

Abstract
Cholinergic control of the activity of human cerebral cortical circuits has long been thought to be accounted for by the interaction of acetylcholine (ACh) with muscarinic receptors. Here we report the discovery of functional nicotinic receptors (nAChRs) in interneurons of the human cerebral cortex and discuss the physiological and clinical implications of these findings. The whole-cell mode of the patch-clamp technique was used to record responses triggered by U-tube application of the nonselective agonist ACh and of the α7-nAChR-selective agonist choline to interneurons visualized by means of infrared-assisted videomicroscopy in slices of the human cerebral cortex. Choline induced rapidly desensitizing whole-cell currents that, being sensitive to blockade by methyllycaconitine (MLA; 50 nm), were most likely subserved by an α7-like nAChR. In contrast, ACh evoked slowly decaying whole-cell currents that, being sensitive to blockade by dihydro-β-erythroidine (DHβE; 10 μm), were most likely subserved by an α4β2-like nAChR. Application of ACh (but not choline) to the slices also triggered GABAergic postsynaptic currents (PSCs). Evidence is provided that ACh-evoked PSCs are the result of activation of α4β2-like nAChRs present in preterminal axon segments and/or in presynaptic terminals of interneurons. Thus, nAChRs can relay inhibitory and/or disinhibitory signals to pyramidal neurons and thereby modulate the activity of neuronal circuits in the human cerebral cortex. These mechanisms, which appear to be retained across species, can account for the involvement of nAChRs in cognitive functions and in certain neuropathological conditions.
Although the psychological effects of nicotine and cigarette smoking have been acknowledged for centuries (for review, see Dani and Heinemann, 1996; Pidoplichko et al., 1997), control of neuronal functions by nAChRs in the human brain remains to be demonstrated. In the brain of nonprimate species, ACh has been shown to interact with nAChRs to mediate synaptic transmission (Zhang et al., 1993; Roerig et al., 1997; Alkondon et al., 1998; Frazier et al., 1998) or to control synaptic transmission mediated by the major inhibitory and excitatory neurotransmitters, GABA and glutamate, respectively (McMahon et al., 1994; Albuquerque et al., 1996; Gray et al., 1996;Lindstrom et al., 1996; Role and Berg, 1996; Alkondon et al., 1997;Bertolino et al., 1997; Léna and Changeux, 1997; Wonnacott, 1997;Radcliffe and Dani, 1998). If such actions of ACh via nAChRs could be detected in the human CNS, then inferences could be made regarding the involvement of these receptors in the fine-tuning of brain activity, which has been shown to depend on complex mechanisms of synaptic integration (for review, see Kandel and Schwartz, 1991). Clearly, with the ever increasing body of evidence of species-related differences in the pharmacological profile and function of many receptors (Galzin et al., 1992; Ferraro et al., 1993; Miyake et al., 1995; Nguyen et al., 1995), understanding the involvement of the human neuronal nAChRs (Pereira et al., 1997; Hilmas et al., 1998; Wevers et al., 1999) on the effects of nicotine and cigarette smoking and, perhaps, even more significantly, on a number of physiological processes and pathological conditions requires the demonstration of nAChR function directly in human brain.
The goals of this study were to identify the nAChR subtypes present in interneurons of the human cerebral cortex and to investigate whether their functional properties resemble those of nAChRs present in interneurons of the CA1 region of the hippocampus (Alkondon et al., 1999). To fulfill this aim, the patch-clamp technique was applied to interneurons of slices from the human temporal lobe. Only a limited number of studies have addressed by means of electrophysiological techniques neuronal function in human brain tissue (Halliwell, 1989;Cummins et al., 1993; Isokawa, 1996; Isokawa et al., 1997; Pereira et al., 1997; Hilmas et al., 1998; Wevers et al., 1999), and, to our knowledge, this is the first to provide evidence that human cerebral cortical interneurons express both α7- and α4β2-like nAChRs and that activation of α4β2-like nAChRs present in preterminal axonal segments and/or in presynaptic terminals of the interneurons triggers a synchronous release of GABA. An understanding of the mechanisms by which nAChRs control the inhibitory tonus in human cerebral cortex is likely to provide new insights into the involvement of these receptors in the behavioral effects of tobacco smoking and in neurological dysfunctions such as Alzheimer's disease, schizophrenia, and epilepsy.
