

Abstract
We have previously shown that activation of protein kinase Cε (PKCε) in male rats induces a chronic, long-lasting change in nociceptors such that a subsequent exposure to proinflammatory mediators produces markedly prolonged mechanical hyperalgesia. This neuroplastic change, hyperalgesic priming, is dependent on activation of cytoplasmic polyadenylation element-binding protein (CPEB), downstream of PKCε, and consequent translation of mRNAs in the peripheral terminal of the nociceptor. Since α calmodulin-dependent protein kinase II (αCaMKII), a molecule implicated in neuroplasticity, is a target of CPEB and can also affect CPEB function, we investigated its role in the transition from acute to chronic pain. Priming induced by direct activation of PKCε can be prevented by inhibition of αCaMKII. In addition, direct activation of αCaMKII induces priming, which was not prevented by pretreatment with PKCε antisense, suggesting that αCaMKII is downstream of PKCε in the induction of priming. Activation of ryanodine receptors (RyRs), which can lead to activation of αCaMKII, also induced priming, in a calcium- and αCaMKII-dependent manner. Similarly, inhibition of the RyR and a calcium buffer prevented induction of priming by PKCε. Unlike activation of PKCε, ryanodine and αCaMKII induced priming in female as well as male rats. Our results demonstrate a contribution of αCaMKII to induction of hyperalgesic priming, a phenomenon implicated in the transition from acute to chronic pain.
Introduction
The inability to reverse chronic pain is due in part to lack of knowledge of its underlying mechanism. We have demonstrated that inflammatory mediators that activate protein kinase Cε (PKCε) can produce a neuroplastic change in isolectin B4-positive [IB4(+)] nociceptors, referred to as hyperalgesic priming, such that subsequent exposure to a pronociceptive inflammatory mediator [e.g., prostaglandin E2 (PGE2)] produces enhanced and markedly prolonged mechanical hyperalgesia (Aley et al., 2000; Parada et al., 2003a, 2005; Reichling and Levine, 2009; Joseph and Levine, 2010). However, once priming is established, although attenuation of PKCε can transiently abrogate its expression, i.e., inhibit the prolongation of PGE2 hyperalgesia, it cannot permanently reverse the underlying neuroplastic change in the nociceptor and terminate the primed condition. To evaluate the mechanisms involved in the induction of priming downstream of PKCε, in subsequent experiments we demonstrated that downregulation of cytoplasmic polyadenylation element-binding protein (CPEB), an RNA-binding molecule that regulates the translation of otherwise dormant mRNAs in peripheral axons (Richter, 2007; Villalba et al., 2011) and that is a downstream target of PKCε (Bogen et al., 2012), also prevents priming (Bogen et al., 2012). In the present study, to explore pathways downstream of CPEB that underlie the induction of hyperalgesic priming, we evaluated the role of α calmodulin-dependent protein kinase II (αCaMKII), a cytoplasmic polyadenylation element-containing species of dormant mRNA (Wu et al., 1998) that is found in sensory axons (VanBerkum and Goodman, 1995; Hiruma et al., 1999; Geddis and Rehder, 2003; Gleason et al., 2003) and that has been implicated in neuroplasticity (Cammarota et al., 2002; Gleason et al., 2003; Yamauchi, 2005; Buard et al., 2010; Coultrap et al., 2010; Jama et al., 2011).
Another aspect of hyperalgesic priming that has remained unexplained is that production of priming by agonists for receptors that signal through PKCε, or by direct activation of PKCε, which both produce priming in male rats, does not produce priming in females (Joseph et al., 2003). Therefore, as a secondary goal of the present experiments, we tested the hypothesis that activation of hyperalgesic priming mechanisms downstream of PKCε can produce priming in females as well as males.
Materials and Methods
Animals.
All experiments were performed on adult male and female Sprague Dawley rats (220–400 g; Charles River Laboratories). Animals were housed, three per cage, under a 12 h light/dark cycle in a temperature- and humidity-controlled room in the animal care facility of the University of California at San Francisco. Food and water were available ad libitum. All nociceptive testing was done between 10:00 A.M. and 5:00 P.M., and the experimental protocols were approved by the Institutional Animal Care and Use Committee at the University of California at San Francisco and adhered to the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. All efforts were made to minimize the number of animals used and their suffering.
Mechanical nociceptive threshold testing.
Mechanical nociceptive threshold was quantified using a Ugo Basile Analgesymeter (Randall-Selitto paw-withdrawal test; Stoelting), which applies a linearly increasing mechanical force to the dorsum of the rat's hindpaw, as described previously (Randall and Selitto, 1957; Taiwo and Levine, 1989; Taiwo et al., 1989). Rats were placed in cylindrical acrylic restrainers designed to provide adequate comfort and ventilation, allow extension of the hind leg from the cylinder, and minimize restraint stress. All rats were acclimatized to the testing procedure, and testing was performed in parallel across groups. Rats were adapted to the restrainer for 1 h before starting each study and for 30 min before experimental manipulations. The nociceptive threshold was defined as the force (in grams) at which the rat withdrew its paw, and the baseline paw-pressure threshold was defined as the mean of the three readings taken before the test agents were injected. Each paw was treated as an independent measure, and each experiment was performed on a separate group of rats. Group data are presented as mean ± SEM change from baseline mechanical nociceptive threshold.
Drugs and their administration.
The following chemicals were used in this study: PGE2 (direct-acting hyperalgesic inflammatory mediator), ryanodine [a ryanodine receptor (RyR) activator], dantrolene sodium salt (an RyR inhibitor), and the intracellular calcium transport inhibitor 3,4,5-trimethoxybenzoic acid 8-(diethylamino) octyl ester (TMB-8), all from Sigma-Aldrich; αCaMKII recombinant (activated αCaMKII; New England Biolabs); the CaMKII inhibitor peptide CaM2INtide (GenScript); the PKCε-specific translocation inhibitor peptide (PKCε-I) PKCεV1–2, (Johnson et al., 1996; Khasar et al., 1999) (Calbiochem); the PKCε activator ψεRACK (Biomatik); and the protein translation inhibitors cordycepin 5′-triphosphate sodium salt (Sigma-Aldrich) and rapamycin (EMD Chemicals). The selection of the drug doses used in this study was based on doses determined during our previous studies (Taiwo et al., 1990; Ouseph et al., 1995; Khasar et al., 1999; Aley et al., 2000; Parada et al., 2005; Ferrari et al., 2013b).
Stock solutions of PGE2 in absolute ethanol (1 μg/μl) were diluted in 0.9% NaCl (1:50, Cfinal = 0.2 μg/μl) immediately before injection. The ethanol concentration of the final PGE2 solution was ∼2%, and the injection volume was 5 μl. Stock solutions of cordycepin (10 μg/μl, dissolved in a 1:1 mixture of 0.9% NaCl and absolute ethanol) or rapamycin (20 μg/μl, dissolved in absolute DMSO) were further diluted in 0.9% NaCl or distilled water, respectively, immediately before injection. The ethanol or DMSO concentration in the final solutions was ∼2%.
