

Abstract
Homopentameric α7 nicotinic receptors have a high affinity for acetylcholine (ACh), are permeable to Ca2+ ions, and are abundant in hippocampal interneurons. Although nicotinic agonists evoke inward currents and Ca2+ transients in stratum radiatum interneurons, the role of endogenous ACh in modulating synaptic integration by interneurons is incompletely understood. Many cholinergic axonal varicosities do not have postsynaptic specializations, but α7 receptors frequently occur close to synaptic GABAA receptors. These observations raise the possibility that α7 nicotinic receptors activated by ACh released from cholinergic axons modulate GABAergic transmission in interneurons. We show that agonists of α7 receptors profoundly depress GABAergic IPSCs recorded in stratum radiatum interneurons in the CA1 region of the hippocampus. This depression is accompanied by a small increase in GABA release. α7 nicotinic receptor agonists also depress GABA- or muscimol-evoked currents in interneurons, indicating that the major effect is a postsynaptic modulation of GABAA receptors. The depression of GABA-evoked currents is abolished by chelating Ca2+ in the recorded interneuron and attenuated by inhibitors of PKC. We also show that stimuli designed to release endogenous ACh from cholinergic axons evoke an α7 receptor-dependent heterosynaptic depression of GABAergic IPSCs in interneurons. This heterosynaptic modulation is amplified by blocking cholinesterases. These results reveal a novel mechanism by which cholinergic neurons modulate information processing in the hippocampus.
Introduction
The cholinergic innervation of the hippocampus from the medial septal nucleus/diagonal band complex (Mesulam, 2004a,b) plays an important role in rhythmogenesis (Lee et al., 1994) and is implicated in a variety of cognitive processes. Degeneration of cholinergic afferents contributes to memory impairment in Alzheimer's disease (Davies and Maloney, 1976; Mesulam, 2004a), and cholinesterase inhibitors are among the only treatments available (Cummings, 2004; Hogan et al., 2004).
ACh acts on metabotropic muscarinic receptors and ionotropic nicotinic receptors (nAChRs). Activation of nAChRs has been shown to improve performance in certain cognitive tasks (Newhouse et al., 2004). Homopentameric α7 nAChRs are an abundant subtype in the brain (Couturier et al., 1990), have a high Ca2+ permeability, and exhibit rapid desensitization (Bertrand et al., 1993; Seguela et al., 1993; Hogg et al., 2003). They are densely expressed in the hippocampus, especially in interneurons (Freedman et al., 1993). Their number has been shown to decrease in schizophrenia (Freedman et al., 1995), and a polymorphism in the gene encoding α7 has been linked to a processing deficit associated with this disease (Freedman et al., 1997). α7 nAChRs have attracted considerable attention as a potential target in the treatment of schizophrenia and dementia (Ripoll et al., 2004).
Although the available anatomical data must be interpreted with caution (Herber et al., 2004), the expression of α7 nAChRs is consistent with the finding that exogenous agonists directly excite interneurons, especially in stratum radiatum (Alkondon et al., 1997a; Jones and Yakel, 1997; McQuiston and Madison, 1999; Ji and Dani, 2000; Buhler and Dunwiddie, 2002). Exogenous nicotinic receptor agonists also evoke local Ca2+ transients in the dendrites of interneurons, consistent with the high Ca2+ permeability of α7 nAChRs (Khiroug et al., 2003; Vizi et al., 2004). How endogenous ACh released from cholinergic fibers acts on interneurons is less clear, but electrical stimuli have been reported to evoke synaptic currents mediated by nAChRs (Alkondon et al., 1998; Frazier et al., 1998b).
Anatomical studies have shown that nAChRs frequently occur at GABAergic synapses, both presynaptically and postsynaptically (Fabian-Fine et al., 2001; Kawai et al., 2002; Zago et al., 2006). At ultrastructural resolution, postsynaptic α7 nAChRs have been reported both within the synapse and in a perisynaptic annulus, implying an intimate relationship with postsynaptic GABAA receptors (Fabian-Fine et al., 2001). α7 nAChRs, however, tend to occur relatively far from the sites of ACh release: most varicosities on cholinergic axons in the hippocampus are nonsynaptic (Umbriaco et al., 1995). This anatomical arrangement suggests that ACh carries a diffuse signal in the hippocampus (Descarries et al., 2004) that might modulate inhibitory neurotransmission among interneurons. Exogenous α7 nAChR agonists enhance transmitter release at various synapses (Seguela et al., 1993; McGehee et al., 1995; Gray et al., 1996; Alkondon et al., 1997a; MacDermott et al., 1999). However, little is known about postsynaptic effects of α7 nAChRs at GABAergic synapses.
We report here that α7 receptor activation by either exogenous agonists or endogenous ACh release inhibits GABAA receptor-mediated currents in hippocampal interneurons. These results point to a novel candidate mechanism by which the cholinergic innervation of the hippocampus exerts its cognition-modulating effects.
Materials and Methods
Four-week-old male guinea pigs or 8-week-old male Sprague Dawley rats were killed in accordance with the United Kingdom Animals (Scientific Procedures) Act of 1986. Horizontal hippocampal slices (300–350 μm thick) were cut on a vibrating tissue slicer (VT1000S; Leica, Wetzlar, Germany) in an ice-cold oxygenated sucrose-based solution containing (in mm) 75 sucrose, 70 NaCl, 2 KCl, 1 NaH2PO4, 26.2 NaHCO3, 5.6 MgCl2, 0.5 CaCl2, and 25 glucose, equilibrated with 95% O2/5% CO2, pH 7.4, adjusted to 315 mOsm. After recovering in an interface storage chamber in a sucrose-free solution, they were placed on a perfusion stage mounted on an upright microscope equipped with infrared differential interference contrast imaging. The perfusion medium contained (in mm) 119 NaCl, 2.5 KCl, 1.3 MgCl2, 2 CaCl2, 26.2 NaHCO3, 1 NaH2PO4, and 22 glucose and was equilibrated with 95% O2/5% CO2, pH 7.4.
CA1 pyramidal cells or stratum radiatum interneurons were recorded in whole-cell voltage clamp using pipettes filled with a solution containing (in mm) 120 CsCl, 10 HEPES, 2 EGTA or 10 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA), 8 NaCl, 0.2 MgCl2, 2 Mg-ATP, 0.3 Na3-GTP, and 5 5-N-(2,6-dimethylphenyl-carbamoylmethyl)triethylammonium bromide (QX-314 Br), pH 7.2, 290 mOsm. The pipette resistance was 3–4 MΩ. For perforated-patch recordings, 8 MΩ electrodes were used and filled with the above intracellular solution (with EGTA), to which amphotericin B was added (final concentration 75–150 μg/ml prepared from a stock in DMSO). The electrode tip was dip filled with an amphotericin-free solution for 15–30 s and subsequently backfilled with the amphotericin-containing intracellular solution. QX-314 Br was present in the intracellular solution, and depolarizing voltage steps were intermittently delivered to evoke action currents to verify that the patch was not accidentally ruptured. The series resistance (<15 MΩ and <100 MΩ in whole-cell and perforated-patch configurations, respectively) was monitored throughout the experiment using a −5 mV step command, and cells showing unstable series resistance (>15% change) were rejected.