MATERIALS AND METHODS
Preparation of human cerebral cortical slices and visualization of interneurons in the slices. Specimens (10- to 12-mm-wide) of human lateral neocortex were obtained from the temporal or frontal cortical lobe of 13 male and 9 female patients (age, 8–52 years) undergoing resections as treatment for their intractable seizures. Tissue used in the present study was presumed not to be part of the seizure focus and was removed as part of a standard surgical procedure to access adjacent structures containing the focus. In some cases, the seizure focus appeared to be originating from the neocortex, whereas in others, mesial temporal structures were more likely to be involved. This information was derived from a combination of depth and surface recordings from implanted electrodes. The tissue used in this study did not show any histopathological abnormalities and came from areas that did not include the epileptic focus. However, whether the epileptic focus was in the neocortex or in mesial temporal structures, there is still a possibility, albeit very remote, that the circuitry of the otherwise apparently normal neocortical tissue was altered in some subtle way because of the occurrence of persistent seizures. Written consent was obtained from each patient or relatives of the patients regarding the use of the surgical brain samples. Within 2 min after removal from the brain, the samples of tissue were placed in well carbogenated, ice-cold artificial CSF (ACSF), which had the following composition (in mm): NaCl, 125; NaHCO3, 25; KCl, 2.5; NaH2PO4, 1.25; CaCl2, 2, MgCl2, 1; and glucose, 25. Preparation of the slices and subsequent recordings were easier in samples obtained from the younger patients. Within 30 min after removal of the specimens, slices of 250 μm thickness were prepared using a vibratome according to the procedure described earlier for rat hippocampal slices (Alkondon et al., 1999). Slices were cut tangentially to the outer surface of the cortical specimen, and, therefore, corresponded very closely to coronal or sagittal sections of whole brain. Slices were stored at room temperature in ACSF, which was bubbled with 95% O2 and 5% CO2. Neurons in the slices were visualized by means of infrared-assisted videomicroscopy (Alkondon et al., 1999). Neurons were also acutely dissociated by mechanical means from some slices according to the procedure described elsewhere (Barbosa et al., 1996). The protocol for the use of human tissue was approved by the Institutional Review Board of the University of Maryland (Baltimore, MD).
Electrophysiological recordings. Using an LM-EPC7 patch-clamp system (List Electronic, Darmstadt, Germany), whole-cell currents were recorded according to the standard patch-clamp technique (Hamill et al., 1981) from the soma of interneurons that were visualized by means of infrared-assisted videomicroscopy in slices of the human cerebral cortex. The signals were filtered at 2 kHz and either recorded on a video cassette recorder tape for later analysis or directly sampled by a microcomputer using the pClamp 6 program (Axon Instruments, Foster City, CA). The slices were superfused with ACSF at 2 ml/min. Atropine (1 μm) was added to the ACSF to block muscarinic receptors. Patch pipettes were pulled from borosilicate glass capillary (1.2 mm outer diameter), and when filled with the internal solutions had resistances between 3 and 6 MΩ. The series resistance ranged from 10 to 20 MΩ. For voltage-clamp recordings, the internal solution consisted of (in mm) either EGTA, 10; HEPES, 10; CsCl, 80; CsF, 80, pH was adjusted to 7.3 with CsOH, 340 mOsm, referred to subsequently as the Cl−, F−-containing internal solution, or EGTA 10; HEPES, 10; Cs-methane sulfonate 130; CsCl 10; MgCl2, 2; QX-314, 5; and biocytin 0.25%, pH adjusted to 7.3 with CsOH; 340 mOsm, referred to subsequently as the methanesulfonate-containing internal solution. The membrane potentials were corrected for liquid junction potential. All recordings were performed at room temperature (20–22° C). The peak amplitudes of agonist-evoked whole-cell currents were determined using the pClamp 6 program. Postsynaptic currents (PSCs) were analyzed using the Continuous Data Recording program (Dempster, 1989). Antagonists were applied via bath superfusion, and agonists were delivered to a large field of neurons via a modified U-tube (Alkondon et al., 1999). The tip of the modified U-tube had a diameter between 100 and 160 μm, and the lower rim of the U-tube tip was placed close to the neuron under study, almost touching the slice. The hydrodynamics of the U-tube tended to delay the onset and prolong the rising phase of agonist-elicited responses (Alkondon et al., 1999). Chemical processing of biocytin-filled neurons was done according to the procedure described elsewhere (Svoboda et al., 1999), and postrecording reconstruction of the image of the biocytin-filled neurons was achieved using the Neurolucida program (MicroBrightField, Colchester, VT)
RESULTS
Morphology of neurons in human cerebral cortical slices
Infrared-assisted videomicroscopy was used for the morphological identification of neurons studied in slices from human cerebral cortex (Fig. 1). Typically, two types of neurons were found. Neurons with a pyramid-like cell soma and a single thick tapering apical dendrite (Fig. 1A) were identified as pyramidal neurons; other neurons were classified as nonpyramidal neurons, the presumed GABAergic interneurons (Fig.1B). Most of the nonpyramidal neurons were multipolar or bipolar, and postrecording reconstruction of the image of biocytin-filled nonpyramidal neurons revealed that their morphology was consistent with that of interneurons (Fig. 1C,D). In the human cerebral cortical slices examined, interneurons were seen more frequently than pyramidal neurons, and therefore were the major target of investigation. It is likely that the short delay (1–2 min) in receiving the cortical samples in the surgery room may have contributed to the death of neurons, particularly the anoxia-prone pyramidal neurons.