Activation of αCaMKII was performed in vitro, and a dose of 25 ng (2.5 μl) of the activated αCaMKII was injected on the dorsum of the rat hindpaw. αCaMKII was diluted in 1× NEBuffer for PK (50 mm Tris-HCl, 10 mm MgCl2, 0.1 mm EDTA, 2 mm DTT, 0.01% Brij 35, pH 7.5 at 25°C) supplemented with 200 μm ATP, 1.2 μm calmodulin, and 2 mm CaCl2 and incubated for 10 min at 30°C before injection.
Drugs were administered on the dorsum of the hindpaw via a beveled 30 gauge hypodermic needle attached to a Hamilton microsyringe by a short length of polyethylene (PE-10) tubing. The administration of activated αCaMKII, CaM2INtide, PKCε-I, ψεRACK, ryanodine, dantrolene sodium salt, TMB-8, and the protein translation inhibitors was preceded by a hypotonic shock to facilitate cell permeability to these agents (2 μl of distilled water, separated by a bubble of air to avoid mixing in the same syringe), to get compounds into the nerve terminal (Borle and Snowdowne, 1982; Burch and Axelrod, 1987).
Oligodeoxynucleotide antisense to PKCε and αCaMKII.
Oligodeoxynucleotide (ODN) antisense (AS) to PKCε mRNA, shown previously to decrease PKCε in dorsal root ganglion neurons (Parada et al., 2003a) when administered intrathecally, was synthesized by Invitrogen. The ODN AS, 5′-GCCAGCTCGATCTTGCGCCC-3′, was directed against a unique sequence of PKCε. The ODN mismatch (MM), 5′-GCCAGCGCGATCTTTCGCCC-3′, is the AS sequence with two bases (denoted in boldface) switched. A search of the EMBL and NCBI GenBank Rattus norvegicus databases identified no homologous sequences.
The ODN AS sequence for the α-subunit of CaMKII, 5′-GGTAGCCATCCTGGCACT-3′ (Invitrogen), was directed against a unique region of the rat mRNA sequence. The corresponding NCBI GenBank accession number and ODN position within the mRNA sequence are NM_012920 and 33–50, respectively. That this ODN AS can be used to downregulate the expression of αCaMKII has been shown previously (Churn et al., 2000). The ODN MM sequence 5′-GGTAGCCATAAGGGCACT-3′ corresponds to the AS sequence with three bases mismatched (denoted in boldface).
Before use, the ODNs were lyophilized and reconstituted in 0.9% NaCl to a concentration of 2 μg/μl. During each injection, rats were briefly anesthetized with 2.5% isoflurane in 95% O2. A 30 gauge hypodermic needle was inserted into the subarachnoid space on the midline, between the L4 and L5 vertebrae. A total of 40 μg of ODN in a volume of 20 μl per rat was slowly injected. Proper intrathecal injections were systematically confirmed by checking for a sudden flicking of the tail, a reflex that is evoked by subarachnoid space access and bolus injection (Mestre et al., 1994). This method of injecting into the intrathecal space has proven to be very accurate and reproducible, as revealed by intrathecal injections of vital dyes and radioligands such as methylene blue and [3H] morphine (Mestre et al., 1994; Bilsky et al., 1996). The animals regained consciousness ∼1 min after the injection. The use of ODN AS to manipulate the expression of proteins in nociceptors, important for their role in nociceptor sensitization, is well supported by previous studies by others (Song et al., 2009; Su et al., 2011; Quanhong et al., 2012; Sun et al., 2013) as well as our group (Parada et al., 2003a; Ferrari et al., 2010, 2012; Bogen et al., 2012).
Statistics.
In all experiments, the dependent variable was paw-withdrawal threshold, expressed as the percentage change from baseline. No significant difference in the mechanical nociceptive thresholds was observed before the injection of the priming stimuli (ψεRACK, activated αCaMKII, or ryanodine, depending on the experiment) and immediately before PGE2 injections (average mechanical nociceptive thresholds before priming stimuli were 118.8 ± 0.7 g; average mechanical nociceptive thresholds before PGE2 injections were 118.1 ± 0.6 g; n = 170 paws; paired Student's t test, t(52) = 0.7035, p = 0.4849). As noted in the figure legends, two-way repeated-measures ANOVA, followed by the Bonferroni's post hoc test, was performed to compare the magnitude of the hyperalgesia induced by the priming stimulus or by PGE2 injection in different conditions, and p values <0.05 were considered statistically significant. Data are presented as mean ± SEM.
Results
αCaMKII inhibition prevents induction of priming
To determine whether αCaMKII has a role in the induction of hyperalgesic priming, we tested whether intrathecal treatment with ODN AS to αCaMKII mRNA would prevent the development of PKCε agonist ψεRACK-induced hyperalgesic priming. Since the AS treatment does not totally knock down the expression of αCaMKII (Churn et al., 2000), we combined AS treatment with intradermal injection of the CaMKII inhibitor CaM2INtide, at the site of nociceptive testing, to minimize activation of αCaMKII by PKCε. We found that intrathecal treatment with αCaMKII AS combined with local inhibition of CaMKII prevented the development of priming induced by injection of ψεRACK; PGE2-induced hyperalgesia, tested 1 week after the treatment with the AS plus inhibitor for CaMKII has finished (11 d after ψεRACK injection), was significantly less at the fourth hour in the AS plus inhibitor-treated group compared with the group treated with αCaMKII MM (p < 0.001; Fig. 1). This result supports the hypothesis that αCaMKII is important for the induction of hyperalgesic priming. In addition, a control experiment was performed to confirm the inhibitory effect of CaM2INtide against αCaMKII. The injection of activated αCaMKII (25 ng) on the dorsum of the hindpaw induced significant mechanical hyperalgesia that lasted at least 1 week. However, administration of αCaMKII activated in the presence of the inhibitor did not induce significant change in the mechanical threshold, showing the efficacy of the inhibitor against αCaMKII (p < 0.0001, activated αCaMKII group compared with the groups treated with αCaMKII plus inhibitor or vehicle; Fig. 2A).
