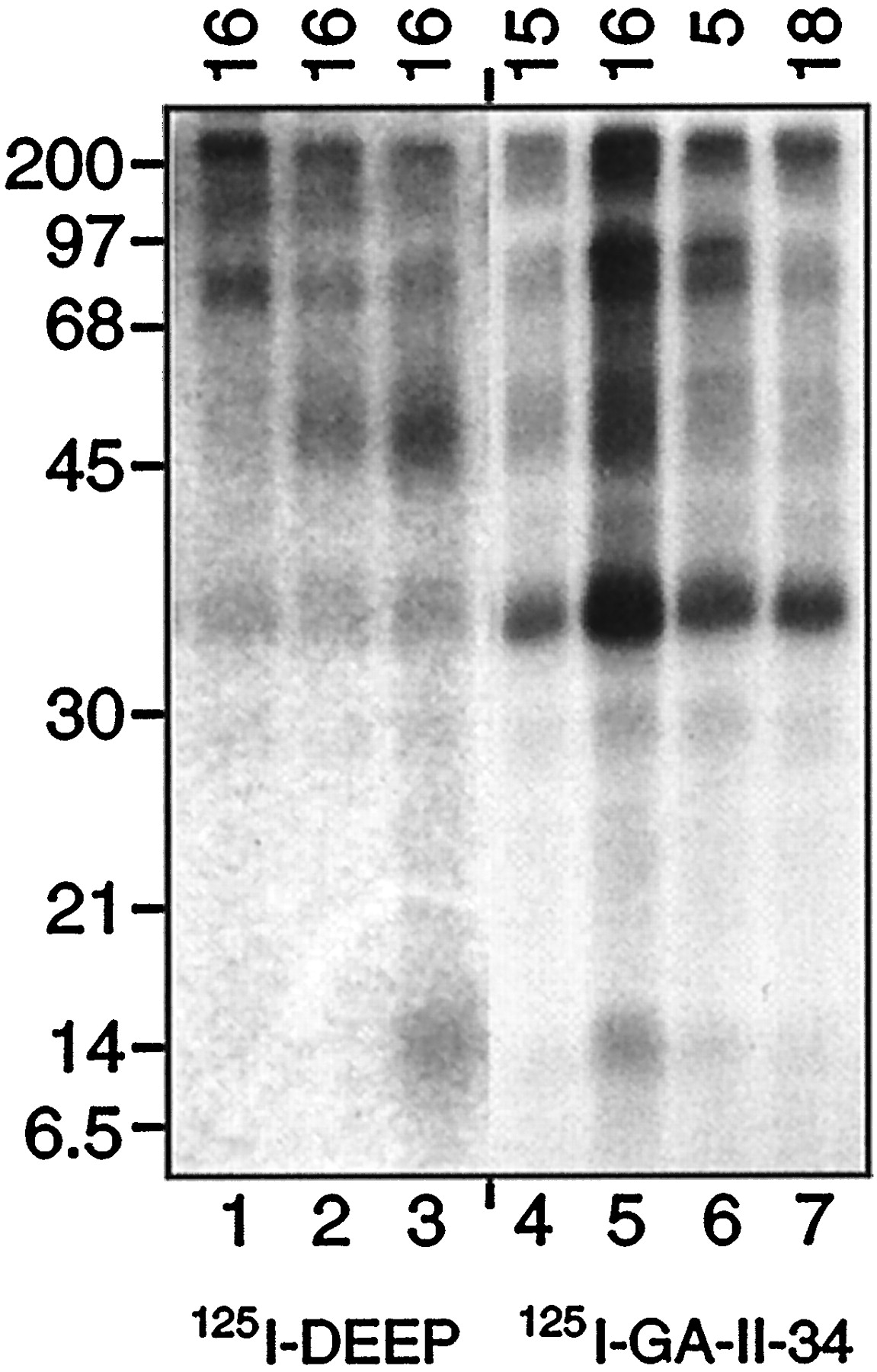
To verify the site of [125I]GA II 34 incorporation, we analyzed photolabeled tryptic fragments by epitope-specific immunoprecipitation. [125I]GA II 34–photolabeled membranes were treated to in situproteolysis with 10 μg/ml trypsin, membranes were separated from supernatants by centrifugation, and aliquots of solubilized membranes were immunoprecipitated with serum 15, 16, 5, or 18 (Fig.4). [125I]DEEP-labeled membranes were protease-treated and precipitated with serum 16 in parallel. [125I]RTI 82–labeled samples were not included in this analysis because the low molecular weight proteolytic fragments originate from more C-terminal regions of the protein (near transmembrane domain 4–6) and do not immunoprecipitate with antibody 16 (see Fig. 6) (Vaughan, 1995; Vaughan and Kuhar, 1996).