Photomicrographs of neurons in human cerebral cortical slices. A, B,Typical examples of infrared-assisted videomicroscopic images of a pyramidal neuron (A) and a nonpyramidal neuron/interneuron (B) in human cerebral cortical slices. C, Image of a biocytin-filled interneuron visualized in a formaldehyde-fixed human cerebral cortical slice.D, Neurolucida drawing of the biocytin-filled neuron shown in C.
Electrophysiological recordings from interneurons in human cerebral cortical slices
In the absence of the Na+-channel blocker tetrodotoxin (TTX), spontaneously occurring PSCs were recorded from the soma of all voltage-clamped interneurons sampled (Fig.2A,C). These PSCs were of sufficient strength to depolarize unclamped areas of the dendrites in the neurons and initiate action potentials. Backpropagating action potentials were then recorded as fast current transients (FCTs) superimposed on the PSCs (Fig. 2A,D). The high frequency of PSCs recorded from most neurons indicated that several synaptic connections in the slices were preserved. PSCs recorded from the human cerebral cortical interneurons were blocked by the GABAA-receptor antagonists bicuculline (Fig.2B) and picrotoxin (data not shown), indicating that these events were GABAergic in nature.

Whole-cell recordings from visually identified interneurons in human cortical slices. A, Samples of spontaneous PSCs and FCTs recorded at −62 mV from a cerebral cortical interneuron. B, Sample recording obtained from the same interneuron after 5 min exposure to bicuculline (10 μm).C, Individual PSCs are shown on an expanded scale.D, An FCT superimposed on a PSC is shown in expanded scale. E, Sample recording obtained from a neuron during a 6 sec U-tube application of quisqualate (20 μm) at −68 mV. F, Sample recording obtained from a neuron during a 6 sec U-tube application of GABA (20 μm) at −62 mV.Solid bars at the top of the traces indicate duration of the agonist pulses. The Cl−, F−-containing internal solution (see Materials and Methods) was used in most of the experiments (A–D,F), and the methanesulfonate-containing solution (see Materials and Methods) was used in the experiment shown inE.
Application via the U-tube of quisqualate to cerebral cortical interneurons also induced inward currents (Fig. 2E), indicating the presence of AMPA-type receptors in these interneurons. The amplitude of quisqualate-evoked currents ranged from 112 to 310 pA (mean ± SE = 204 ± 43 pA;n = 4 neurons). In contrast, spontaneously occurring glutamatergic currents could not be recorded from the neurons sampled (n = 3), because all the PSCs were sensitive to blockade by the GABAA receptor antagonist bicuculline. Additionally, as alluded to in Materials and Methods, the results described below were obtained under experimental conditions that did not enable us to detect glutamatergic events.
In addition to GABAergic PSCs, whole-cell currents evoked by U-tube application of GABA could also be recorded from human cerebral cortical interneurons (Fig. 2F). The amplitude of GABA-evoked currents at −62 mV ranged from 240 to 560 pA (mean ± SE = 390 ± 73 pA; n = 4 neurons) when using the Cl−, F−-containing internal solution. The success of consistently recording spontaneous GABAergic PSCs and responses evoked by U-tube application of GABA demonstrated the feasibility of studying both presynaptic and postsynaptic mechanisms at GABAergic synapses present on the interneurons in the human cerebral cortex.
Postsynaptic nicotinic currents triggered by activation of α7- and α4β2-like nAChRs present in interneurons of the human cerebral cortex
To identify responses evoked by ACh as being nicotinic, all experiments were performed in the presence of the muscarinic receptor antagonist atropine (1 μm). In addition, nicotinic currents and ACh-evoked PSCs could be recorded separately from a single neuron when the methanesulfonate-containing solution (see Materials and Methods) was used to fill the pipette. Under this experimental condition, GABAergic currents reversed at −44 mV. Thus, in neurons voltage-clamped at −44 mV, responses evoked by nicotinic agonists would be nicotinic currents resulting from activation of nAChRs present on the somatodendritic area of the neuron under study.
U-tube application of the α7 nAChR-selective agonist choline (10 mm) to five of six interneurons voltage-clamped at −44 mV resulted in the activation of inward currents (Fig.3). These currents had peak amplitudes ranging from 12 to 54 pA (mean ± SE = 35.2 ± 9 pA;n = 5 neurons). The amplitude of choline-induced currents remained nearly the same in an experiment in which the agonist was applied to the neuron after a cocktail of blockers containing TTX (200 nm), picrotoxin (100 μm), CNQX (10 μm), and APV (50 μm), confirming that the agonist-induced currents were mediated via direct activation of postsynaptic α7-like nAChRs. Bath application of the α7 nAChR-selective antagonist MLA (50 nm) for 5 min reduced the amplitude of choline-induced currents (Fig. 3), and this effect was reversed after washing of the neurons with MLA-free ACSF.