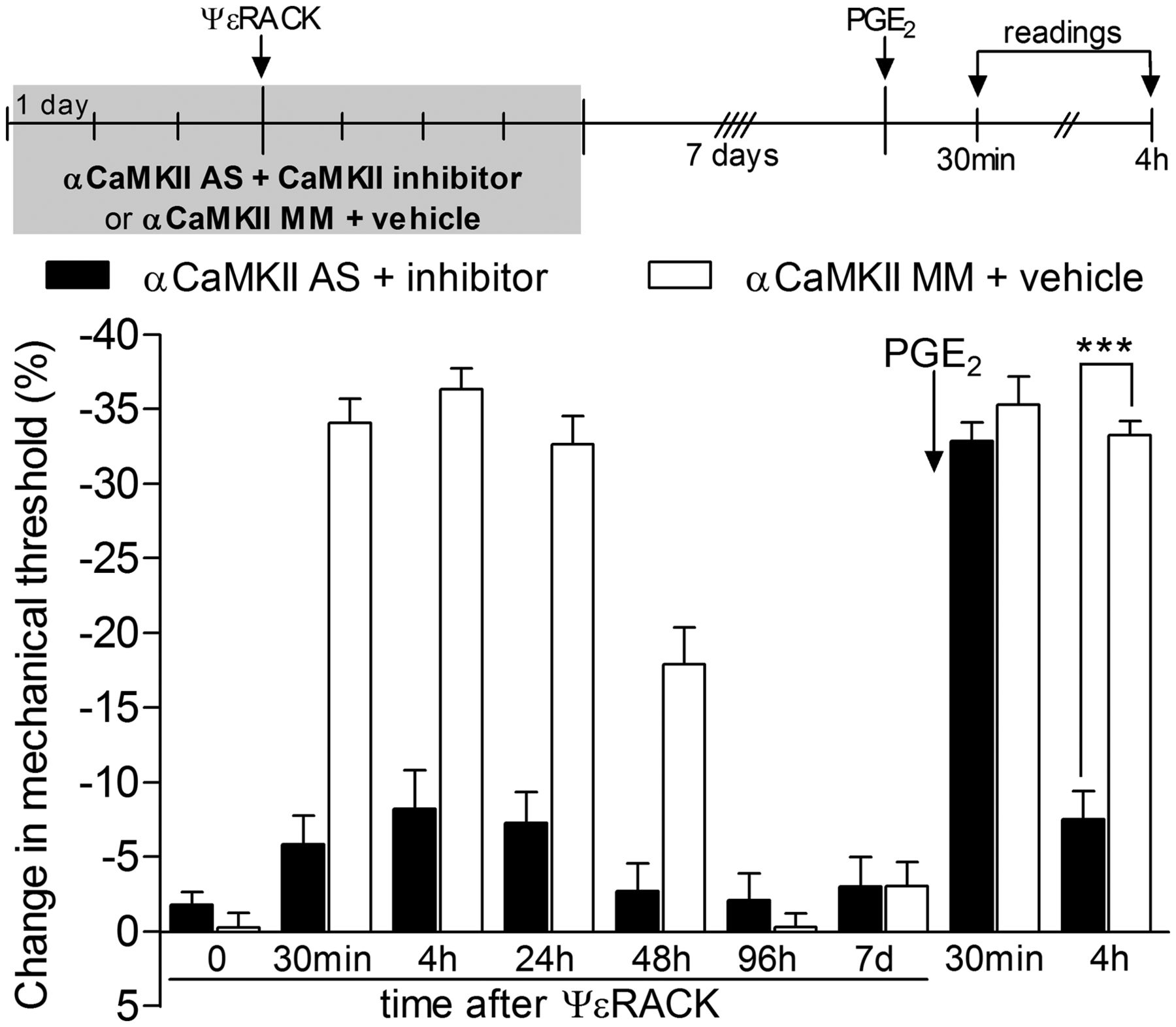
PKCε-induced hyperalgesic priming is αCaMKII dependent. Rats were treated with daily intrathecal injections of ODN AS (filled bars) or mismatch (MM; open bars) for αCaMKII mRNA for 3 consecutive days. In addition, the CaMKII inhibitor (1 μg, filled bars) or vehicle (open bars) was injected on the dorsum of the hindpaw daily. On the third day of ODN AS plus inhibitor or MM plus vehicle treatment, ψεRACK (1 μg) was administered at the same site as the inhibitor or vehicle. Mechanical nociceptive thresholds were then evaluated by the Randall-Sellitto paw-withdrawal test 30 min and 4, 24, 48, and 96 h after ψεRACK administration. The ODN plus inhibitor or vehicle treatments were continued until the return of the mechanical threshold to baseline values (on the seventh day). Repeated-measures ANOVA followed by Bonferroni's post hoc test showed significant mechanical hyperalgesia induced by injection of ψεRACK in both groups (F(5,50) = 31.28; p < 0.0001 compared with baseline thresholds). However, when both groups were compared, ψεRACK-induced mechanical hyperalgesia was significantly attenuated in the AS plus inhibitor group (F(1,50) = 237.8; p < 0.0001). Seven days after the last treatment with ODN AS plus inhibitor or ODN MM plus vehicle (11 d after ψεRACK injection), we tested for hyperalgesic priming by intradermal injection of PGE2 (100 ng) in the same site as ψεRACK and the CaMKII inhibitor (or vehicle). Two-way repeated-measures ANOVA followed by Bonferroni's post hoc test showed that, although the mechanical hyperalgesia was not significantly different in both groups 30 min after PGE2 (p > 0.05), at the fourth hour a significant attenuation in the group previously treated with ODN AS plus inhibitor was observed, when compared with the MM plus vehicle-treated group (***p < 0.001). n = 6 paws per group.

αCaMKII activation induces mechanical hyperalgesia and hyperalgesic priming. A, αCaMKII was activated in vitro in the presence (black bars) or absence (white bars) of the CaMKII inhibitor CaM2INtide. Intradermal injections, on the dorsum of the hindpaws, were performed in different groups of rats (25 ng of αCaMKII). A control group (gray bars) received injection of the vehicles instead of αCaMKII. Mechanical nociceptive threshold was evaluated 30 min, 4 h, 24 h, 96 h, 7 d, and 10 d after injections. Repeated-measures ANOVA followed by Bonferroni's post hoc test showed significant mechanical hyperalgesia induced by αCaMKII activated in the absence of the inhibitor (F(12,78) = 11.04; ***p < 0.0001 when compared with the groups treated with αCaMKII plus inhibitor or vehicles) that was still significant 7 d after the injection (**p < 0.01). Comparison of the groups that received the αCaMKII plus its inhibitor or the vehicles showed no significant statistical difference (F(6.48) = 1.70; p = 0.1412). n = 6 paws per group. B, Rats received intradermal injection of activated αCaMKII (25 ng, black bars) or its vehicle (white bars). No significant difference was observed between the mechanical thresholds before and 10 d after injection of αCaMKII or vehicle, i.e., immediately before PGE2 injection (data not shown). PGE2 (100 ng) was then injected at the same site as αCaMKII or vehicle, and the mechanical nociceptive thresholds were evaluated 30 min and 4 h later. Repeated-measures ANOVA followed by Bonferroni's post hoc test showed that PGE2-induced hyperalgesia was still significant at the fourth hour in the paws pretreated with αCaMKII, whereas in the vehicle-treated paws, the mechanical threshold had already returned to baseline at that time point (***p < 0.001, when comparing both groups at the fourth hour). n = 10 paws in the αCaMKII group; n = 6 paws in the vehicle group.