Cells were recorded at 23–25°C in voltage-clamp mode at a holding potential of −60 mV using an Axopatch 1D amplifier (Molecular Devices, Sunnyvale, CA). Evoked currents (inward currents with a Cl− reversal potential of ∼0 mV) were low-pass filtered (2 kHz) and acquired at 5 kHz on a PC for off-line analysis [LabView (National Instruments, Austin, TX), MiniAnalysis (Synaptosoft, Decatur, GA), and Excel (Microsoft, Seattle, WA)]. Dynamic current clamp was implemented with G-clamp (Kullmann et al., 2004) running under LabView RealTime with an iteration frequency of 20 kHz.
To block fast glutamatergic transmission, NBQX (1,2,3,4-tetrahydro-6-nitro-2,3-dioxo-benzo[f]quinoxaline-7-sulfonamide; 25 μm) and APV (dl-2-amino-5-phosphonopentanoic acid; 50 μm) were routinely added to the perfusion solution. GABAB, group III metabotropic glutamate, muscarinic, α4β2 nicotinic, and A1 adenosine receptors were also blocked by adding to the perfusion medium CGP52432 (5 μm), MSOP (RS-α-methylserine-O-phosphate; 100 μm), atropine (1 μm), DHβE (dihydro-β-erythroidine; 1 μm) and DPCPX (8-cyclopentyl-1,3-dipropylxanthine; 200 nm), respectively.
IPSCs were evoked every 15 s with stimuli (100 μs; 50–250 μA square pulses) delivered via bipolar stainless steel electrodes (FHC, Bowdoinham, ME) positioned in stratum radiatum. Paired-pulse ratio (PPR) was measured as IPSC2/IPSC1 with an intertrial interval of 20 ms. GABAA or nAChR agonists were pressure applied with a Picospritzer II (3–5 MΩ glass pipette; 5–50 ms at 15–20 psi; General Valve, Brookshire, TX). The agonists (100 μm GABA, 10 μm muscimol, 1 mm ACh, or 10 mm choline) were dissolved on the day of the experiments in artificial CSF containing the same mixture of antagonists present in the perfusion medium. For the experiments in Figure 2, 1% dextran Texas Red 3000 molecular weight (MW) was included in the pipette solution to help visualize the main dendrites using a TILLVision imaging system (Till Photonics, Martinsried, Germany). A bipolar stimulating electrode, prepared from theta glass filled with extracellular medium, was used to evoke IPSCs, and a pressure-application pipette containing ACh was positioned near the visualized dendrite to activate nAChRs. For the heterosynaptic depression experiments, a second stimulus electrode was positioned in CA1 stratum radiatum or in the alveus/stratum oriens relatively far (>1 mm) from the recording site.
Where averages are shown, they represent all interneurons recorded. The data are shown normalized to baseline and expressed as mean ± SEM. They were analyzed with nonparametric Wilcoxon paired or unpaired tests. Representative traces were averaged from 5–10 consecutive trials, and for the time-course plots, data points were binned in 30–45 s periods.
Chemicals were purchased from Sigma (St. Louis, MO). Receptor antagonists were purchased from Tocris Cookson (Bristol, UK) except staurosporine (Streptomyces sp.) and bisindolylmaleimide I (BisI; Merck, Darmstadt, Germany), FK506 (A.G. Scientific, San Diego, CA), the protein kinase C (PKC) inhibitory peptide (IP19-36; Alexis, Lausen, Switzerland) and dextran Texas Red 3000 MW (Invitrogen, Carlsbad, CA).
Bath perfusion of 10 mm ACh evoked an increase in holding current in stratum radiatum interneurons, consistent with activation of cation-permeable nAChRs (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Pilot experiments showed that local pressure application of 1 mm ACh in interneurons evoked similar inward currents in rat and guinea pig, which were blocked by 30 nm methyllycaconitine (MLA). Data shown in Figures 1, 2, 4B–E, 5, 6, and 7 and supplemental Figures 1, 3, 4, and 6 (available at www.jneurosci.org as supplemental material) were obtained from rat slices. Data in Figures 3, 4A, and 8 and supplemental Figures 2 and 5 (available at www.jneurosci.org as supplemental material) were obtained from guinea pig slices. The heterosynaptic depression phenomenon was demonstrated in slices from both species.
Results
α7 nAChRs depress IPSCs
Several studies have shown that excitatory actions of α7 nAChR agonists are prominent in interneurons in stratum radiatum (Frazier et al., 1998a; Ji and Dani, 2000; Khiroug et al., 2003), especially those close to stratum lacunosum-moleculare (McQuiston and Madison, 1999). We therefore focused attention on CA1 interneurons in this region, identified using infrared video microscopy (although we also present some data on pyramidal cells). To test whether the effects observed are species specific, we obtained data from rat and guinea pig. No differences were observed, and so data are presented together (see Materials and Methods for the species used for each illustrated experiment). To avoid potential contaminating polysynaptic effects, fast glutamatergic transmission was routinely blocked with selective antagonists, as were GABAB, muscarinic, and nicotinic α4β2 receptors.
We evoked monosynaptic GABAergic IPSCs by delivering electrical stimuli via an electrode positioned in stratum radiatum. Bath-applied ACh (10 mm) reversibly depressed stimulus-evoked IPSCs (eIPSCs; mean depression, 56 ± 6%; recovery to 78 ± 4% of baseline; n = 5; p < 0.001) (Fig. 1A–C). The depression was not accompanied by detectable changes in eIPSC kinetics (supplemental Table 1, available at www.jneurosci.org as supplemental material). The effect of ACh on eIPSCs was completely blocked by the α7-selective blocker MLA (100 nm; ACh in MLA, 95 ± 6% of baseline; n = 5; p = 0.84). MLA also abolished the ACh-evoked increase in holding current. Application of a lower concentration of ACh (300 μm) also evoked a reversible decrease in eIPSCs (29 ± 5% reduction; n = 5; p < 0.01) (Fig. 1D–F), although this was smaller than that seen with 10 mm. Bath perfusion of 100 μm ACh did not significantly depress eIPSCs (data not shown).

ACh depresses eIPSCs in stratum radiatum interneurons. A, Representative eIPSCs recorded from an interneuron in stratum radiatum during baseline (1), during bath application of 10 mm ACh (2), and after washout in the presence of 100 nm MLA, an α7 nAChR-selective antagonist (3). B1, Time course plots showing the effect of ACh on the eIPSC amplitude (B1) and holding current (B2) and blockade of the effect by MLA (same cell as shown in A). C, Summary of data obtained in five cells. ACh, After 10 mm ACh application; Wash, recovery after 10 min washout. The data obtained in the presence of MLA (+ MLA) were renormalized to a new baseline. D, Representative eIPSCs before (1), during (2), and after (3) bath application of 300 μm ACh. E, Time course of ACh effect on eIPSC amplitude (E1) and holding current (E2) (same cell as shown in D). F, Summary of data obtained with 300 μm ACh in 5 cells. **p < 0.01; ***p < 0.001. Ihold, Holding current; norm, normalized.