Nicotinic currents evoked by agonists applied to interneurons in human cortical slices. Traces, Samples of nicotinic currents evoked by choline (10 mm) in the absence and in the presence of the α7 nAChR-selective antagonist MLA (50 nm) and samples of nicotinic currents evoked by ACh (1 mm) in the absence and in the presence of the α4β2 nAChR-selective antagonist DHβE (10 μm). Samples are from two different neurons. Choline-induced current decayed to the baseline level during the application of the agonist, whereas ACh-induced current maintained a steady-state level in the presence of the agonist. Choline failed to induce any current when the slice was superfused for 5 min with MLA (50 nm)-containing ACSF. ACh-induced slowly decaying current was blocked when the slice was superfused for 5 min with DHβE (10 μm)-containing ACSF. The methanesulfonate-containing internal solution was used to fill the patch pipette. Atropine (1 μm) was present in all recording solutions bathing the slice and in the agonist solutions. Membrane potential, −44 mV.Graph, Average current amplitude evoked by choline in the absence (control) and in the presence of MLA (n= 5 neurons), and average current amplitude elicited by ACh in the absence (control) and in the presence of DHβE (n= 3 neurons). Results are mean ± SE.
In three of six neurons, ACh evoked a slowly decaying inward current, which was inhibited by pre-exposing the neurons to DHβE (10 μm) (Fig. 3). This finding suggested that human cerebral cortical interneurons can also express α4β2-like nAChRs.
GABAergic PSCs are triggered by activation of α4β2-like nAChRs in interneurons of human cerebral cortical slices
Voltage clamping the neurons at a wide range of membrane potentials enabled us to demonstrate that nAChRs can also modulate the release of GABA in the human cerebral cortex. Large inward currents (250–2000 pA) (Fig.4A), whose rising phases were much faster than those of whole-cell currents induced by any agonist applied via the U-tube (compare the current shown in top trace of Fig. 4A to those shown in Figs. 2, 3), could be triggered by application of ACh to the interneurons voltage-clamped at −68 mV. By itself, the fast rising phase of these currents would be indicative of responses resulting from synaptically released rather than U-tube applied agonist. When the large, fast rising currents triggered by ACh were analyzed at a higher sampling rate (10 kHz instead of 1 kHz), it became clear that their rising phases were composed of several overlapped events (see multiple peaks in the trace in Fig. 4A, asterisk). If GABA was the synaptic transmitter released by ACh, it would be expected that these currents would reverse at −44 mV in methanesulfonate-containing internal solution (Alkondon et al., 1999). As predicted, the current induced by ACh was dramatically reduced at −44 mV (see middle trace in Fig. 4A); the remaining small current could be attributed to the activation of postsynaptic nAChRs present in the neuron under study. Furthermore, in five neurons voltage-clamped at 0 mV, a membrane potential at which nicotinic and glutamatergic currents reverse, ACh triggered a robust burst of outward-going PSCs. The GABAergic nature of these events was confirmed by their sensitivity to blockade by bicuculline (10 μm) (n = 3) (Fig. 4D). In three other neurons voltage-clamped at 0 mV, ACh only induced PSCs in the absence of TTX; after perfusion of these neurons with TTX (200 nm)-containing ACSF, ACh failed to induce PSCs (Fig. 4C). These findings altogether are consistent with the notion that activation of nAChRs present in interneurons synapsing onto the neurons under study results in the release of GABA.

Characteristics of ACh-induced PSCs in human cerebral cortical interneurons. A, Samples of ACh-induced PSCs recorded at different membrane potentials. U-tube application of ACh (1 mm; A, top trace) induced a large inward current associated with isolated inward-going PSCs at −68 mV. The rising phase of the current sampled at a higher frequency (10 kHz instead of 1 kHz) is illustrated by the trace marked with an asterisk (the faster time scale is applicable only to this trace). Note that this rising phase is composed of multiple peaks and is the result of summation of several PSCs. At the null potential (−44 mV; middle trace) for GABA-mediated currents under the present ionic conditions, ACh produced a small inward current, probably representing the postsynaptic nicotinic response of this cell. At 0 mV (bottom trace), ACh induced a burst of outward going PSCs that also showed summation in the beginning of the agonist pulse. The methanesulfonate-containing internal solution (see Materials and Methods) was used in all the experiments illustrated in this figure. B, Samples of ACh-induced outward PSCs recorded at 0 mV under control condition (top trace), 5 min after exposure of the slices to DHβE (10 μm; middle), and 8 min after exposure of the slices to MLA (50 nm;bottom). C, Samples of ACh-evoked PSCs recorded before and 5 min after bath application of TTX (200 nm). D, Samples of ACh-induced PSCs recorded before and 5 min after bath application of bicuculline (10 μm).
Pharmacological identification of the nAChR subtype(s) involved in triggering the GABAergic PSCs in human cortical neurons was made possible by the use of choline, MLA, and DHβE. In cerebral cortical neurons that responded to ACh at 0 mV with bursts of PSCs, choline was unable to induce PSCs (n = 3 neurons). In the same neurons (n = 3), ACh-evoked PSCs were also insensitive to blockade by MLA (50 nm) (Fig.4B). Instead, they were reversibly inhibited by DHβE (10 μm;n = 4 neurons) (Fig. 4B), indicating that the ACh-induced GABA release was mediated by α4β2-like nAChRs.
