

Abstract
Ca2+ influx through voltage-gated Ca2+ channels (VGCCs) into nerve terminals triggers vesicular fusion and neurotransmitter release. However, it is unknown whether the coupling between VGCCs and synaptic vesicles (SVs) is developmentally regulated. By paired patch-clamp recordings from the mouse calyx of Held synapse, we show here that injection of a Ca2+ buffer with slow binding kinetics (EGTA; 10 mm) potently attenuated transmitter release in young terminals [postnatal day 8 (P8)-P12] but produced little effect in older ones (P16-P18), suggesting that SVs in young synapses are loosely coupled to VGCCs, but the coupling tightens spatially during maturation. Using voltage paradigms that specifically recruit different numbers of VGCCs without changing the driving force for Ca2+, we found that the Ca2+ cooperativity (m), estimated from graded presynaptic Ca2+ currents and transmitter release, was much higher in P8-P12 synapses (m = 4.8-5.5) than that in P16-P18 synapses (m = 2.8-3.0; 1 mm [Ca2+]o), implying that the number of VGCCs or Ca2+ domains required for release of single SVs decreases with maturation. The m value remained significantly different between two age groups at 35°C or in 2 mm [Ca2+]o and was independent of postsynaptic receptor desensitization. We demonstrated that release from P8-P12 terminals involved both N- and P/Q-type VGCCs, but P/Q-type-associated release sites specifically displayed low m values. These results suggest a developmental transformation of the release modality from “microdomain,” involving cooperative action of many loosely coupled N- and P/Q-type VGCCs, to “nanodomain,” in which opening of fewer tightly coupled P/Q-type VGCCs effectively induce a fusion event. Spatial tightening improves the release efficiency and is likely a critical step for the development of high-fidelity neurotransmission in this and other central synapses.
- auditory
- glutamate receptor
- synaptic plasticity
- calcium channels
- development
- synaptic vesicle release
Introduction
In developing central synapses, synaptic modifications likely take place concurrently in both presynaptic terminals and postsynaptic neurons (Li and Sheng, 2003). Although remarkable progress has been made in understanding the organization of postsynaptic receptors and their scaffolding structures (Li and Sheng, 2003; McGee and Bredt, 2003), little is known about changes in the spatial coupling of voltage-gated Ca2+ channels (VGCCs) to vesicular release of neurotransmitter in the nerve terminal. Inaccessibility to tiny nerve terminals of typical central synapses has primarily precluded direct electrophysiological analysis of these two most critical elements engaged in transmitter release. As a result of the large size of presynaptic calyces, the giant calyx of Held synapse in the auditory brainstem permits simultaneous recordings from both presynaptic and postsynaptic elements (Borst et al., 1995; Takahashi et al., 1996), providing an excellent model for studying the mechanisms of fast synaptic transmission. This synapse is an axosomatic glutamatergic synapse critically involved in the sound localization circuit, in which the temporal fidelity of synaptic transmission, even at high frequencies (up to several hundred hertz), must be preserved for the auditory brain to compute interaural timing differences and ultimately determine the audible source in space (Trussell, 1999). Within the first two postnatal weeks, profound adaptations in morphological structure and biophysical properties of a variety of ion channels converge to facilitate the capability of transmission at high rates across this synapse (von Gersdorff and Borst, 2002). Although presynaptic release is one of the most important elements in determining synaptic efficacy, it remains primarily unknown how the calyceal terminal develops its ability to precisely control quantal output during this critical period of development.