Immunoprecipitation of [125I]GA II 34 and [125I]DEEP tryptic fragments. [125I]GA II 34–labeled striatal membranes were treated with 10 μg/ml trypsin, the supernatants were removed, and the resulting membranes were solubilized and immunoprecipitated with antisera 15, 16, 5, or 18 as indicatedalong the top (lanes 4–7). [125I]DEEP-labeled membranes were treated in parallel with 0, 1, or 10 μg/ml trypsin (lanes 1–3, respectively) and precipitated with serum 16. Samples were electrophoresed on 14% gels followed by autoradiography.
The results show that the 14 kDa fragment labeled with [125I]GA II 34 was precipitated by serum 16 (Fig.4, lane 5) but not by antisera 15, 5, or 18 (lanes 4, 6, 7). Some remaining unproteolyzed protein and the 45 kDa fragment also precipitate with serum 16; this is seen more clearly in Figure 5. Figure 4, lanes 2and 3, shows the serum 16 immunoprecipitation of [125I]DEEP-labeled fragments generated by 1 and 10 μg/ml trypsin, respectively. The 45 and 14 kDa fragments produced by these treatments both precipitate with serum 16, and previous experiments showed that the 14 kDa [125I]DEEP-labeled fragment was not recognized by DAT antisera 15, 5, or 18 (Vaughan and Kuhar, 1996). These results provide strong evidence that the 14 kDa fragments, although labeled with structurally distinct compounds, are originating from the same region of the DAT primary sequence. The specificity of serum 16 recognition of the [125I]GA II 34–labeled fragments is shown in Figure 5, which demonstrates that peptide 16 blocks serum 16 precipitation of the 14 and 45 kDa fragments as well as unproteolyzed DAT, whereas an irrelevant peptide (peptide 12) has no effect. The 35 kDa band present in all samples in Figure 4 is a nonspecific contaminant that was less pronounced in other experiments (Fig. 5).

Specificity of precipitation of [125I]GA II 34 tryptic fragments. [125I]GA II 34–labeled striatal membranes were treated with 10 μg/ml trypsin, the supernatants were removed, and the membranes were solubilized. The resulting samples were immunoprecipitated with antiserum 16 that contained either no addition (no add’n), 50 μg/ml peptide 16 (+ pep 16), or 50 μg/ml peptide 12 (+ pep 12), as indicated along the top. An aliquot of the solubilized input sample (Total) is shown in the left lane.
DISCUSSION
There is currently very little understanding of the molecular basis of dopamine transport or the mechanisms by which uptake blockers inhibit transport. The structure of the DAT is believed to consist of 12 TMDs with intracellularly oriented N and C terminals and a large N-glycosylated extracellular loop between TMDs 3 and 4 (for review, see Amara and Kuhar, 1993). Specific residues that contribute to function include aspartic acid 79 in TMD 1, which is essential for dopamine (DA) transport and contributes more modestly to cocaine binding, and serines 356 and 359 in TMD 7, which contribute to DA transport but not to cocaine binding (Kitayama et al., 1992). More generalized domains identified by analysis of dopamine–norepinephrine transporter chimeras have also indicated that N-terminal transmembrane domains are essential for DA translocation, whereas cocaine binding was more associated with TMDs 6–8 in the central third of the polypeptide (Buck and Amara, 1994; Giros et al., 1994). Binding-site studies using photoaffinity labels based on GBR 12935 and cocaine have shown that these two structurally different dopamine uptake inhibitors interact with different regions of the DAT primary sequence (Vaughan, 1995;Vaughan and Kuhar, 1996). [125I]DEEP, a GBR 12935 analog, becomes incorporated in a region of the protein near TMDs 1 and 2, whereas [125I]RTI 82, a tropane-based cocaine analog, becomes incorporated closer to the C terminal, near TMDs 4–7 (Vaughan, 1995; Vaughan and Kuhar, 1996). Incorporation of both compounds is closely associated with transmembrane regions (Vaughan and Kuhar, 1996), indicating that the ligand binding sites and/or transport inhibition mechanisms involve transmembrane-spanning structure, possibly a translocation pore or channel. Taken together these studies suggest that binding of GBR-like compounds in or near TMD 1 may block uptake by interfering with a transport process involving aspartic acid 79 and that cocaine and its analogs interact with the central region of the protein. These data are also at least partially compatible with a recently proposed three-dimensional computer model of DAT based on cocaine SAR that suggested that TMDs 1, 7, and 9–11 are involved in cocaine binding (Edvardsen and Dahl, 1994). However, computer modeling of dopamine uptake inhibitors structurally unrelated to cocaine has not yet been attempted, and more precise understanding of the DAT three-dimensional structure and molecular basis of action is currently unknown.