Activation of αCaMKII induces priming
To determine whether activation of αCaMKII is not only necessary for the induction of but also sufficient to induce hyperalgesic priming, we injected activated αCaMKII (25 ng) or its vehicle intradermally on the dorsum of the hindpaw at the site of nociceptive testing. Activated αCaMKII, but not its vehicle, produced hyperalgesia that was prominent 4 h after its injection and was no longer detectable 10 d later (Fig. 2A, black bars). When the nociceptive threshold had returned to baseline, we injected PGE2 (100 ng) at the site on the dorsum of the hindpaw where we had previously injected activated αCaMKII or its vehicle. When injected in the paw previously treated with αCaMKII vehicle, PGE2 induced hyperalgesia that was present 30 min later but was no longer detectable at 4 h. In contrast, when PGE2 was injected in the paw previously treated with αCaMKII, robust PGE2-induced hyperalgesia was still present 4 h later (p < 0.001 when both groups are compared at the fourth hour; Fig. 2B) without attenuation, a signature feature of the primed state (Reichling and Levine, 2009).
Prolongation of PGE2 hyperalgesia in rats primed by activation of αCaMKII is prevented by inhibiting PKCε and local protein translation
A key feature of hyperalgesic priming is that the prolonged PGE2 hyperalgesia in the primed nociceptor is PKCε dependent (Aley et al., 2000; Parada et al., 2003a; Reichling and Levine, 2009). To confirm that this is also true for the neuroplastic change induced by injection of activated αCaMKII, we administered the PKCε inhibitor (PKCε-I), 5 min before PGE2, in rats previously treated (2 weeks before) with activated αCaMKII. As with priming induced by carrageenan, tumor necrosis factor α (TNFα), or interleukin 6 (IL-6), PGE2-induced hyperalgesia in rats previously treated with activated αCaMKII was also attenuated by the PKCε inhibitor (p < 0.001; Fig. 3). Thus, prior treatment with active αCaMKII produces a neuroplastic change in the nociceptor such that subsequent injection of PGE2 into its peripheral receptive field induces mechanical hyperalgesia, with the following two signature features of classic hyperalgesic priming: (1) marked prolongation of PGE2-induced hyperalgesia and (2) PKCε dependence of the prolongation of PGE2-induced hyperalgesia (Aley et al., 2000; Parada et al., 2003a; Reichling and Levine, 2009).

Prolongation of PGE2-induced mechanical hyperalgesia in αCaMKII primed rats is PKCε dependent. Activated αCaMKII (25 ng) was injected intradermally in the rat hindpaw. Two weeks later, when the mechanical thresholds had returned to baseline (paired Student's t test showed no difference in the mechanical thresholds between both groups; t(10) = 1.340, p = 0.2099; data not shown), the PKCε-specific translocation inhibitor peptide PKCεV1–2 (PKCε-I, 1 μg, filled bars), or its vehicle (control, open bars), was administered in the same site. Five minutes later, PGE2 (100 ng) was injected, and the mechanical nociceptive thresholds were evaluated 30 min and 4 h later. Significant attenuation of the prolonged PGE2-induced hyperalgesia, measured at 4 h, was observed in the paws treated with PKCε-I (***p < 0.001), when compared with the vehicle-treated group, without affecting hyperalgesia at the 30 min time point (two-way repeated-measures ANOVA followed by Bonferroni's post hoc test; F(1,10) = 27.36, p = 0.0004, both groups compared at 30 min). n = 6 paws per group.
We have also previously shown that hyperalgesic priming induced by activation of PKCε is dependent on protein translation in the peripheral terminal of the nociceptor (Ferrari et al., 2013b). Thus, to further confirm that the neuroplastic change induced by injection of activated αCaMKII is hyperalgesic priming, we injected the protein translation inhibitors cordycepin (1 μg; Fig. 4A) or rapamycin (1 μg; Fig. 4B), or their respective vehicles, intradermally at the site of nociceptive testing on the dorsum of the hindpaw previously injected with activated αCaMKII. Thirty minutes later, PGE2 was injected at the site of nociceptive testing. We found that both protein translation inhibitors, but not their vehicle, injected before PGE2 inhibited the ability of PGE2 to induce prolonged hyperalgesia induced by the previous injection of activated αCaMKII (p < 0.0001 when the cordycepin and rapamycin groups were compared with their vehicles; Fig. 4, A and B, respectively).

Prolongation of PGE2-induced mechanical hyperalgesia in αCaMKII primed rats is dependent on local protein translation. Activated αCaMKII (25 ng) was injected intradermally on the dorsum of the hindpaw of different groups of rats. Mechanical nociceptive threshold evaluation showed significant hyperalgesia 30 min and 4 h after injection and mechanical thresholds similar to baseline values on the day of the test with PGE2 (2 weeks after αCaMKII injection; data not shown). A, B, Injection of PGE2 (100 ng) at the same site as αCaMKII was preceded (30 min before) by injection of cordycepin (A, 1 μg, filled bars) or rapamycin (B, 1 μg, filled bars). Control groups received vehicle in the PGE2 injection site (open bars). Repeated-measures ANOVA followed by Bonferroni's post hoc test showed significant hyperalgesia, evaluated 30 min and 4 h after injection in the groups treated with vehicle; however, in the groups treated with cordycepin or rapamycin, the magnitude of the PGE2-induced hyperalgesia was significantly decreased at the fourth hour (A, F(1,10) = 44.42, ***p < 0.0001; B, F(1,10) = 73,25, ***p < 0.0001; when the protein translation inhibitor groups are compared with the vehicle groups), indicating an effect of protein translation inhibitors on the prolongation of the PGE2-induced hyperalgesia in paws primed with activated αCaMKII. n = 6 paws per group.
Induction of priming by αCaMKII is not PKCε dependent
We next tested the hypothesis that αCaMKII is downstream of PKCε in the induction of hyperalgesic priming, since it can be activated by CPEB (Wu et al., 1998; Huang et al., 2002), which is, in turn, activated by PKCε (Bogen et al., 2012). If αCaMKII is downstream of PKCε in the induction of priming, then pretreatment with ODN AS to mRNA for PKCε should not prevent the development of priming by activated αCaMKII, as it does for priming induced by activation of cell-surface receptors for TNFα and IL-6 (Parada et al., 2003b; Dina et al., 2008, 2011). We found that treating animals with ODN AS for PKCε in a protocol that prevented priming induced by TNFα and IL-6 (Parada et al., 2003b; Dina et al., 2008, 2011) did not prevent the development of priming induced by activated αCaMKII (p = 0.1784 when the ODN AS-treated group was compared with the ODN MM-treated group; Fig. 5).