The relationship between Ca2+ influx and quantal output (Q) at this and other synapses can be approximated by the following equation: Q ∝ [Ca2+]m, where the parameter m is defined as Ca2+ cooperativity. Historically, m, or Hill coefficient, has been interpreted as the minimal number of Ca2+ ions that must cooperatively bind to the sensor of the release machinery to trigger a fusion event (Dodge and Rahamimoff, 1967). Numerous studies in a variety of synapses have shown that the m value usually falls in the 3-4 range, meaning that slight alterations in Ca2+ entry into the terminal can profoundly affect synaptic strength as a result of the nonlinearity defined by m. Depending on experimental conditions, the m value derived from analysis of the input-output relationship of any given synapse provides specific information about the coupling of Ca2+ to vesicular release. For example, the m value obtained by pairing photolysis uncaging Ca2+ within the terminal itself and measurement of transmitter release most likely reveals the “molecular” cooperativity at the sensor, because this approach bypasses VGCCs and directly targets the binding sites for Ca2+ (Bollmann et al., 2000; Schneggenburger and Neher, 2000). If the input-output relationship is based on changes in the number of VGCCs engaged in triggering release, the m value is then an indirect readout of the spatial interaction between synaptic vesicles (SVs) and Ca2+ ions near the inner mouth of open channels (i.e., Ca2+ domain) (Borst and Sakmann, 1999; Gentile and Stanley, 2004) and hence may be referred as the “Ca2+ channel/domain” cooperativity. Intuitively, one can envision that when VGCCs become spatially closer to a SV, the minimal number of Ca2+ domains required to elevate the intracellular Ca2+ to the fusion threshold declines. Therefore, Ca2+ cooperativity has distinct conceptual meanings in mirroring the coupling nature of Ca2+ and vesicular release under different experimental circumstances.
To probe developmental plasticity in transmitter release, we have examined the spatial coupling properties of VGCCs and synaptic vesicles at the calyx of Held synapse using brainstem slices taken from postnatal mice before and after the onset of hearing [i.e., postnatal day 11 (P11)/P12]. By loading Ca2+ buffers with distinctive binding kinetics into calyces and by measuring Ca2+ channel/domain cooperativity with different voltage paradigms, in the absence and presence of subtype-specific toxins for VGCCs, we found that the coupling between VGCCs and synaptic vesicles tightens as maturation progresses. Such changes lead to a switch in release modality from “microdomain,” involving the cooperative action of many loosely coupled N- and P/Q-type VGCCs, to “nanodomain,” in which opening of fewer tightly coupled P/Q-type VGCCs is sufficient to induce a fusion event.
Materials and Methods
Slice preparation. Mice were housed in the facility certified by the Canadian Council of Animal Care and used for this study according to a protocol approved by the Hospital for Sick Children Animal Care Committee. Brainstem slices were prepared from P8-P18 mice (CD1/C57 black) as described previously (Forsythe and Barnes-Davies, 1993). After decapitation with a small guillotine, brains were immediately immersed into semifrozen artificial CSF (aCSF) containing the following (in mm): 125 NaCl, 2.5 KCl, 10 glucose, 1.25 NaH2PO4, 2 Na-pyruvate, 2 myo-inositol, 0.5 ascorbic acid, 26 NaHCO3, 1 MgCl2, and 2 CaCl2 at a pH of 7.3 when oxygenated (95% O2 and 5% CO2), followed by rapid dissection. Transverse slices of the auditory brainstem containing the medial nucleus of the trapezoid body were cut at a thickness of 150-300 μm using a microtome (VT100S; Leica, Nussloch, Germany) followed by incubation at 37°C for 1 h before experimentation. For paired voltage-clamp recordings, thin slices (150-200 μm) were prepared slightly angled (∼15° tilt away from the coronal plane) during slicing to minimize presynaptic axon length and reduce voltage-clamp errors. Most experiments were performed at room temperature (20-22°C) except for one set performed at 35°C (see Fig. 5) using an inline heater with feedback thermistor (TC-324B; Warner Instruments, Hamden, CT).

Comparison of Ca2+ cooperativity independent of gating kinetics of VGCCs and Ca2+ driving force. A, Example recordings of presynaptic Ca2+ currents (Pre-ICa) in response to voltage steps with increasing plateau duration (top; -70 to +60 mV; rise and fall time, 0.2 ms; plateau duration, from 0 to 0.5 ms in 0.1 ms increments) and EPSCs from a P10 (middle) and a P17 (bottom) synapse in 1 mm [Ca2+]o. B, Area integral of Pre-ICa and EPSCs from recordings in A are normalized (norm) and plotted on a log-log scale. Solid lines are linear regressions of paired data for two synapses and m values given. C, A summary plot of cooperativity m values from two age groups under four given experimental conditions (insets). Single or double asterisks in C indicate statistical significance (p < 0.05) between the P8-P12 and P16-P18 groups or within the same age group. D, The 10-90% rise and decay time of the maximal Pre-ICa evoked by APD are summarized for two age groups of synapses. Error bars represent SE.