The present study extends our knowledge of DAT structure and function by providing strong evidence that the benztropine compound [125I]GA II 34 becomes incorporated in the DAT primary sequence in the same N-terminal region as does [125I]DEEP. Based on their size and reactivity with serum 16, the [125I]GA II 34– and [125I]DEEP-labeled 14 kDa fragments generated in this study would be expected to contain putative transmembrane domains 1 and 2, as well as some flanking hydrophilic structure. The presence of integral membrane structure within these polypeptide sequences is verified by their retention in membranes after protease treatment and indicates that binding of [125I]GA II 34 is also closely associated with transmembrane-spanning helices. We had localized previously the [125I]DEEP incorporation site more precisely to a 4 kDa sequence containing TMDs 1 and 2, with little to no flanking structure, indicating that [125I]DEEP incorporation occurs either in one of these TMDs or in the six intervening extracellular amino acids (Vaughan, 1995). Although we did not characterize [125I]GA II 34 fragments smaller than 14 kDa in this study, the mapping patterns of [125I]DEEP and [125I]GA II 34 were indistinguishable (Fig. 4), presenting the possibility that [125I]GA II 34 may be incorporated in the same TMD 1–2 region as is [125I]DEEP. This region contains the aspartic acid 79 essential for DA transport and is the most highly conserved region of the Na+-Cl−–coupled neurotransmitter transporter family, displaying 43% identity throughout the family as a whole and 70% identity and 85% homology in the cocaine-sensitive monoamine transporters (Amara and Kuhar, 1993). This extensive conservation may indicate that this region performs a transport function common to all members of this family, and the finding that [125I]GA II 34 as well as [125I]DEEP interact with this conserved region provides additional evidence highlighting its importance in transport and transport inhibition. Conversely, the targeting of [125I]GA II 34 to N-terminal helices is in distinct contrast to the more C-terminal recognition site of the cocaine analog [125I]RTI 82, which has been localized to a 16 kDa region encompassing TMDs 4–7 (Vaughan and Kuhar, 1996). The binding sites for these three DAT photoaffinity labels are shown schematically in Figure 6, which summarizes and compares the results of the present and previous peptide-mapping studies.

Schematic representation of DAT photoaffinity label binding sites. The middle line represents the DAT primary amino acid sequence, with positions of predicted TMDs indicated by filled rectangles; positions of antibody epitopes are shown by open, numbered rectangles; consensus N-glycosylation sites are indicated by open circles; and lysine and arginine residues (potential tryptic cleavage sites) are shown by tic marks. Thehorizontal bars above the protein sequence indicate the origins of the 14 kDa fragments labeled with [125I]GA II 34 and [125I]DEEP and a 16 kDa fragment labeled with [125I]RTI 82 (from Vaughan and Kuhar, 1996).
These photolabeling results confirm our earlier pharmacological studies that suggested that benztropine and cocaine analogs interact with different DAT-binding domains (Newman et al., 1994, 1995). These earlier conclusions were based on the dissimilar cocaine and benztropine SAR profiles and on the distinctive noncocaine-like behavioral profile of benztropines in animal models of cocaine use. For the benztropines, optimal binding to DAT is achieved with the 3-position of the tropane ring having a [bis(4′-fluorophenyl)methoxy] substituent in the α or axial stereochemistry (Newman et al., 1994,1995). The [bis(4′-fluorophenyl)methoxy] substituent also contributes to the highly potent uptake inhibition seen in the GBR-type compounds (van der Zee et al., 1980). In contrast, for cocaine and WIN-type compounds, the opposite stereochemistry at the 3-position leads to optimal binding (Carroll et al., 1992a). Furthermore, for the 2-carbomethoxy-substituted benztropine analogs, optimal binding affinity for DAT is achieved with the opposite enantiomer from cocaine; i.e., for benztropine, the (+)-2-carbomethoxy-[4′(bis-fluorophenyl)methoxy]tropane is active, in contrast to cocaine and WIN 35,428 in which the active form is the (−)-2-carbomethoxy-3-benzoyloxy or phenyl-substituted tropanes (Meltzer et al., 1994). In addition, the SAR profile of a series of N-substituted 2-carbomethoxy-3α-[bis(4′-fluorophenyl)methoxy]tropanes was found to be similar to that of the GBR compound series (Meltzer et al., 1996).