Dynamics of ACh-induced GABAergic PSCs
During a 6 sec U-tube application of ACh (1 mm), an initial summated PSC response was followed by a long-lasting (30–120 sec) increase in the frequency of isolated PSCs (Fig.5A,D). The increased frequency was also associated with an increase in the amplitude of the PSCs (compare the traces in Fig. 5B,C). The effect on PSCs was specific to ACh, because neither GABA nor a pulse of ACSF was able to induce such changes (Fig. 5D).

Specificity of ACh effect and time course of ACh-induced PSCs. A, Sample of recordings obtained from a human cortical interneuron exposed for 6 sec to ACh at −62 mV. The ACh-induced, summated PSCs are shown expanded and truncated so that the isolated PSCs that remained well after the agonist pulse can be seen.B, Control PSCs from a small segment of the recording shown on an expanded scale. C, ACh-induced PSCs shown on an expanded scale. D, Summary of the changes in the frequency of PSCs with time caused by a 6 sec, U-tube application of various agents. The patch pipette was filled with the Cl−, F−-containing internal solution.
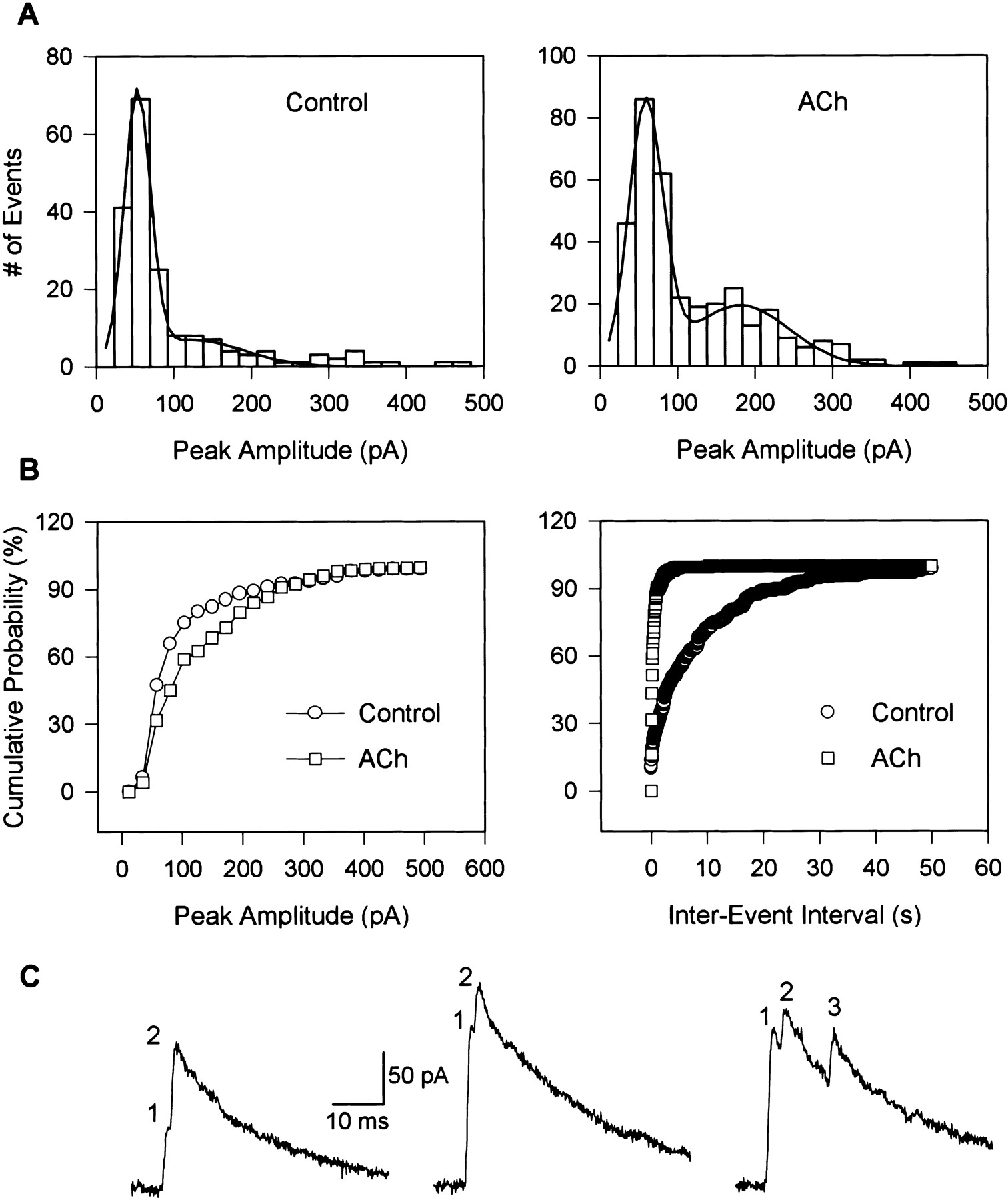
The distribution of the peak amplitude of the PSCs recorded under control conditions was similar to that of ACh-evoked PSCs (Fig.6). Both distributions were fitted by a Gaussian function with two peaks, one in the range between 50 and 59 pA (mean ± SE = 53.9 ± 0.7 pA in control and 55.4 ± 1.9 pA in ACh; n = 4), and the other in the range between 87 and 181 pA (mean ± SE = 117.5 ± 12.4 pA in control and 137.9 ± 16.5 pA in ACh; n = 4). To quantify the differences between control and ACh-evoked PSCs, the area under the curves was analyzed. As shown in Table1, ACh significantly increased the area under the curve for the second peak, indicating that large amplitude events were markedly increased by the agonist. This effect is also illustrated in the cumulative probability plots of peak amplitudes of PSCs recorded under control condition and after exposure of the neuron to ACh (Fig. 6B).