Electrophysiology. All recordings were made with a dual-channel amplifier (MultiClamp 700A; Axon Instruments, Union City, CA). aCSF was supplemented with bicuculline (10 μm) and strychnine (1 μm) to block inhibitory inputs during recording. For recording of presynaptic Ca2+ currents, tetrodotoxin (0.5 μm), tetraethylammonium (TEA; 10 mm), and 4-aminopyridine (0.3 mm) were added to block Na+ and K+ channels, respectively, while [Ca2+]o was adjusted to 1 or 2 mm. For experiments with ω-agatoxin-IVA and ω-conotoxin-GVIA, slices were first perfused with aCSF containing cytochrome c (500 μg/ml) before the desirable amount of either toxin was added (Iwasaki and Takahashi, 1998; Wu et al., 1999). Patch electrodes typically had resistances of 4-6 and 2.5-3 MΩ for presynaptic and postsynaptic recordings, respectively. For paired voltage-clamp recordings, presynaptic and postsynaptic series resistances were 6-15 MΩ (<10 MΩ in the majority of recordings) and 4-10 MΩ, respectively, and compensated to 90% with cells showing higher resistances being omitted from analysis. Intracellular recording solution for presynaptic Ca2+ currents contained the following (in mm): 110 CsCl2, 40 HEPES, 0.5 or 10 EGTA, 1 MgCl2, 2 ATP, 0.5 GTP, 12 phosphocreatine, 20 TEA, 3 K-glutamate, pH adjusted to 7.3 with CsOH. Intracellular solution for postsynaptic recordings contained the following (in mm): 97.5 K-gluconate, 32.5 CsCl, 5 EGTA, 10 HEPES, 1 MgCl2, 30 TEA, and 3 lidocaine N-ethyl bromide, pH 7.2. The holding potential was -70 and -60 mV for presynaptic terminals and postsynaptic neurons, respectively. Presynaptic Ca2+ currents were evoked by the various voltage command protocols indicated in the text, and leak subtraction was done with the on-line P/4 protocol. For experiments in which real action potentials (APs) were used as presynaptic voltage-command waveforms (see Fig. 1), we first recorded APs from calyces, at a sampling rate of 50 kHz and a filtering frequency of 4 kHz, in current-clamp configuration by stimulating afferent axon fibers using a bipolar platinum electrode. After manually removing stimulation artifacts preceding the APs, the digitized values were used as voltage-command templates and fed back into amplifier as stimulation files (Axon Text File) through pClamp 8 software (Axon Instruments) at the same frequency as their acquisition (50 kHz). For action potential-like (APL; -70 to +60 mV steps; 100 μs incremental increases in repolarization time) and action potential-duration (APD; -70 to +60 mV steps; 100 μs incremental increases in plateau time) paradigms, on- and off-time of command voltage were set at 200 μs that resembled rise and repolarization time courses of action potentials and also minimized artifacts of leak subtraction. For presynaptic current-clamp recordings, pipettes were filled with a solution containing the following (in mm): 97.5 K-gluconate, 32.5 KCl, 0.5 or 10 EGTA or 1 BAPTA, 40 HEPES, 1 MgCl2, and 3 K-glutamate, pH 7.3. Reagents were from Sigma (St. Louis, MO), Tocris Cookson (Bristol, UK), and Alomone Labs (Jerusalem, Israel).

Profile of Ca2+-dependent transmitter release in response to action potentials recorded from the developing calyx of Held. A, Action potentials recorded from young and older synapses. B, C, Examples of presynaptic Ca2+ currents (Pre-ICa) and EPSCs from postsynaptic neurons (Post-EPSC) simultaneously recorded from P11 (B) and P17 (C) synapses in response to the same set of voltage command waveforms in A. Note that APM evoked ICa but not release from young synapse (B, arrow) and that results in B and C were all obtained in 1 mm [Ca2+]o. D, Pooled Ca2+ current areas for both young and older test groups in response to both API (open bars) and APM (filled bars) waveforms. Abs, Absolute. Error bars represent SE.