Although these results are consistent with an action of α7 nAChRs on GABAergic signaling, MLA at 100 nm has been reported to act on other subtypes of nAChRs (Mogg et al., 2002). Moreover, the concentration of ACh required to attenuate eIPSCs was higher than expected from the EC50 at α7 nAChRs (Alkondon et al., 1997b; Papke and Porter Papke, 2002). A possible explanation is that ACh is hydrolyzed by extracellular cholinesterases, reducing the effective concentration reaching the nAChRs. Furthermore, some α7 nAChRs may have been desensitized by the slow wave of ACh penetrating the slice. We therefore delivered ACh locally via pressure application while evoking IPSCs (Fig. 2A). Brief pressure application of ACh (pipette concentration, 1 mm) reversibly depressed eIPSCs (36 ± 7% depression; p < 0.01; n = 9) (Fig. 2B) as well as evoking fast inward currents (Fig. 2C). Notably, the depression of eIPSCs evolved more slowly than the ACh-evoked inward currents and persisted for several minutes after return of the holding current to baseline (see Fig. 2C, example traces). A low concentration of MLA (10 nm) fully blocked both the inward currents and the modulation of eIPSCs (ACh in MLA, 101 ± 1% of baseline; n = 6; p = 0.82) (Fig. 2B–D), consistent with the sensitivity of α7 nAChRs to this blocker.

Brief applications of ACh reversibly depress eIPSCs in interneurons. A, Differential interference contrast (left) and epifluorescence (right) images showing the arrangement of stimulating electrode (Stim.), pressure-application pipette (ACh; 1 mm), and recording pipette (Rec.). Scale bar, 20 μm. B1, eIPSCs recorded from an interneuron during baseline (1), 3 min after pressure application of 1 mm ACh (2), and after a 10 min recovery period (3), and subsequently in the presence of 10 nm MLA before (4) and after (5) ACh application. B2, Time course of ACh effect on eIPSCs and blockade of the effect by MLA, showing the times at which the traces in B1 were obtained (1–5). IPSCs were evoked every 15 s (traces, averages of 10 successive trials at the times indicated). Arrowheads indicate pressure application of ACh, 2 s before IPSCs were evoked, repeated 10 times. C1, Representative traces showing ACh-evoked inward current (10 ms pulse at 10 psi) in control conditions (left) and in the presence of 10 nm MLA (right). C2, Time course of the ACh effect on the holding current measured immediately before (open symbols) and at a time corresponding to the peak of (filled symbols) the ACh-evoked inward currents in the same neuron. D, Summary of data obtained in nine cells under control conditions and six cells in MLA. ACh, 3 min after the last ACh application; Wash, recovery 10 min after the last ACh application; the data obtained in the presence of MLA (+ MLA) were renormalized to a new baseline. *p < 0.05; **p < 0.01. Ihold, Holding current; norm, normalized.
To test further whether α7 nAChRs accounted for the depression of eIPSCs, we applied two selective agonists. Choline is a low-affinity but relatively nondesensitizing agonist at α7 nAChRs, with an EC50 ∼20-fold greater than for ACh (Alkondon et al., 1997b; Papke and Porter Papke, 2002). We first verified that pressure-applied choline (pipette concentration, 10 mm; 10–15 ms at 10 psi) elicited a robust inward current in all interneurons tested (613 ± 36 pA; n = 5; data not shown), which was blocked by 30 nm MLA. Having established that choline activates nAChRs with a pharmacological profile consistent with α7, we asked whether choline could decrease the amplitude of eIPSCs. Bath-applied choline (1 mm) reversibly depressed eIPSCs in stratum radiatum interneurons (22 ± 2% inhibition; n = 4; p < 0.01) (Fig. 3A). This effect was blocked by MLA (data not shown). When repeated in pyramidal neurons, the effect of choline was much smaller (8.6 ± 1.5% inhibition; n = 6) albeit significant at p < 0.01 (Fig. 3A), consistent with a lower density of α7 nAChRs (Alkondon et al., 1998; Frazier et al., 1998a).
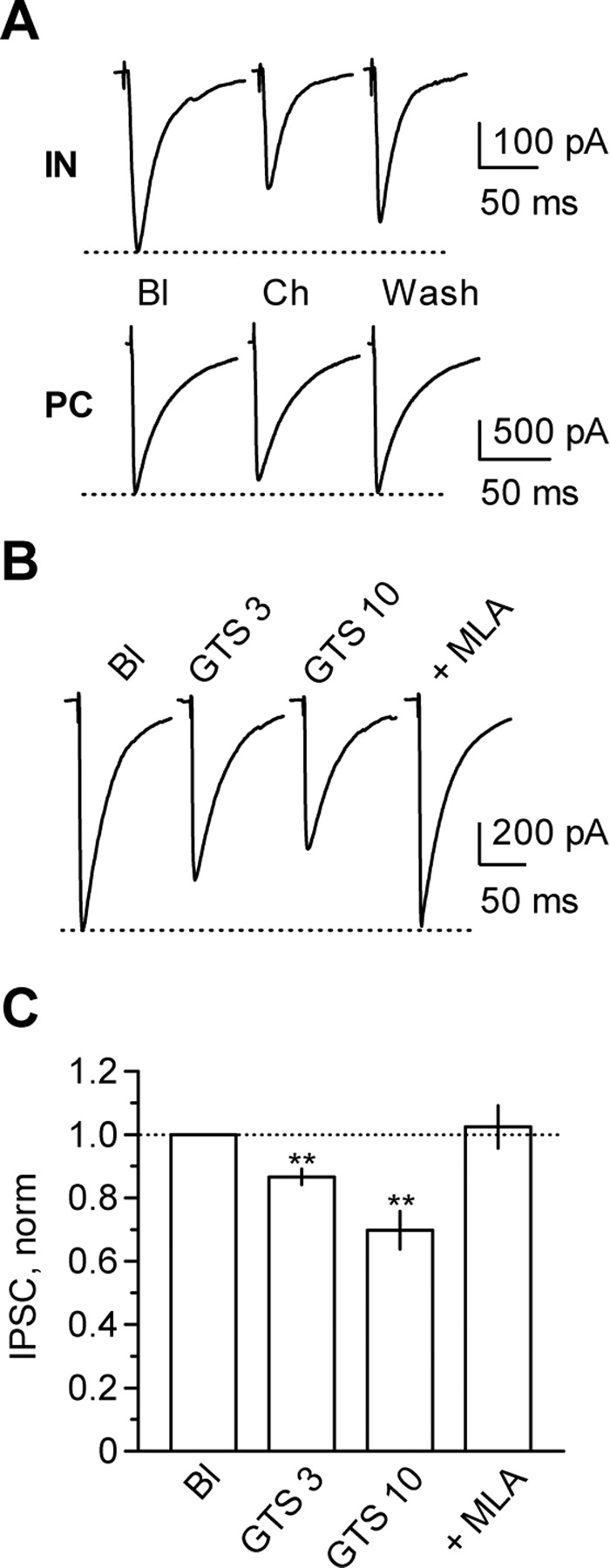
Selective α7 nAChR agonists depress eIPSCs in interneurons to a larger extent than in pyramidal cells. A, Representative eIPSCs recorded in stratum radiatum interneurons (IN) and pyramidal cells (PC) in the CA1 region before (Bl), during (Ch), and after (Wash) bath application of 1 mm choline. B, Representative traces showing concentration-dependent depression of eIPSCs by the selective α7 nAChR agonist GTS and inhibition of the GTS effect by 30 nm MLA (+ MLA). C, Averaged results showing the effect of 3 and 10 μm GTS and reversal of the effect of 10 μm GTS by 30 nm MLA (Bl, n = 8; GTS 3, n = 6; GTS 10, n = 8; + MLA, n = 7). **p < 0.01. norm, Normalized.