Analysis of ACh-induced PSCs. A,Distribution of peak amplitudes of PSCs before (184 events;left) and after a 6 sec U-tube application of ACh (1 mm, 348 events; right) to a human cerebral cortical interneuron. PSCs were sampled at 5 kHz. Thus, multi-peak events appeared as single-peak events in this analysis. The Gaussian fit of the histogram is shown as a solid line. Membrane potential, 0 mV. B, Cumulative probability plots of peak amplitude histograms and of interevent intervals before and after a 6 sec exposure of the interneuron to ACh (1 mm). Data are from the same experiment as in A. C,Examples of PSCs that show more than one peak (indicated bynumbers at the top) either during the rising phase or during the decay phase of the currents. Traces were sampled at 20 kHz in order to reveal multiple peaks.
Characteristics of GABAergic PSCs recorded from human cortical interneurons
The frequency of PSCs was quantitatively analyzed by collecting control and ACh-induced events for ∼1000 sec (in four 250 sec segments) and 120 sec (in four 30 sec segments), respectively. Averaging the results obtained from four different experiments revealed that the frequency of PSCs was significantly enhanced by ACh (Table 1). The substantial increase in the PSC frequency induced by ACh is also evident in cumulative probability plots of the interevent intervals (Fig.6B).
Overlapping PSCs recorded under control condition and after exposure of the neurons to ACh were also analyzed for the purpose of understanding the mechanism of action underlying the effect of ACh on the amplitude of the PSCs. Typically, overlapping PSCs occur when more than one synaptic release site is activated during the time course of a single PSC event. If the activation of multiple synaptic sites is not synchronized, the resulting events would have more than one peak. Examples of such multi-peak events obtained in our experiments are illustrated in Figure 6C. ACh increased significantly the percentage of multi-peak events (Table 1).
ACh-induced GABAergic PSCs in neurons acutely dissociated from human cerebral cortical slices
Spontaneously occurring synaptic currents that were sensitive to blockade by TTX (200 nm) could be recorded frequently from neurons acutely dissociated from slices of the human cerebral cortex (Fig. 7A), indicating the existence of functional synaptic boutons associated with the surface of the acutely dissociated neurons. Thus, to gain insights regarding the possible location of nAChRs that are responsible for induction of GABAergic PSCs after activation, we performed experiments on neurons acutely dissociated from the human cortical slices. In the absence of TTX, application of ACh to acutely dissociated neurons (n = 4) voltage-clamped at −62 mV induced a rapid burst of summated PSCs (Fig. 7B) that were abolished in the presence of bicuculline (10 μm). In the presence of TTX (200 nm), neurons showed either no response (n = 3) or inward nicotinic currents (n = 4) (Fig. 7C) when briefly exposed to ACh. The inward nicotinic currents were the result of activation of nAChRs present on the neurons from which recordings were obtained. In addition, each of two neurons that responded to ACh with PSCs in the absence of TTX showed no response to ACh in the presence of TTX (Fig.7B). The inability of ACh to induce PSCs in acutely dissociated neurons perfused with TTX suggests that nAChRs located in close proximity to and/or in presynaptic terminals can cause a depolarization that is sufficient to trigger an action potential, and, subsequently, the release of GABA.

ACh-induced PSCs in acutely dissociated human cerebral cortical neurons. A, Image of a neuron acutely dissociated by mechanical means (Barbosa et al., 1996) from a human cerebral cortical slice. B, Sample recordings obtained from a human cerebral cortical neuron exposed to ACh before (B1) and after (B2) its perfusion with TTX (200 nm)-containing external solution.C, Sample recording obtained from a neuron that showed an inward nicotinic current in response to ACh (1 mm) in the presence of TTX. Membrane potential = −62 mV. The Cl−, F−-containing internal solution was used to fill the recording pipette. The bathing and the agonist-containing solutions had atropine (1 μm).