Induction of hyperalgesic priming by αCaMKII is not PKCε dependent. Rats were treated with ODN AS (filled bars) or MM (open bars) for PKCε mRNA for 10 consecutive days. Activated αCaMKII (25 ng) was injected on the dorsum of the hind paw on the third day of ODN AS or MM treatment. Three days after the last ODN treatment (on the 13th day), PGE2 (100 ng) was injected at the same site as activated αCaMKII (paired Student's t test showed no significant difference between the mechanical thresholds in both groups before αCaMKII administration and immediately before PGE2 injection: t(5) = 0.8660, p = 0.4261 for the AS group and t(5) = 0.7559, p = 0.4838 for the MM group, respectively). Two-way repeated-measures ANOVA followed by Bonferroni's post hoc test showed that in both the AS and the MM groups the PGE2-induced mechanical hyperalgesia was still present at the fourth hour, indicating that PKCε is not necessary for the induction of priming by αCaMKII (F(1,10) = 2.10; p = 0.1784, when comparing both groups; NS). n = 6 paws per group.
PKCε-induced hyperalgesic priming is dependent on activation of the RyR
Since αCaMKII activation is calcium dependent (Hanson and Schulman, 1992; Pereda et al., 1998; Coultrap and Bayer, 2012) and in other forms of neuroplasticity this calcium is derived from activation of RyR (Cheng et al., 2010; Adasme et al., 2011; Stutzmann and Mattson, 2011), we determined whether blocking RyRs would prevent the induction of hyperalgesic priming by activation of PKCε. In this study, we found that pretreatment with the RyR inhibitor dantrolene prevented the development of priming by injection of the PKCε activator ψεRACK (p < 0.001; Fig. 6, gray bars). In addition, we also observed significant attenuation of ψεRACK-induced priming by the pretreatment with a calcium chelator, TMB-8 (p < 0.001; Fig. 6, filled bars), suggesting that calcium release by activation of the RyR plays a role in the induction of priming.

PKCε-induced hyperalgesic priming is dependent on the RyR. Rats received intradermal injection of vehicle (white bars), dantrolene (1 μg, gray bars), or TMB-8 (1 μg, black bars) on the dorsum of the hindpaw. Ten minutes later, the PKCε activator ψεRACK (1 μg) was injected at the same site and the mechanical nociceptive thresholds were evaluated (data not shown). Daily injections of vehicle, dantrolene, or TMB-8 continued for 4 d, until the mechanical thresholds had returned to baseline. Two days after the last injection (6 d after ψεRACK), PGE2 (100 ng) was administered at the same site, and the mechanical thresholds were evaluated 30 min and 4 h later. Two-way repeated-measures ANOVA followed by Bonferroni's post hoc test showed significant mechanical hyperalgesia induced by PGE2 in all groups (F(2,30) = 24.01; p < 0.0001 when compared with baseline mechanical thresholds). However, in the groups that received dantrolene or TMB-8, the PGE2-induced hyperalgesia was significantly attenuated at the 4 h time point (***p < 0.001 for both groups when comparing 30 min and 4 h time points), in contrast with the vehicle-treated group (NS; p > 0.05). n = 6 paws per group.
Ryanodine produces priming
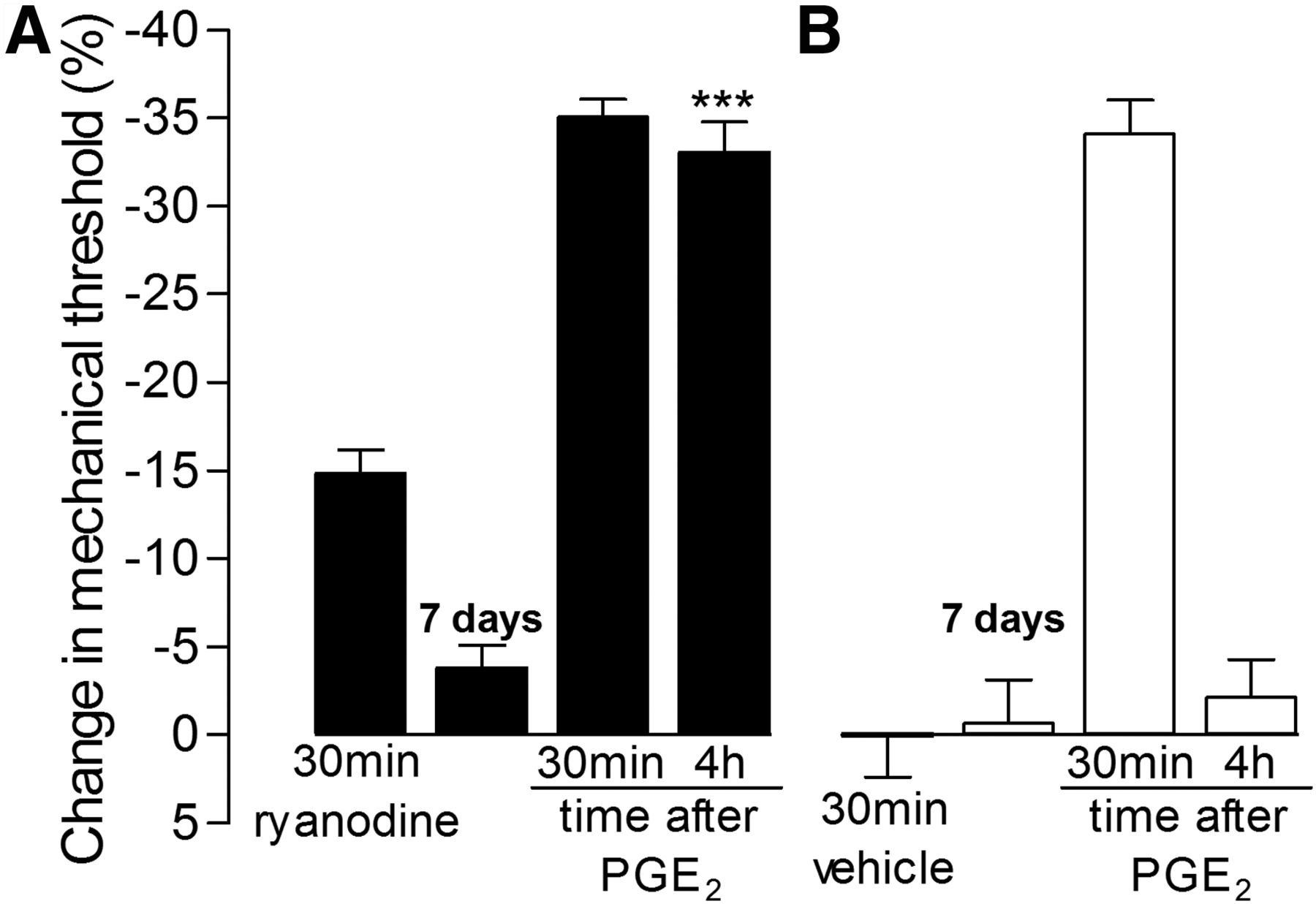
αCaMKII is a calcium-dependent kinase (Hanson and Schulman, 1992; Pereda et al., 1998; Coultrap and Bayer, 2012) that has been shown to associate with the RyR (Shakiryanova et al., 2007, 2011; Wong et al., 2009), which can, when activated, induce localized high concentrations of calcium (Ehrlich et al., 1994; MacKrill, 1999, 2012). Thus, since activation of the RyR can also activate αCaMKII (Shakiryanova et al., 2007, 2011; Wong et al., 2009), we determined whether ryanodine, an RyR agonist (Meissner, 1986; Sattelle et al., 2008), could also induce priming. To determine whether activation of the RyR is sufficient to induce hyperalgesic priming, we injected ryanodine (1 μg) or its vehicle intradermally on the dorsum of the hindpaw. Ryanodine produced mechanical hyperalgesia, measured 30 min after its injection, which was no longer significant 7 d later (Fig. 7, filled bars). PGE2 (100 ng) was then injected at the site on the dorsum of the hindpaw where previously ryanodine or its vehicle had been injected. When PGE2 was injected in paws previously treated with ryanodine vehicle, it induced hyperalgesia that could be detected 30 min later but was back to baseline at 4 h. In contrast, when injected in the paw previously treated with ryanodine, PGE2-induced hyperalgesia was still present 4 h later without attenuation, characteristic of the primed state (p < 0.001, compared with the vehicle-treated group at the fourth hour; Fig. 7, filled bars). Therefore, to test the hypothesis that the ability of ryanodine to induce priming was mediated by its ability to induce an increase in calcium, we pretreated rats with the calcium buffer TMB-8 (1 μg) 5 min before injection of ryanodine. We found that prior injection of TMB-8 not only inhibited the hyperalgesic effect of ryanodine (p < 0.001) but also prevented induction of priming, since the injection of PGE2 at the same site of ryanodine, 1 week later, did not produce prolonged hyperalgesia in the group treated with TMB-8, in contrast to the group treated with vehicle, in which the PGE2-induced hyperalgesia was still present at the fourth hour (p < 0.001; Fig. 8).