α7 nAChRs are also activated by the highly potent and selective agonist 3-(4-hydroxy,2-methoxybenzylidene)anabaseine [4OH-GTS21 (GTS)] (Papke et al., 2000). Bath application of this agonist elicited a concentration-dependent reduction of eIPSC amplitude in interneurons: 13 ± 3% (n = 6; p < 0.01) and 30 ± 6% (n = 8; p < 0.01) for 3 and 10 μm GTS, respectively. This reduction was reversed by addition of 30 nm MLA (recovery to 102 ± 7% of baseline; n = 7; p = 0.73) (Fig. 3B,C).
We thus conclude that α7 nAChRs robustly depress GABAergic eIPSCs recorded in hippocampal interneurons.
α7 nAChR-mediated depression of IPSCs is postsynaptically mediated
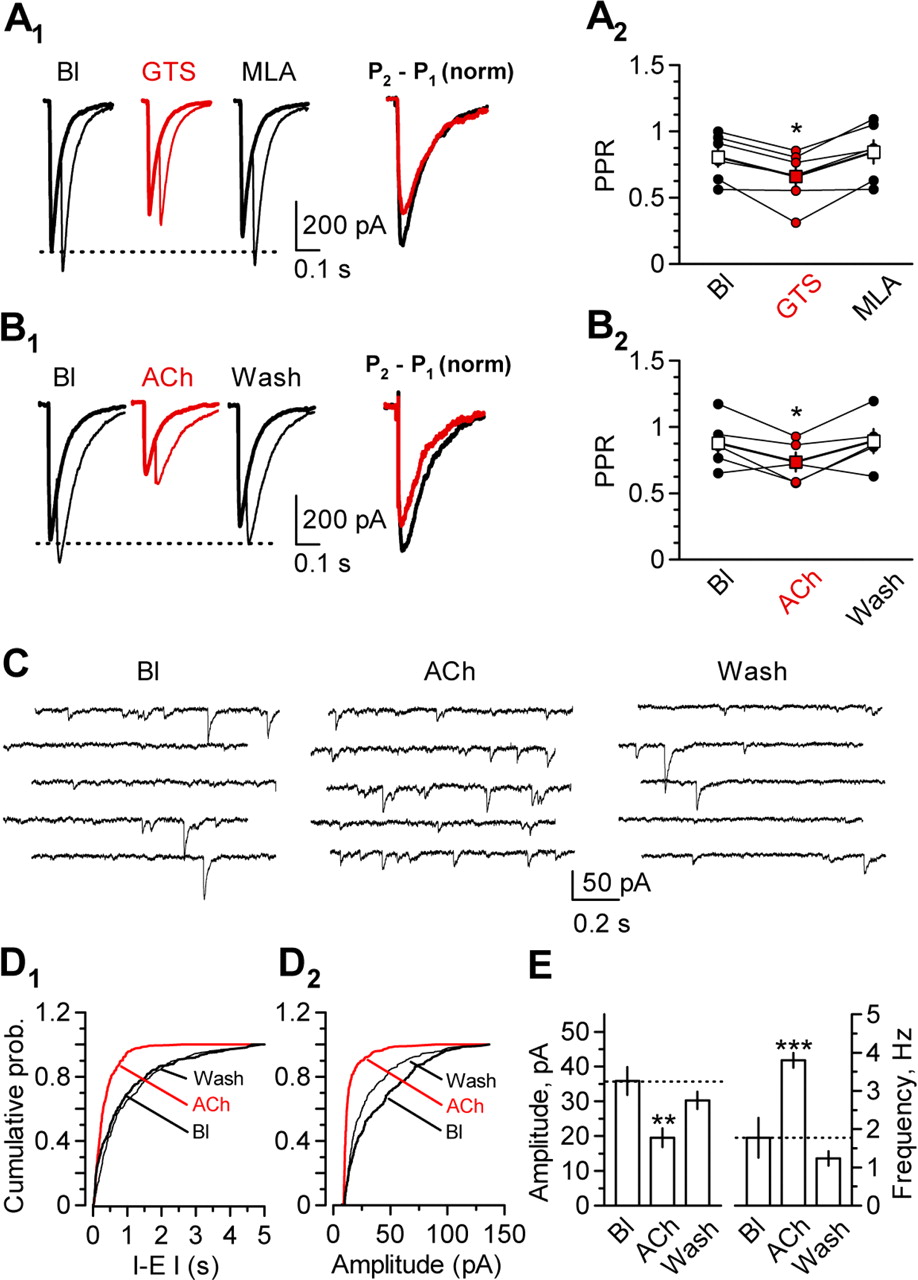
nAChRs have previously been reported to affect neurotransmitter release, although in the opposite direction from that required to explain the inhibition of eIPSCs (Seguela et al., 1993; McGehee et al., 1995; Gray et al., 1996). We asked whether the α7 nAChR-mediated depression of eIPSCs was accompanied by a change in PPR, which is sensitive to manipulations that alter transmitter release probability (Manabe et al., 1993). We observed a small, albeit significant, decrease in PPR with 10 μm GTS. The PPR decreased on average from 0.81 ± 0.07 to 0.65 ± 0.08 (n = 6; p < 0.05) and recovered to 0.84 ± 0.09 in the presence of MLA (n = 6; p = 0.27 for baseline vs MLA). This change in PPR is consistent with an increase in release probability, although it accompanied a net decrease in the amplitude of the first eIPSC of 25 ± 6% (n = 6; p < 0.01) (Fig. 4A). A similar result was observed after bath application of 10 mm ACh: the PPR decreased from 0.97 ± 0.11 to 0.75 ± 0.06 (n = 5; p < 0.05) and recovered after washout to 0.99 ± 0.11 (p = 0.34). This change in PPR accompanied a reduction of the first eIPSC of 56 ± 6% (p < 0.001) (Fig. 4B).