Data analysis. Data were acquired on-line, filtered at 4 kHz, digitized at 50 kHz, and analyzed off-line using the pClamp 8 software package and Excel 2000 (Microsoft, Redmond, WA). For cooperativity analysis, peak amplitudes or area integrals for both Ca2+ currents and EPSCs were detected using analysis windows that began at the onset of Ca2+ currents and EPSCs and ended at the points that contain entire area of Ca2+ tail current and the area up to the 10% decay point of EPSC peak amplitude. Current peaks and integrals were normalized to the maximal current (100%). The logarithms of peak or area values were then plotted against each other on linear axes and normalized to their respective maximal currents within its linear range according to the following equation using either current peaks or integrals: log(ICa/max ICa × 100) versus log(IEPSC/max IEPSC × 100). Least-squares linear regression was performed using Excel 2000 to obtain the slope value defined as m, the cooperativity. The m values were virtually identical when amplitude or area was chosen for analysis, but only results based on area integrals are presented. Because only a subset of data situated in the initial linear range of the log[ICa] versus log[IEPSC] plot (i.e., low input and output) was used to determine this parameter for each synapse, postsynaptic receptor saturation unlikely played a significant role in m. The amplitude of EPSCs included for analysis were usually well below the maximum of the dynamic range. Statistical tests of significance were two-tailed, unpaired Student's t tests assuming unequal variances with a p value cutoff of <0.05. Data were expressed as the mean ± SE from a population of synapses (n).
Results
Developmental difference in action potential-evoked release of transmitter
The quantal output of any given synapse is critically dependent on the shape and size of presynaptic APs, which ultimately determine the amount of Ca2+ influx and transmitter release (Llinas et al., 1982; Augustine, 1990; Sabatini and Regehr, 1997; Borst and Sakmann, 1999). To examine the effect of AP waveform on Ca2+ currents and transmitter release in developing calyx of Held synapses, we first made current-clamp recordings of APs from immature (P8-P12) and nearly mature (P16-P18) calyces in response to axonal stimulation (Fig. 1A). We confirmed that the AP waveform of P8-P12 calyces showed slower rise and repolarization times as well as wider width than that of P16-P18 calyces, as reported previously for the developing calyx of Held synapse in rat (Taschenberger and von Gersdorff, 2000; von Gersdorff and Borst, 2002). Subsequently, we used these waveforms as command templates to perform voltage-clamp recordings of presynaptic Ca2+ currents (see Materials and Methods) paired simultaneously with recordings of EPSCs in 1 mm extracellular Ca2+, which is near the physiological concentration of Ca2+ in the brain fluid (Jones and Keep, 1988). This condition also helped improve the quality of the voltage clamp by reducing the amplitude of Ca2+ currents and EPSCs and minimized the desensitization of postsynaptic glutamate receptors. When alternating AP waveforms from young (API; amplitude, 110 mV; half width, 0.6 ms) and older (APM; amplitude, 110 mV; half width, 0.3 ms) calyces were given to voltage-clamped calyces, we found that Ca2+ currents were effectively evoked by these two protocols in two populations of synapses (Fig. 1B, C). The total charge integral of Ca2+ current evoked by either waveform was not different between the two age groups (APM, 193 ± 24 vs 230 ± 41 pA·ms; API, 549 ± 18 vs 555 ± 52 pA·ms for P8-P12 and P16-P18 group, respectively) (Fig. 1D). Surprisingly, P8-P12 calyces usually failed to release glutamate in response to APM (no detectable EPSCs in 13 of 15 calyces) (Fig. 1B, right), whereas P16-P18 calyces (eight of eight calyces) released consistently with either voltage paradigm, of which API produced greater EPSC than APM, as expected (Fig. 1C). These observations raised the possibility that the coupling of VGCCs to vesicular release is fundamentally different in the two age groups of synapses.