Ryanodine induces hyperalgesic priming. A, B, Rats received intradermal injection of ryanodine (A, 1 μg, filled bars) or vehicle (B, open bars) on the dorsum of the hindpaw, and the mechanical nociceptive thresholds were evaluated 30 min later. Repeated-measures ANOVA showed significant mechanical hyperalgesia induced by ryanodine, but not by vehicle (F(1,10) = 46.08; p < 0.0001 when the ryanodine group is compared with the vehicle group). Testing for priming with intradermal injection of PGE2 (100 ng) was performed 7 d later, when the mechanical nociceptive threshold had returned to pre-ryanodine baseline (paired Student's t test showed no significant difference between the mechanical thresholds before and 7 d after ryanodine or vehicle administration; t(5) = 0.1784, p = 0.1048 for the ryanodine group; t(5) = 0.4385, p = 0.6793 for the vehicle group). Repeated-measures ANOVA followed by Bonferroni's post hoc test showed significant hyperalgesia induced by PGE2 injected at the same site as ryanodine or vehicle at 30 min. And, in contrast to the vehicle group, in which the hyperalgesia was no longer present 4 h after PGE2 injection, in the paws that received ryanodine 1 week before, the PGE2-induced hyperalgesia was unattenuated (***p < 0.001, when compared with the vehicle group). n = 6 paws per group.

Ryanodine-induced priming is calcium dependent. A, B, Rats received intradermal injection of the calcium buffer TMB-8 (A, 1 μg, filled bars) or its vehicle (B, open bars) on the dorsum of the hindpaw. Ten minutes later, ryanodine (1 μg) was injected at the same site and the mechanical nociceptive thresholds were evaluated after 30 min. Two-way repeated-measures ANOVA followed by Bonferroni's post hoc test showed significant mechanical hyperalgesia induced by ryanodine in the control group, as opposed to the group treated with TMB-8, in which the hyperalgesia was significantly attenuated (F(1,20) = 15.67; ***p < 0.001 when both groups are compared 30 min after ryanodine injection). Seven days later, when the mechanical thresholds were back to baseline (paired Student's t test showed no significant difference between the mechanical thresholds before and 7 d after ryanodine plus TMB-8 or ryanodine plus vehicle administration; t(5) = 0.6547, p = 0.5416 and t(5) = 0.7565, p = 0.3701, respectively), testing for priming was performed by intradermal injection of PGE2 (100 ng) at the same site as ryanodine. Repeated-measures ANOVA followed by Bonferroni's post hoc test showed that the group treated with TMB-8, but not with vehicle, before ryanodine injection had a significant decrease in the PGE2-induced hyperalgesia at the fourth hour (F(1,10) = 92.90; ***p < 0.001 when the TMB-8-treated group is compared with the vehicle-treated group at the fourth hour). n = 6 paws per group.
αCaMKII is downstream of RyR in the induction of hyperalgesic priming
To test whether the underlying mechanism of the priming produced by ryanodine is dependent on αCaMKII activation, rats pretreated with ODN AS (filled bars) or MM (open bars) for αCaMKII for 3 consecutive days received coinjection of ryanodine (1 μg) and the CaMKII inhibitor CaM2INtide (1 μg) (filled bars) or its vehicle (open bars) on the dorsum of the hindpaw (Fig. 9). The daily treatment with ODN AS or MM plus CaMKII inhibitor or vehicle continued for 3 more days, since it has been shown that priming takes 72 h to develop (Bogen et al., 2012). Four days after the ODN treatment was discontinued, PGE2 was injected at the same site as ryanodine, CaMKII inhibitor, or vehicle. We found that in the rats that had the activation of αCaMKII prevented by the treatment with ODN AS and the CaMKII inhibitor, but not in the group treated with MM and vehicle, the PGE2-induced hyperalgesia was significantly attenuated at the fourth hour, suggesting that the priming induced by ryanodine is dependent on αCaMKII activation (p < 0.001; Fig. 9).

αCaMKII is downstream of the RyR in the induction of hyperalgesic priming. Rats were treated with ODN AS (A, filled bars) or MM (B, open bars) for αCaMKII for 6 consecutive days. On the third day of ODN AS or MM treatment, ryanodine (1 μg) was coinjected with the CaMKII inhibitor CaM2INtide (A, 1 μg, filled bars), or vehicle (B, open bars), on the dorsum of the hindpaws. Mechanical thresholds were evaluated 30 min later. Ryanodine induced mechanical hyperalgesia in both groups, without significant statistical difference between the groups (two-way repeated-measures ANOVA followed by Bonferroni's post hoc test; F(1,20) = 2.04; p = 0.1841). The daily treatment with ODN AS plus CaMKII inhibitor or MM plus vehicle continued for 3 more days. PGE2 (100 ng) was injected, 4 d after the ODN AS or MM treatment was discontinued, at the same site as the injection of ryanodine plus CaMKII inhibitor or vehicle. Repeated-measures ANOVA showed that the treatment with αCaMKII AS plus CaMKII inhibitor prevented the induction of priming by ryanodine, since the PGE2-induced hyperalgesia, although similar in both groups at the 30 min time point, was significantly attenuated in the AS plus inhibitor group but not on the MM plus vehicle group (***p < 0.001, when both groups are compared at the fourth hour). n = 6 paws per group.