It has also recently been demonstrated that high-affinity binding of cocaine and WIN compounds at DAT does not require a basic nitrogen (Madras et al., 1996; Meltzer et al., 1997). Replacement of the tropane nitrogen with oxygen maintains high DAT-binding affinity (Meltzer et al., 1997), as do nonbasic nitrogen (N-sulfonylamide) derivatives of cocaine analogs (Kozikowski et al., 1994). In contrast, for (+)-2-carbomethoxy-benztropine compounds, replacement of the tropane nitrogen with an oxygen markedly reduced DAT-binding affinity (Meltzer et al., 1997), complementing our findings that the benztropine compounds also require a basic nitrogen for high-affinity binding to DAT (Agoston et al., 1997a). Although a non-nitrogenous analog of GBR 12909 has not been reported, it has been found that one but not the other terminal piperazine nitrogen could be replaced with a methine group without significant diminution of DAT-binding affinity (Dutta et al., 1993, 1996). The required nitrogen on which the propylphenyl side chain is attached is analogous to the tropane nitrogen in our benztropine series.
An important consideration is that the arylazidoiodo functionality in [125I]GA II 34 and [125I]DEEP extends from the terminal nitrogen by an alkyl chain, whereas this group is placed at the 2-position on the tropane ring of [125I]RTI 82. Given the differing placement of the functionality that forms the covalent bond to the peptide residue, we cannot eliminate the possibility that the binding domains of the pharmacophores of these agents may be partially or substantially overlapping. Differing orientation of the functional groups may result in covalent attachment of the ligands to peptide residues that are distant in terms of primary sequence but close together three dimensionally. The limitations of the present assay do not allow us to discriminate between the possibility that the various photoaffinity ligands bind to entirely distinct sites on the protein. Nevertheless, we believe that the results of the present study coupled with the SAR studies, described above, are indicative of divergence between binding domains for benztropine and the cocaine or WIN compounds.
Although the functional correlates of these differing binding domains are speculative, they suggest the potential for separable pharmacologies for these drugs. Because we have demonstrated that benztropine compounds do not induce cocaine-like behavioral profiles in animal models of psychostimulant abuse (Newman et al., 1994; Agoston et al., 1997a; Katz et al., 1997), the contention that different binding properties may be effecting the behavioral pharmacology of these compounds is compelling. Support for this hypothesis also comes from studies on the proposal of GBR 12909 and its decanoate analog as potential treatments for cocaine abuse. GBR 12909 is a potent and selective dopamine uptake inhibitor but, unlike cocaine, fails to maintain self-administration in rats over a prolonged period of time (Tella et al., 1996). These studies may indicate that simple blockade of DA uptake is insufficient to explain behavioral reinforcement and that additional properties of uptake blockers, possibly related to binding-site differences, may affect downstream events. Further, although GBR 12909 has been reported to be self-administered in nonhuman primates, the decanoate analog of GBR 12909 attenuates the self-administration of cocaine in rhesus monkeys (Baumann et al., 1994;Rothman and Glowa, 1995; Glowa et al., 1996).
Recent studies in dopamine transporter knock-out mice that maintain reinforcing behavior of cocaine provide further evidence that simple inhibition of dopamine reuptake at the DAT may not be an exclusive neurochemical correlate to abuse liability (Caine, 1998; Rocha et al., 1998; Sora et al., 1998). Therefore, clearly, factors other than binding-domain differences contribute to the pharmacological profile of all of these compounds. However, support for the development of cocaine-abuse treatments based on subtle and yet divergent molecular mechanisms at the DAT is growing (Newman, 1998). Molecular and peptide-mapping studies of DAT have provided evidence of differential sites of activity for substrate translocation and cocaine binding (Kitayama et al., 1992; Buck and Amara, 1994; Giros et al., 1994), as well as of different sites of action for different structural classes of dopamine uptake blockers (Vaughan, 1995; Vaughan and Kuhar, 1996). The present study is the first demonstration that not only do structurally divergent dopamine uptake inhibitors access different binding domains on DAT but compounds that share the tropane ring show differential binding. These differing binding sites may play a role in the distinctive pharmacological profiles of these compounds and provide a template for design of potential therapeutics. We are currently developing residue-specific affinity compounds to attempt to identify the specific amino acids that form the ligand-binding domain. These studies will extend our insight into the structure and function of DAT and its role in psychostimulant reinforcement.
Footnotes
This work was supported by the Intramural Research Program of the National Institute on Drug Abuse, National Institutes of Health, and by United States Public Health Service Grant DA 08870 to J.R.L. G.E.A. was fully funded by a National Institutes of Health Intramural Research Training Award fellowship. We thank Dr. Barry Hoffer for his support and editorial assistance.
Correspondence should be addressed to Dr. Amy Hauck Newman, Psychobiology Section, National Institute on Drug Abuse-Intramural Research Program, National Institutes of Health, 5500 Nathan Shock Drive, Baltimore, MD 21224.
Dr. Vaughan’s present address: Department of Biochemistry and Molecular Biology, University of North Dakota School of Medicine, Grand Forks, ND 58202.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}