Evidence for opposing presynaptic and postsynaptic effects of α7 nAChRs on eIPSCs. A, Paired-pulse ratio measurement and effect of α7 nAChR activation. A1, Sample traces show IPSCs evoked by either one (thick line) or two (thin line) stimuli, before (Bl) and during (red traces) bath application of 10 μm GTS and after washout and bath application of 100 nm MLA. The superimposed traces on the right were obtained by subtracting the first eIPSC from the paired-stimulus response to reveal the shape of the second eIPSC [P2 − P1, normalized (norm)]. The traces were then normalized by the amplitude of the first eIPSC and are shown before (black) and during (red) GTS application. The decreased relative amplitude of the second eIPSC indicates a decrease in PPR. A2, Results pooled from six experiments, showing a decrease in PPR during GTS application (red symbols), which accompanied a reduction of the first eIPSC by 24.5 ± 5.9% (n = 6; p < 0.01; circles represent individual experiments; squares represent means). B1, Representative traces obtained in one interneuron before (Bl), during (ACh; red), and after (Wash) bath application of 10 mm ACh. B2, Results pooled from five experiments showing the effect of ACh on PPR. ACh reduced the eIPSC amplitude by 56 ± 6% (n = 5; p < 0.001; circles represent individual experiments; squares represent means). C, Representative traces showing miniature IPSCs recorded in an interneuron in the presence of TTX (2 μm) before (Bl) and during ACh application (ACh) and after 10 min washout (Wash). In each condition, traces represent a continuous recording period of 5 s. D, Cumulative probability (prob.) plots for the interevent interval (I-E I; D1) and the amplitude (D2) from the cell shown in C at baseline (Bl), during ACh application (ACh; red line), and after washout (Wash). E, Averaged results obtained from seven cells showing the effect of ACh on the mIPSCs amplitude (left) and frequency (right). *p < 0.05; **p < 0.01; ***p < 0.001.
The change in PPR is strongly suggestive of a presynaptic increase in GABA release probability, consistent with previous reports on glutamate release. Consistent with this, ACh application also reversibly increased the frequency of spontaneous IPSCs (sIPSCs) by 193 ± 18% (from 3.4 ± 0.3 to 6.3 ± 0.3 Hz; n = 7; p < 0.05) (supplemental Table 2, available at www.jneurosci.org as supplemental material). This increase in frequency was accompanied by a 9 ± 3% decrease in sIPSC amplitude (from 59 ± 7 to 49 ± 7 pA; n = 7; p < 0.05). Both the increase in frequency and the decrease in amplitude were reversible after wash (2.6 ± 0.5 Hz, p = 0.06; 63 ± 4 pA, p = 0.33). In the presence of 100 nm MLA, neither the frequency nor the amplitude of sIPSCs was affected by ACh [baseline (Bl), 3.6 ± 1.1 Hz, 61 ± 7 pA; ACh, 3.4 ± 0.8 Hz, 61 ± 8 pA; n = 7; p = 0.25 and p = 0.09, respectively) (supplemental Table 2, available at www.jneurosci.org as supplemental material). In seven other cells, we further confirmed that ACh enhanced GABA release by recording action potential-independent miniature IPSCs (mIPSCs) in the presence of tetrodotoxin (TTX; 2 μm) (Fig. 4C): bath application of ACh (10 mm) elicited a reversible 317 ± 87% increase in mIPSC frequency (from 1.8 ± 0.5 Hz in Bl to 3.8 ± 0.2 Hz in ACh; n = 7; p < 0.01) (Fig. 4D1,E, right). This increase in frequency was accompanied by a 54 ± 4% decrease in mIPSCs amplitude (from 36 ± 4 pA in Bl to 19 ± 3 pA in ACh; n = 7; p < 0.01) (Fig. 4D2,E, left). After 10 min washout, both the amplitude and the frequency of the mIPSCs returned to baseline values (Fig. 4C–E).
The alternative explanation for the reduction in eIPSC amplitude is that, in parallel with a modest presynaptic enhancement of GABA release, α7 nAChRs also cause a more profound postsynaptic decrease in the response to GABA. We tested this by measuring the effect of bath-applied ACh on currents evoked in hippocampal interneurons by pressure application of GABA (100 μm; 5–10 ms at 10 psi), to bypass presynaptic transmitter release mechanisms. We verified that GABA-evoked currents were fully blocked by 100 μm picrotoxin at the end of the experiment (data not shown). GABA-evoked currents were profoundly depressed by 10 mm ACh (55 ± 1%; n = 5; p < 0.001) (Fig. 5A,B). When applied twice in the same cell, ACh evoked a robust, reversible, and reproducible depression of the GABA-evoked current (depression, 56 ± 4% and 53 ± 5% for the first and second ACh applications, respectively; n = 6; p = 0.083 for comparison), with full recovery after washout (98 ± 2 and 96 ± 1% for the two applications; p = 0.79). MLA (100 nm) completely blocked the depression of the GABA current when added to the perfusion solution before the second application of ACh (Fig. 5A,B).

ACh produces an α7 nAChR-mediated and concentration-dependent reduction of GABA-evoked currents. A, Time course plot showing the effect of ACh (10 mm) on the agonist-evoked GABA current amplitude (A1) and holding current (A2) and blockade of the effect by MLA. B, Averaged results from five experiments. The data obtained in the presence of MLA were renormalized to a new baseline. C, Representative traces showing currents evoked by GABA application before (black) and during (red) bath application of 0.1 (n = 4), 0.3 (n = 4), and 10 mm (n = 5) ACh (each trace is normalized to the peak amplitude of the GABA current before ACh application). D, Pooled results showing ACh-evoked depression of GABA currents. ***p < 0.001. Ihold, Holding current; norm, normalized.
The depression of GABA-evoked currents was concentration dependent: 300 μm ACh produced a 17 ± 3% depression (n = 4; p < 0.001), whereas 100 μm ACh was ineffective (1.0 ± 2.5%; n = 4; p = 0.34) (Fig. 5C,D). These results are consistent with the effects of different concentrations of ACh on eIPSCs (Fig. 1). As mentioned above, a possible explanation for the failure to evoke a depression of GABA-evoked currents with 100 μm ACh is that cholinesterase activity greatly lowers the effective concentration reaching the receptors. We therefore repeated the 100 μm ACh application in the presence of the cholinesterase blocker eserine (5 μm). This revealed a robust depression of the GABA-evoked current (22 ± 4%; n = 7; p < 0.05), accompanied by a small increase in holding current (Fig. 6).

Cholinesterase blockade reveals depression of GABA-evoked currents by a low concentration of ACh. A, Representative GABA-evoked currents recorded during baseline (1) and in the continued presence of eserine before (2) and during (3) bath application of ACh and after washout (4). B, Time course plot showing the effect of ACh on the GABA-evoked current amplitude (B1) and holding current (B2), before and after addition of eserine [Ese; same cell as (A)]. C, Summary data obtained in five and seven cells in the absence and presence of eserine, respectively. The data obtained in the presence of eserine were renormalized to a new baseline. ACh, After 100 μm ACh application; Wash, recovery after 10 min washout. *p < 0.05. Ihold, Holding current; norm, normalized.