Hyperalgesic priming in females
In previous studies in which hyperalgesic priming was induced by agents that act at receptors on the nociceptor terminal (e.g., TNFα or IL-6; Aley et al., 2000; Parada et al., 2003b; Dina et al., 2008, 2011) or by direct activation of PKCε, using the PKCε-selective agonist ψεRACK (Aley et al., 2000), while priming was induced in male rats, the same experimental protocol did not induce priming in female rats (Joseph et al., 2003). To determine whether the sexual dimorphism in induction of priming is caused by sex differences in male and female rats proximally in the intracellular pathway leading to the production of priming, activated αCaMKII (25 ng; Fig. 10A, gray bars) or ryanodine (1 μg; Fig. 10B, gray bars) was injected intradermally on the dorsum of the hindpaw of the female rat. At the same time after their injection that induced priming in male rats, both activated αCaMKII and ryanodine induced prolonged PGE2 hyperalgesia in the female rat (Fig. 10, A and B, respectively; p < 0.001 in both cases compared with the control groups). Thus, activation of molecules downstream but not upstream of PKCε produces hyperalgesic priming in the female as well as the male rat.

αCaMKII and ryanodine induce priming in female rats. A, Female rats received intradermal injection of activated αCaMKII (25 ng, gray bars) or its vehicle (white bars). Mechanical nociceptive threshold was evaluated before and 30 min, 4 h, 24 h, and 10 d after their injection. Two-way repeated-measures ANOVA followed by Bonferroni's post hoc test showed significant hyperalgesia in the αCaMKII-treated group but not in the vehicle-treated group (F(1,40) = 162.26; ***p < 0.001), with no significant (NS) difference between both groups on the 10th day. Test for priming was then performed with intradermal injection of PGE2 (100 ng) on day 10. Repeated-measures ANOVA showed that PGE2-induced hyperalgesia was still significant at the fourth hour in the αCaMKII-treated paws, whereas in the vehicle-treated group, the mechanical threshold had already returned to baseline values at that time point (F(1,10) = 26.61; ***p < 0.001 when both groups are compared at the fourth hour). n = 6 paws per group. B, Ryanodine (1 μg, gray bars) or vehicle (white bars) was injected intradermally on the dorsum of the hindpaw of female rats, and the mechanical thresholds were evaluated 30 min later. Significant hyperalgesia was observed in the ryanodine-treated group but not in the vehicle-treated paws 30 min after injection (***p < 0.001, when ryanodine group is compared with the vehicle group). Seven days after ryanodine or vehicle injection, PGE2 (100 ng) was injected at the same site (paired Student's t test showed no significant difference between the mechanical thresholds before and 7 d after ryanodine or vehicle administration: t(5) = 1.225, p = 0.2752 and t(5) = 1.035, p = 0.3481, respectively). Two-way repeated-measures ANOVA followed by Bonferroni's post hoc test showed that the PGE2-induced mechanical hyperalgesia was still present 4 h after injection in the rats that previously received ryanodine, whereas in the group that received vehicle, the nociceptive thresholds had returned to baseline at that time point (F(1,10) = 55.24; ***p < 0.001). n = 6 paws per group.
Discussion
Although chronic pain remains one of the most costly items in the health care budget of developed countries (de Girolamo, 1991; Langley et al., 2010; Langley, 2011; Anastassopoulos et al., 2012; Gaskin and Richard, 2012), progress toward its effective management, much less reversal of the responsible underlying neuroplastic changes in the pain pathway, has experienced limited improvement. To approach this problem, we have developed a model of the transition from acute to chronic pain in which, after recovery from an inflammatory insult that produces PKCε-dependent nociceptor sensitization, a subsequent exposure to an inflammatory mediator with a receptor on the primary afferent nociceptor produces enhanced, and markedly prolonged, PKCε-dependent hyperalgesia. Hyperalgesic priming is mediated by changes in intracellular signaling pathways that underlie mechanical hyperalgesia induced by inflammatory mediators, such as PGE2 (Aley et al., 2000; Parada et al., 2005; Ferrari et al., 2013a). Since hyperalgesic priming is still present, unattenuated, for months after its induction (Aley et al., 2000; Parada et al., 2005), it is likely caused by a phenotypic switch in primary afferent nociceptors. However, given the continuous turnover of most cellular proteins, it is difficult to conceive that a simple switch in the coupling of a G-protein-coupled receptor to a different second-messenger signaling pathway (Parada et al., 2005; Khasar et al., 2008; Dina et al., 2009; Ferrari et al., 2013a) is sufficient to explain the endurance of the plasticity associated with hyperalgesic priming.
In a follow-up study of the mechanism of hyperalgesic priming, we have shown that downregulation of the level of expression of CPEB, an RNA-binding molecule that regulates the translation of otherwise dormant mRNAs (Richter, 2007; Villalba et al., 2011), that is almost exclusively expressed by IB4(+) nociceptors (the subset of nociceptors in which hyperalgesic priming occurs; Joseph and Levine, 2010), and that can be coimmunoprecipitated with PKCε, by the intrathecal administration of ODN AS targeting its mRNA, can prevent (Bogen et al., 2012), but not reverse (our unpublished observations), the neuroplastic change in the primary afferent nociceptor that underlies hyperalgesic priming. Because our ODN AS to CPEB mRNA only decreased CPEB by 28% in peripheral nerves, we considered that we might not be inhibiting peripheral translation to a degree required to produce permanent reversal of priming, which may need to be far more complete than that needed to prevent the development of priming. Importantly in this regard, we have recently shown that local injection of protein translation inhibitors (i.e., rapamycin and cordycepin) reversed as well as prevented the induction of priming (Ferrari et al., 2013b), suggesting a role of local protein synthesis in maintaining the neuroplastic change in the nociceptor in our model of the transition from acute to chronic pain. To begin to explore the nature of the dormant mRNA species in the peripheral axon of the primary afferent nociceptor, whose protein product mediates the contribution of CPEB to hyperalgesic priming, we evaluated the possible role of αCaMKII. The translation of αCaMKII is regulated by CPEB (Wu et al., 1998). Moreover, it has been reported to be in the axons of sensory neurons (VanBerkum and Goodman, 1995; Hiruma et al., 1999; Geddis and Rehder, 2003; Gleason et al., 2003) and has been implicated in neuroplasticity in neurons in the CNS involved in learning and memory (Cammarota et al., 2002; Gleason et al., 2003; Yamauchi, 2005; Buard et al., 2010; Coultrap et al., 2010; Jama et al., 2011), making it a candidate for one of the dormant mRNA species contributing to the neuroplastic changes associated with hyperalgesic priming. In this study, we found that inhibition of αCaMKII was able to prevent the induction of priming by ψεRACK (Fig. 1) and that the direct activation of αCaMKII induced priming independent of PKCε (Fig. 5). These results suggest αCaMKII as a downstream target of PKCε in the events responsible for the induction of hyperalgesic priming. Moreover, similar to PKCε-induced priming (Bogen et al., 2012), αCaMKII-induced priming was also dependent on protein translation (Fig. 4), suggesting an interaction of αCaMKII and CPEB in our model of transition from acute to chronic pain.