DISCUSSION
The present study reports two major findings: (1) that human cerebral cortical interneurons express postsynaptic, functional α7-like, and α4β2-like nAChRs, and (2) that activation of preterminal and/or presynaptic α4β2-like nAChRs triggers GABA release. Modulation by nAChR activation of functional GABAergic inputs to human cerebral cortical interneurons suggests that neuronal nAChRs have a central role in disinhibitory circuits in the human brain.
The existence of functional nAChRs in human cerebral cortical interneurons
At the level of infrared-assisted videomicroscopy, all of the neurons studied were determined to be interneurons, on the basis of the morphological features of their soma and primary dendrites, which were not consistent with those of pyramidal neurons. Also, postrecording reconstruction of the biocytin-filled neurons showed that the morphological features of these cells fit the morphological criteria expected for interneurons (Kawaguchi, 1993). Accordingly, pyramidal neurons were distinguished from interneurons solely on the basis of their morphology (Fig. 1).
Two subtypes of nAChRs were identified on interneurons of the human cerebral cortex. The ability of choline to induce nicotinic currents that decayed to the baseline during the agonist pulse (Fig. 3) and the blockade of these currents by MLA are strong evidence that a human α7-like nAChR (Peng et al., 1994) is present on the surface of the interneurons, most likely on the somatodendritic region. The slower rate of desensitization of the ACh-evoked currents and their blockade by DHβE argue in favor of the presence of a human α4β2-like nAChR (Gopalakrishnan et al., 1996) on the interneurons. Although previous autoradiography studies have indicated that nAChRs are present in the perikaryon and dendrites of a variety of interneurons in the human neocortex (Zilles et al., 1989), this is the first demonstration of the presence of functional nAChRs in human cerebral cortical interneurons.
Location of nAChRs in human cerebral cortical interneurons
Based on the ability of choline and ACh to evoke nicotinic currents in the presence of TTX and of a number of ligand-gated ion channel antagonists, it can be concluded that both α7- and α4β2-like nAChRs are located on the somatodendritic regions of the interneurons. Additionally, several experimental observations support the concept that in preterminal axon segments and/or in presynaptic terminals of the interneurons, agonist interaction with α4β2-like nAChRs results in DHβE-sensitive GABA release. First, the delay for the onset of the ACh-triggered GABAergic PSCs was equal to or shorter than that for the onset of ACh-induced whole-cell nicotinic currents (compare delay in Fig. 4 with that shown in Fig. 3). Such a rapid action is possible only when the nAChRs are located close to the agonist-delivering U-tube. Thus, considering that interneurons are scattered in the cortical slices, it is very unlikely that ACh-evoked PSCs would have been the result of activation of somatodendritic nAChRs located on interneurons synapsing onto the interneuron under study. In agreement with the concept that preterminal and/or presynaptic nAChRs accounted for the ACh-evoked release of GABA was the finding that ACh could also trigger GABAergic PSCs in acutely dissociated neurons that retained fragments of incoming axon segments.
Physiological relevance of nAChRs in the human cerebral cortex
Based on the present results, we suggest that, in the human cerebral cortex, neuronal nAChRs can be involved in inhibitory and disinhibitory mechanisms (Fig. 8). In a cerebral cortical circuit composed of GABAergic interneurons synapsing onto the soma or proximal axon segments of pyramidal neurons, activation of nAChRs present on the interneurons would result in the inhibition of action potential firing by the pyramidal neurons. In that case, nAChRs could function as filtering devices that enhance the signal-to-noise ratio in the human cerebral cortical neuronal circuitry. Likewise, activation of nAChRs present on interneurons that synapse onto the dendrites of pyramidal neurons can cause shunting of dendritic excitatory inputs. On the other hand, inhibition of interneurons via nAChR-mediated GABA release from other interneurons would facilitate afferent excitatory signaling to pyramidal neuron dendrites by reducing GABAergic inhibition of these structures. By means of this disinhibitory mechanism, nAChR activation could lead to synaptic strengthening similar to that seen in long-term potentiation, the putative cellular substrate of learning and memory. Now we can envision the participation of neuronal nAChRs in cholinergic functions and dysfunctions that have heretofore been largely ascribed to ACh acting via functional muscarinic receptors in the human cerebral cortex (Raiteri et al., 1990; Russo et al., 1993; Levey, 1996; Nobili and Sannita, 1997; Rye, 1997).

Proposed scheme of the involvement of nAChRs in the control of the overall activity of a hypothetical neuronal circuit in the human cerebral cortex. Sites a, b, andc are presumed to contain nAChRs.
Mechanism and dynamics of nAChR-mediated GABA release in the human cerebral cortex
The present data demonstrate that several GABAergic synapses can be stimulated by nAChR activation in a synchronized fashion. If exogenously applied agonist caused a temporal and spatial summation of GABAergic PSCs, it is conceivable that endogenously released transmitter would have the same effect. Because timing of GABA release is a critical factor in controlling the final output of a neuron, nAChR-mediated GABA release may be an important mechanism by which the cholinergic system regulates the activity of neurons in human cerebral cortical circuits.