A potential confounding factor in the above experiments is that prolonged whole-cell recording may have perturbed the signaling cascade from α7 nAChRs to GABAA receptors. To minimize this source of artifact, we examined the effect of nAChRs noninvasively, by applying the amphotericin B perforated-patch recording method (Horn and Marty, 1988). With this recording method, 10 μm GTS produced an even larger depression of eIPSC amplitude than with whole-cell recording [43 ± 7%; n = 3; p < 0.001 (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) compared with 30 ± 6%; n = 8 (Fig. 3B,C); p < 0.05 for comparison of perforated-patch vs whole-cell]. The reduction was fully reversed by 100 nm MLA. This implies that whole-cell recording may underestimate the magnitude of the depression of eIPSCs and is again consistent with a postsynaptic site of action.
Because α7 nAChR activation leads to opening of a cation-selective conductance in the interneurons, it is reasonable to ask whether the decrease of GABAA receptor-mediated currents could simply be an electrical shunting artifact. This is highly unlikely, because the time course of the increase in holding current after application of ACh was clearly dissociated from the time course of the depression of eIPSCs or GABA-evoked currents (Figs. 1, 2, 5, 6). We nevertheless tested this hypothesis by injecting simulated synaptic conductances while recording from interneurons in dynamic current-clamp mode. ACh (10 mm) had no detectable effect on simulated IPSPs, whereas stimulus-evoked IPSPs recorded in parallel were profoundly and reversibly decreased (for additional discussion, see supplemental Fig. 3 and legend, available at www.jneurosci.org as supplemental material).
We thus conclude that ACh, choline, and GTS act on α7 nAChRs to depress GABAergic transmission and that this effect can be explained by a postsynaptic action on GABAA receptors that outweighs a small presynaptic enhancement in GABA release. However, the results do not exclude the possibility that α7 nAChRs act indirectly, by exciting surrounding neurons and causing the release of other neurotransmitters, which influence GABAA receptors on the recorded neuron. Among neurotransmitters released from neighboring interneurons, GABA itself could conceivably accumulate in the extracellular space, leading to receptor desensitization. Such a mechanism has been suggested to contribute to the depression of GABAergic transmission by kainate receptors (Frerking et al., 1999). We therefore repeated the application of 10 mm ACh while blocking action potential generation with TTX (2 μm) (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). An almost identical depression of GABA-evoked currents was obtained (61 ± 13%, compared with 56 ± 3% without TTX; p = 0.24), confirming that the effect of α7 nAChR activation did not depend on spontaneous activity in the slice. These results show that postsynaptic α7 nAChRs decrease GABA currents directly and argue against any involvement of presynaptic structures or sodium channels, which may play a role in choline-evoked modulation of GABAA receptor-mediated currents in hippocampal neurons (Alkondon et al., 2000).
The α7 nAChR action on GABAA receptors involves Ca2+ and PKC
In all of the above experiments, whether the agonist was applied by bath or by pressure application, the α7 nAChR-mediated depression of GABAA receptor-mediated currents developed over several minutes. We examined this further by pressure applying the GABAA receptor agonist muscimol (10 μm; 5–10 ms at 10 psi), either alone or simultaneously with choline (10 mm; 10–20 ms at 10 psi), delivered via a second pipette. After pressure application of choline, the GABAA receptor-mediated current decreased progressively to 73 ± 2% of baseline (n = 3; p < 0.001) over several minutes and recovered only slowly (although fully) after terminating the choline coapplication (104 ± 2% of baseline; p = 0.053) (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). The time course of the depression of muscimol-evoked currents by choline was similar to that of the depression of GABA-evoked currents by pressure-applied ACh (Fig. 2B). This suggests that a slow postsynaptic transduction cascade is responsible for the effect, because choline is unlikely to accumulate slowly and persist at high concentrations for several minutes after highly localized pressure application.
A potential candidate messenger that mediates the effect of ACh, choline, or GTS is Ca2+ entering through α7 nAChRs (Mulle et al., 1992). However, direct depolarization to −20 mV for 30 s to evoke Ca2+ influx via voltage-gated Ca2+ channels was without effect on GABA currents (data not shown). Nevertheless, by analogy with the role of NMDA receptors in long-term potentiation, the synaptic localization of α7 receptors (Fabian-Fine et al., 2001; Kawai et al., 2002) may allow Ca2+ entering via the nAChRs to gain privileged access to an effector cascade that alters GABAA receptors. We tested the role of Ca2+ ions by repeating the ACh application while recording from hippocampal interneurons with a pipette containing a high concentration of the Ca2+ chelator BAPTA (10 mm). We first verified that pressure application of ACh evoked inward currents in interneurons recorded with a BAPTA-containing pipette, similar to those evoked in cells recorded with EGTA, arguing that Ca2+ chelation does not impair ion flux through nAChRs. In contrast to the situation observed with 2 mm EGTA (Fig. 7A,C,E), bath application of ACh failed to depress GABA-evoked currents and instead induced a small albeit significant increase (16 ± 8%; n = 5; p < 0.05) (Fig. 7B,D,F). ACh application was, however, accompanied by an increase in holding current (19.0 ± 4.1%), comparable with that seen in the experiments with EGTA (21 ± 7%; n = 5; p = 0.64 for comparison between EGTA and BAPTA), implying that the cation flux through α7 nAChRs could be dissociated from the depression of GABAA receptor-mediated currents. The effect of BAPTA further argues against shunting and/or desensitization as the explanation for the α7 nAChR agonist-evoked depression of GABAA receptor-mediated currents.

Depression of GABAA receptor-mediated currents requires postsynaptic Ca2+ elevation and involves PKC. A, B, Representative traces showing GABA-evoked currents recorded with an intracellular solution containing 2 mm EGTA (A) or 10 mm BAPTA (B) at baseline (1), during bath application of 10 mm ACh (2), and after washout of ACh (3). C, D, Time course of the effect of ACh on GABA-evoked currents (C1, D1) and on the holding current (C2, D2) for the cells shown in A and B. E, F, Averaged data pooled from five experiments with 2 mm intracellular EGTA (E) and five experiments with 10 mm intracellular BAPTA (F). G, Summary of the effects of kinase and calcineurin inhibitors on the ACh-mediated depression of GABA-evoked currents. The inhibition of GABA currents observed with ACh application alone (from Fig. 5B) is shown on the left for comparison. Staurosporine (+ Staur; 200 nm; n = 6), bisindolylmaleimide I (+ Bis I; 1 μm; n = 5), PKC inhibitory peptide (+ IP19-36; 4 μm added to the patch-pipette solution; n = 6), genistein (+ Gen; 30 μm; n = 3), and FK506 (added at 0.5 μm in the patch-pipette solution; n = 3) were used. *p < 0.05; **p < 0.01; ***p < 0.001. Ihold, Holding current; norm, normalized.