An important unanswered question is how activation of αCaMKII, which is known to be downstream of CPEB (Wu et al., 1998; Huang et al., 2002), induces translation-dependent priming in nociceptors. Of note in this regard, αCaMKII has been reported not only to be downstream of CPEB (Wu et al., 1998; Huang et al., 2002) but also to function upstream, regulating the function of CPEB by phosphorylation at the regulatory site threonine 171 (Atkins et al., 2004). Such positive feedback, in which peripheral protein translation is able to signal to dormant αCaMKII mRNA in sensory neurons axons and to enhance CPEB signaling by αCaMKII activity, could help explain why it is so difficult to reverse hyperalgesic priming and supports the suggestion that prevention may be a more tractable therapeutic option than reversal of chronic pain.
Given that αCaMKII is activated by calcium and that activation of the RyR, found on mitochondria (Beutner et al., 2001; Ryu et al., 2011; Yi et al., 2012) and the endoplasmic reticulum (Hasselbach and Migala, 1988; Witcher et al., 1991; Zalk et al., 2007; Currie, 2009; Kho et al., 2012; Van Petegem, 2012), the former which has been shown to be in high concentration of peripheral terminals of nociceptors (Morris and Hollenbeck, 1995; Chada and Hollenbeck, 2003, 2004; Ramírez and Couve, 2011), is capable of activating αCaMKII (Wong et al., 2009; Shakiryanova et al., 2011), we tested the hypothesis that activation of the RyR can also induce hyperalgesic priming. We found that ryanodine, the classic agonist at the RyR, does induce hyperalgesic priming (Fig. 7). To confirm that the effect of ryanodine was, in fact, attributable to an associated increase in calcium, we used the calcium chelator TMB-8 to buffer the increase in calcium induced by activation of the RyR. We found that administration of TMB-8, before ryanodine, prevented induction of hyperalgesic priming by ryanodine (Fig. 8) but not that induced by activated αCaMKII (data not shown). The most parsimonious explanation for these findings is that increases in calcium by activation of the RyR, which is capable of producing a focal high concentration of calcium and which is found in close proximity to αCaMKII, activates αCaMKII to produce hyperalgesic priming. And the fact that αCaMKII can be both upstream and downstream of CPEB could provide a form of positive feedback that might underlie the long-term perseverance of hyperalgesic priming, which can only be permanently reversed by inhibition of protein translation in the peripheral terminal of the nociceptor (Ferrari et al., 2013b).
An important, shortcoming of our model has been its failure to explain the transition from acute to chronic pain in females (Joseph et al., 2003). With access to mechanisms involved in hyperalgesic priming downstream of PKCε, we evaluated whether their direct activation can induce priming in the female rat as it does in the male. In contrast to the failure of activation of proximal mechanisms to induce hyperalgesic priming, such as TNFα, IL-6, and inflammatory mediators induced by carrageenan, acting at cell-surface receptors, or by the PKCε activator ψεRACK (Aley et al., 2000; Parada et al., 2003b; Dina et al., 2008, 2011), direct activation of distal mechanisms (i.e., αCaMKII or RyR) did induce priming in female rats. Confirmation of similarities in the mechanism for hyperalgesic priming in the female and male rat comes from our finding that PGE2-induced prolonged hyperalgesia is mediated by PKCε and that translation inhibitors reverse hyperalgesic priming in the female rat (data not shown) as it does in the male (Ferrari et al., 2013b). What remains to be elucidated here are signaling pathways upstream of αCaMKII that can produce hyperalgesic priming in the female.
In conclusion, in this study we have further pursued the downstream mechanism mediating hyperalgesic priming, a model of the transition from acute to chronic pain that can be induced by inflammatory mediators such as TNFα and IL-6 or by activation of PKCε in the peripheral terminal of the nociceptor and involves protein translation of dormant mRNA in the nociceptor. We now provide evidence that αCaMKII, whose mRNA species is a downstream target of CPEB, plays a key role in the induction of hyperalgesic priming. That peripheral administration of translation inhibitors permanently reverses hyperalgesic priming induced by activation of αCaMKII suggested that αCaMKII could function upstream of CPEB. Based on these findings, we suggest the existence of a self-propagating positive feedback loop between αCaMKII and CPEB that is responsible for the neuroplastic changes in the nociceptor under primed conditions that could contribute to chronic pain (Fig. 11).

Schematic diagram of the proposed mechanisms underlying the induction of hyperalgesic priming in male and female rats. In male but not female rats, activation of TNFα and IL-6 receptors or direct activation of PKCε in the peripheral terminal of the primary afferent nociceptor triggers a cascade of events that culminate in hyperalgesic priming. CPEB, a downstream target of PKCε, activates αCaMKII in the induction of priming. In the PKCε-induced priming, αCaMKII is downstream of CPEB. However, since αCaMKII is also able to affect CPEB activity (dashed arrow) (Atkins et al., 2004), the maintenance of the primed state is hypothesized to be a loop in which αCaMKII is both upstream and downstream of CPEB. Importantly, activation of αCaMKII induces priming in male and female rats (events below the dotted line are involved in priming in both sexes). Direct activation of RyR also produces priming in male and female rats by releasing calcium (Ehrlich et al., 1994; MacKrill, 1999, 2012) and activating αCaMKII (Shakiryanova et al., 2007, 2011; Wong et al., 2009). In italics are shown the inhibitors of the mediators involved in the induction of priming, with the respective steps on the pathway where they act: PKCε AS, CPEB AS, αCaMKII AS and the CaMKII inhibitor CaMINtide; the RyR inhibitor dantrolene; the calcium chelator TMB-8; and the protein translation inhibitors cordycepin and rapamycin. In addition, estrogen is the critical mediator that prevents the development of hyperalgesic priming in female rats, acting at the level of PKCε (Joseph et al., 2003).
Footnotes
This study was supported by the National Institutes of Health.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Jon D. Levine, University of California, San Francisco, 521 Parnassus Avenue, San Francisco, CA 94143-0440. jon.levine{at}ucsf.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}