In addition to increasing the frequency of GABAergic PSCs recorded from human cerebral cortical interneurons, ACh also increased the amplitude of isolated GABAergic PSCs. Similar results have been obtained from rat hippocampal CA1 interneurons (Alkondon et al., 1999) and from neurons of the rat interpeduncular nucleus (Léna et al., 1993). The increase in the amplitude of PSCs by ACh can be attributed to the recruitment of several synaptic sites, as evidenced by the appearance of a greater number of multi-peak events after exposure of the neurons to ACh (Fig. 6, Table 1). The contribution of increased quantal release (recruitment of more vesicles) is also possible.
Possible clinical implications of the finding that nAChRs control GABA release in the human cerebral cortex
Neuronal nAChRs appear to be involved in many pathological conditions including schizophrenia, Parkinson's disease (PD), Alzheimer's disease (AD), and epilepsy (Whitehouse et al., 1988;Whitehouse and Kalaria, 1995; Gerlach et al., 1996; Leonard et al., 1996; Morens et al., 1996; Newhouse et al., 1996; Ben-Shlomo, 1997;Freedman et al., 1997; Gotti et al., 1997), all of which, rather than being attributed to the failure of a single-neurotransmitter system, are characterized by a complex set of imbalances of modulatory functions in neuronal circuits in the CNS (Hietala and Syvälahti, 1996; Hagan et al., 1997; Weinberger, 1997). Based on our findings, numerous mechanisms could explain the participation of neuronal nAChRs in the pathogenesis of these neurological disorders. The ability to gate auditory sensory information, for instance, which is impaired in patients with schizophrenia, depends on the GABAergic inhibitory input to the neurons involved in this physiological process (Weinberger, 1997; van Kammen et al., 1998), and, as such, can be associated with dysfunctions of neuronal nAChRs in presynaptic GABAergic neurons. In addition, glutamate excitotoxicity—one of the possible mechanisms involved in the origin and/or progression of AD and PD (for review, seeDalack et al., 1998)—could be controlled by the nAChR-induced GABA release from interneurons in the human cerebral cortex. A reduced activity of nAChRs in interneurons that synapse onto pyramidal neurons or a loss of such nAChR-containing interneurons may also explain the participation of neuronal nAChRs in epileptogenesis, and, in itself, account for the relationship between mutations of genes coding for nAChR subunits and different forms of epilepsy (Elmslie et al., 1997;Kuryatov et al., 1997). Additionally, it is possible that by filtering the output of pyramidal neurons, activation of nAChRs in GABAergic interneurons of the human cerebral cortex can limit the impact of extraneous stimuli, thereby increasing attention and, consequently, improving memory retention. This may represent one of the mechanisms underlying some of the symptoms associated with nicotine addiction, particularly the improvement in long-term memory recall in humans who smoke (Mangan and Golding, 1983; Peeke and Peeke, 1984; Parkin et al., 1998; Stein et al., 1998) and in laboratory animals treated with nicotine (Changeux et al., 1998; Levin and Simon, 1998; Picciotto, 1998; Arroyo-Jiménez et al., 1999).
It is noteworthy that modulation of GABA release by nAChR activation is a process retained across species and different brain areas, because it has also been reported to occur in the rat interpeduncular nucleus, rat dorsal motor nucleus of the vagus, CA1 field of the rat hippocampus, mouse thalamus, chick lateral spiriform nucleus, and mouse brain synaptosomes (McMahon et al., 1994; Alkondon et al., 1997, 1999;Bertolino et al., 1997; Léna and Changeux, 1997; Lu et al., 1998). Therefore, the knowledge gained from animal models regarding the participation of neuronal nAChRs in different physiological processes and pathological conditions can have direct bearing on the understanding of the functions mediated by these receptors in the human brain.
In summary, the evidence that human cerebral cortical interneurons can express at least one of two types of nAChRs, an MLA-sensitive, α7-like nAChR, and a DHβE-sensitive, α4β2-like nAChR, and that activation of preterminal and/or presynaptic α4β2-like nAChRs triggers the release of GABA supports the contention that nAChRs can modulate the activity of neuronal networks in the human cerebral cortex.
Footnotes
This study was supported by United States Public Health Service Grants NS25296 and ES05730, and by PRONEX 0888/96 (Brazil). The generous help rendered by Mr. Thomas Jemski from the Illustration Department of the University Maryland School of Medicine, the technical assistance of Mr. Benjamin Cumming, Mrs. Barbara Marrow, and Ms. Mabel Zelle, and the superb work of Mrs. Bhagavathy Alkondon in the processing and drawing of biocytin-filled neurons are gratefully acknowledged.
Correspondence should be addressed to Dr. Edson X. Albuquerque, Department of Pharmacology and Experimental Therapeutics, University of Maryland School of Medicine, 655 West Baltimore Street, Baltimore, MD 21201. E-mail: ealbuque{at}umaryland.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}