The results above imply that α7 nAChR activation normally depresses GABAA receptor-mediated currents via a Ca2+-dependent intracellular cascade. Experimental manipulations of several intracellular signaling mechanisms, principally involving protein phosphorylation, have been shown to modulate the function and/or number of GABAA receptors expressed in neurons or cultured neurons [for review, see Kittler and Moss (2003) and Luscher and Keller (2004)]. To determine which pathway is involved in the α7 nAChR-mediated inhibition of GABAergic signaling in interneurons, we repeated the application of ACh in the presence of different inhibitors (each bath applied for at least 20 min before testing the effect of ACh) (Fig. 7G). Genistein (30 μm), a tyrosine kinase inhibitor, did not prevent the inhibition of GABA-evoked currents (52 ± 8%; n = 3) [in a separate study, we have verified that this concentration is sufficient to interfere with NMDA receptor-mediated signaling in stratum radiatum interneurons, implying that the drug penetrated the slice (K. P. Lamsa and D. M. Kullmann, unpublished results)]. In contrast, bath application of staurosporine (200 nm), a broad-spectrum protein kinase inhibitor, markedly attenuated the ACh-evoked inhibition of GABA currents (20 ± 9%; n = 6; p < 0.01 for comparison with ACh effect without staurosporine). A possible explanation for the effect of staurosporine is that it interfered with PKC, activation of which has been shown to negatively modulate GABAA receptors (Sigel and Baur, 1988; Brandon et al., 2000). We therefore applied ACh after preincubating in BisI (1 μm), a more selective PKC inhibitor. This treatment also attenuated the effect of ACh (17 ± 3%; n = 5; p < 0.01 for comparison with ACh alone). We further tested the role of PKC by including the specific inhibitory peptide IP19-36 (4 μm), which corresponds to the autoinhibitory domain of the enzyme (Smith et al., 1990), in the pipette solution. This also significantly attenuated the effect of ACh on GABA-evoked currents (23 ± 6%; n = 6; p < 0.05 for comparison to ACh alone). The phosphatase calcineurin has also been reported to mediate long-term depression of GABA IPSPs in the CA1 region of the hippocampus (Lu et al., 2000; Wang et al., 2003). However, when calcineurin was inhibited by including 0.5 μm FK506 in the patch pipette, the action of ACh on GABA currents was no different from in the control experiments (44 ± 16%; n = 3; p = 0.55). These results are most simply explained by the hypothesis that Ca2+ entering through α7 nAChRs activates PKC, which in turn acts on GABAA receptors in interneurons. Nevertheless, none of the PKC blockers fully abolished the effect of ACh, suggesting an additional PKC-independent pathway leading from Ca2+ influx via α7 nAChRs to GABAA receptor modulation.
Endogenous ACh release evokes heterosynaptic depression of GABAergic signaling
What is the physiological relevance of the phenomena described in the present study? The cholinergic projection to the hippocampus is diffuse and enters via the fornix/fimbria (Mesulam, 2004b), although some intrinsic cholinergic neurons also exist (Frotscher et al., 1986). We attempted to detect a role for endogenous ACh release by performing a two-pathway experiment, to determine whether activation of cholinergic axons could affect a test eIPSC. We monitored a monosynaptic eIPSC in stratum radiatum interneurons evoked by stimulating locally and delivered a train of high-frequency stimuli (20 pulses at 100 Hz) via a second stimulating electrode positioned in stratum radiatum relatively far from the interneuron, with the aim of evoking ACh release from cholinergic axons. This evoked a small but significant and reversible depression of eIPSC amplitude (16 ± 5% depression; n = 11; p < 0.01) (Fig. 8A). Interestingly, this depression developed over 3–4 min, an onset rate similar to that observed after either bath or local pressure application of agonists. The phenomenon was completely blocked by 100 nm MLA (94.6 ± 4.9% of baseline; n = 7; p = 0.31), confirming that it was mediated by α7 nAChRs (Fig. 8A,B).

Endogenous ACh release evokes α7 nAChR-mediated depression of eIPSC in CA1 interneurons. A, Representative eIPSCs obtained from one interneuron, before (1) and 3 min after (2) tetanic stimulation (arrow in B; 20 pulses at 100 Hz) delivered via a second electrode placed in stratum radiatum and 10 min after conditioning (3). Top, Without MLA. Bottom, Results obtained during continued perfusion of 100 nm MLA. B, Time course of heterosynaptic depression (n = 11) and blockade of the effect by MLA (n = 7), showing the times at which the traces in A were obtained (1–3). C, Representative eIPSCs obtained from one neuron, before (1) and after (2) tetanic stimulation (arrow in D) delivered to stratum oriens and after recovery (3). Tetanic stimulation (arrow) was delivered a second time in the presence of the anticholinesterase eserine (4). 5, Recovery. D, Time course of heterosynaptic depression before and after eserine application (Ese), showing times corresponding to the traces in C (n = 9). E, Summary of the extent of heterosynaptic depression expressed as the product of depression and duration (AUC) for the data shown in B and D. str. rad, Stratum radiatum stimulation (n = 11); str. or, stratum oriens stimulation (n = 13); str. or + Ese, repeated in eserine (n = 9). MLA completely blocked heterosynaptic depression evoked by stratum oriens stimulation (+ MLA; n = 4). *p < 0.05; **p < 0.01. norm, Normalized.
We repeated the experiment with the conditioning electrode positioned in the alveus/stratum oriens, where the septohippocampal projection enters the hippocampus, in an attempt to recruit more cholinergic afferent fibers (Fig. 8C,D). We obtained an estimate of the extent of depression by taking the product of its amplitude and duration [area under the curve (AUC)]. This parameter was 2.14 ± 0.60 when the conditioning stimulation was delivered in the alveus/stratum oriens versus 0.96 ± 0.25 when delivered in stratum radiatum (Fig. 8E). To determine whether ACh hydrolysis limits the activation of nAChRs, we repeated the conditioning stimulation train (delivered to stratum oriens) in the presence of eserine (5 μm). The depression was further enhanced and prolonged in the presence of eserine (AUC, 5.01 ± 1.90; n = 9; p < 0.05) but completely blocked in the presence of MLA (Fig. 8D,E). This finding is consistent with the hypothesis that the endogenous agonist liberated by stimulus trains and acting on α7 nAChRs is indeed a substrate of cholinesterases, most likely ACh released from the cholinergic afferent projection from basal forebrain structures. Finally, we repeated the two-pathway experiment in a second species. This experiment gave a similar heterosynaptic depression (23 ± 6%; n = 4; p < 0.001) (supplemental Fig. 6A,B,E, available at www.jneurosci.org as supplemental material), which was partly reversible (92 ± 4% of baseline; p = 0.12). When 10 mm BAPTA was included in the intracellular solution, however, high-frequency stimulation delivered in stratum oriens failed to depress eIPSCs (101 ± 1%; n = 6; p = 0.37) (supplemental Fig. 6C–E, available at www.jneurosci.org as supplemental material), further supporting a postsynaptic locus for the heterosynaptic modulation of GABAergic transmission.
Discussion
The main finding of this study is that α7 nAChRs modulate IPSCs by an action on GABAA receptors and that this effect is achieved not only by applying exogenous agonists but also by activating cholinergic axons in the hippocampus. This action of α7 nAChRs was seen in two species and in all stratum radiatum interneurons tested.
Despite their abundance in the hippocampus, the role of α7 nAChRs has been elusive. Most studies have focused either on direct depolarizing actions of ACh on interneurons or on the enhancement of neurotransmitter release, although postsynaptic modulatory effects have also been reported (Alkondon et al., 2000; Fisher and Dani, 2000). However, there are only very few reports that synaptically released ACh can achieve either of these effects. Indeed, only two actions of endogenous ACh on hippocampal nAChRs have been recorded with electrophysiological methods to date. First, three studies have reported that fast inward currents sensitive to α7 nAChR blockers can be elicited in interneurons (Alkondon et al., 1998; Frazier et al., 1998b; Hefft et al., 1999). Second, two other studies have reported a modulation of excitatory transmission that is consistent with a presynaptic switch in the mode of glutamate exocytosis (Maggi et al., 2003, 2004). By showing that nAChRs robustly downregulate GABAA receptors in all interneurons tested, the present study identifies a novel role in modulating inhibitory transmission among interneurons.
The complete postsynaptic cascade leading from nAChR activation to modulation of GABAA receptor-mediated currents remains to be elucidated. We found an obligatory role for an increase in [Ca2+]i, most simply explained by Ca2+ influx via α7 nAChRs, which have a high Ca2+ permeability (Bertrand et al., 1993; Seguela et al., 1993; Fucile, 2004). However, we cannot rule out a role for Ca2+ release from intracellular stores. Ca2+-evoked modulation of GABAA receptors has been described previously (Desaulles et al., 1991; Mulle et al., 1992; Stelzer and Shi, 1994; Wang and Stelzer, 1996; Akopian et al., 1998; Rossi et al., 2003), although this has not previously been shown to occur consequent to release of endogenous ACh. Several mechanisms have been proposed to underlie the dynamic regulation of GABAA receptors, mainly involving protein kinases and phosphorylases, leading to altered phosphorylation status of GABAA receptors and/or internalization (Kittler and Moss, 2003; Luscher and Keller, 2004).
We observed a profound, albeit incomplete, reduction in the effect of ACh on GABA currents when it was applied in the presence of either the broad-spectrum kinase blocker staurosporine or the more specific PKC blockers bisindolylmaleimide I and IP19-36. This is consistent with evidence that activators of PKC attenuate currents mediated by recombinant (Sigel and Baur, 1988) and native (Brandon et al., 2000) GABAA receptors. However, the present results do not distinguish between instructive and permissive roles for PKC. Whether the effect of ACh is principally attributable to direct phosphorylation of β subunits of GABA receptors (Browning et al., 1990) or to PKC-triggered internalization (Chapell et al., 1998) remains to be determined. If the main effect of nAChRs is to promote the internalization of receptors, the fact that the effect is slow and reversible suggests that this occurs against a background of constitutive cycling of receptors in and out of the synapse. Interestingly, the effect of α7 nAChR agonists on postsynaptic GABAergic signaling via activation of PKC is similar to that of exogenous activation of TrkB receptors (Tanaka et al., 1997; Brunig et al., 2001; Henneberger et al., 2002; Jovanovic et al., 2004), serotonin receptors (Feng et al., 2001), and muscarinic receptors (Brandon et al., 2002), suggesting substantial convergence of signaling.
Although staurosporine, bisindolylmaleimide I, and IP19-36 inhibited the α7 nAChR-mediated modulation of GABAA receptor-mediated currents to a similar extent, none of them fully blocked the effect, suggesting that another pathway may also contribute. Neither the tyrosine kinase blocker genistein nor the calcineurin blocker FK506 inhibited the action of ACh. A full characterization of the pathway leading from α7 nAChRs to GABAA receptors and of the role of Ca2+ stores and other GABA receptor-interacting proteins and adaptors (Luscher and Keller, 2004) is beyond the scope of this study.
The decrease in eIPSCs by electrical stimulation of cholinergic axons seen here was relatively small (15–25%) relative to the effect of exogenous agonists (up to 50% depression). However, the heterosynaptic depression of eIPSCs measured here underestimated the dynamic range available to this form of modulation because the stimulating electrode was positioned in the slice relatively far from the recorded cell to minimize the activation of monosynaptically coupled interneurons. Because many cholinergic afferents are likely to have been cut during tissue slicing, this probably compromised the ability to induce ACh release from all local varicosities.
It is, of course, surprising that α7 nAChRs should simultaneously evoke both a presynaptic increase in transmitter release and a postsynaptic downregulation of GABAA receptors, because the two effects would be expected to oppose one another. However, it is possible that the two are dissociable by parameters that we have not tested here, for instance in their temporal profiles or in their interaction with other influences on transmitter release and receptor activation.
The hippocampal GABAergic interneuron network is spontaneously active even when glutamatergic transmission is blocked and performs several functions, including rhythm setting (Cobb et al., 1995; Whittington et al., 1995). GABAergic transmission among interneurons is highly sensitive to several neurotransmitters acting via presynaptic and axonal receptors (Semyanov and Kullmann, 2000, 2001; Cossart et al., 2001), suggesting that modulation of this form of communication is important for the normal function of the hippocampus. Several of these modulatory phenomena specifically affect GABAergic transmission to interneurons, as opposed to pyramidal neurons (for review, see Semyanov, 2003). The modulation of postsynaptic GABAA receptors revealed in the present study also obeys this principle and is consistent with the higher density of α7 nAChRs in interneurons than in pyramidal cells. Because most cholinergic axons arise from extrinsic structures rather than intrinsic ACh-releasing neurons (Frotscher et al., 1986) the phenomenon reported here provides a possible novel mechanism for an afferent projection to the hippocampus to affect synaptic transmission among interneurons. The evidence implicating α7 nAChRs in cognition and schizophrenia further argues that modulation of GABAA receptors by ACh release plays an important role in the information-processing functions of the hippocampus.
During revision of this paper, Zhang and Berg (2007) reported findings that are broadly in agreement with our study.
Footnotes
-
This work was supported by the Medical Research Council. We are indebted to R. L. Papke for the gift of 4OH-GTS21, to R. Fabian-Fine, R. L. Papke, and C. J. Frazier for early discussions on the project, and to D. A. Rusakov and members of the laboratory for comments on this manuscript. Some experiments were performed at the Biozentrum, and we are very grateful to A. Lüthi for providing the experimental facilities. We are also grateful to P. H. Kullmann for help implementing the dynamic current-clamp method.
- Correspondence should be addressed to Dimitri M. Kullmann, Institute of Neurology, Queen Square, London WC1N 3BG, UK. d.kullmann{at}ion.ucl.ac.uk





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}