Age-dependent reduction in EGTA-induced attenuation of transmitter release
It has been shown that many VGCCs (in 2 mm [Ca2+]o) are required to trigger release of single synaptic vesicles in young calyces (Borst and Sakmann, 1996). In line with this fact, our observation that a substantial Ca2+ influx (in 1 mm [Ca2+]o) failed to evoke vesicular release in P8-P12 calyces implies that many of the VGCCs may not be closely coupled to individual synaptic vesicles at this developmental stage. However, given the increased release efficacy in P16-P18 synapses, we postulate that presynaptic coupling may tighten during maturation to account for this difference between the two groups of synapses. To test this, we first performed experiments to compare the effectiveness of Ca2+ chelators, EGTA and BAPTA, on reducing quantal output. Despite similar equilibrium dissociation constants, these two buffers exhibit very different forward-rate constants (∼160-fold difference) for binding Ca2+ ions, allowing some inference to be made, based on their differing effectiveness in attenuating synaptic strength, with respect to the distance between VGCCs and vesicles in nerve terminals (Adler et al., 1991; Borst et al., 1995; Naraghi and Neher, 1997; Meinrenken et al., 2002). The rationale of this approach was that if VGCCs were tightly coupled to vesicles, slow buffers such as EGTA should not be able to intercept Ca2+ ions before they reach the Ca2+ sensor of the release machinery, therefore minimally altering synaptic strength. However, EGTA would effectively attenuate release if VGCCs were physically distant from synaptic vesicles, providing sufficient time for EGTA to bind Ca2+ ions in transit. In contrast, fast buffers such as BAPTA should be able to intercept Ca2+ ions and decrease synaptic strength independent of the tightness of VGCCs and synaptic vesicle coupling. For this set of experiments, we sealed a patch electrode containing EGTA (10 mm) or BAPTA (1 mm) onto the presynaptic calyx and collected control EPSCs evoked through axonal stimulation (Fig. 2Ai). The concentrations of two buffers chosen for these experiments were based on previous experimental results and theoretical simulation (Borst et al., 1995; Borst and Sakmann, 1996; Naraghi and Neher, 1997; Meinrenken et al., 2002). After EPSC baseline measurements, breakthrough of the presynaptic terminal membrane and current-clamp recording mode were sequentially established (Fig. 2Aii). After ∼2-3 min allowing diffusion and equilibration of the Ca2+ buffer within the calyx without afferent stimulation, recording of EPSCs were then resumed (Fig. 2Aiii). When 10 mm EGTA was diffused into presynaptic calyces from developing synapses across a P10-P18 age span, we found a strong age dependence in the effectiveness of EGTA on reducing quantal output, being very robust at P10 (∼70%) to marginal at P18 (∼20%) (mean, 57 ± 4% for P10-P12; 22 ± 4% for P16-P18) (Fig. 2B, D). On the contrary, loading of BAPTA (1 mm) did not show such a marked age dependence with 43 ± 8 and 43 ± 5% reduction for P10-P12 and P16-P18 calyces, respectively (Fig. 2D). The degrees of attenuation by these buffers are consistent with those reported previously for young synapses in the rat (P8-P10) (Borst et al., 1995; Borst and Sakmann, 1996). However, our data reveal a strong correlation between the extent of EGTA-induced attenuation and the developmental stage, suggesting that the physical distance between VGCCs and synaptic vesicles indeed tightens with development.

Age-dependent changes in spatial coupling between VGCCs and synaptic vesicles. A, Paired recording configurations for Ca2+ buffer injections. Postsynaptic whole-cell voltage-clamp recordings coupled with: i, presynaptic cell-attached mode for baseline measurements; ii, break-in of presynaptic membrane and buffer infusion in voltage-clamp mode (2-3 min); slight positive pressure was given to ensure thorough dialysis of buffers into the nerve terminal while whole-cell configuration were monitored in voltage-clamp mode; and iii, presynaptic current-clamp mode at its resting potential (approximately -70 mV). EPSCs were evoked through stimulation of the presynaptic axon fiber bundles with a bipolar platinum electrode placed adjacent to the midline of the slice, and presynaptic APs were monitored. APs usually had an overshoot above +10 mV without series resistance and capacitance compensation, and their waveform remain unchanged over the recorded time (data not shown). Stimulation voltage was set 20-30% above threshold. Ca2+ chelators (BAPTA or EGTA) were supplemented into the K+-based intracellular solution and dialysed directly into calyces through the patch pipettes. MNTB, Medial nucleus of the trapezoid body. B, The amplitudes of EPSCs before and after 10 mm EGTA injections were plotted against time for a P12 (top) and a P18 (bottom) synapse, respectively. Raw traces (insets) with (black line) or without (blue line) EGTA are superimposed for comparison. C, The extent of EGTA-induced block was plotted against the age of synapses. The block percentage was calculated as the ratio between the mean amplitude of EPSCs after EGTA injections/the mean amplitude of baseline EPSCs (100%) over the duration shown. The solid line represents least-squares linear regression of all data. D, A summary plot comparing the extent of block induced by EGTA (10 mm) and BAPTA (1 mm) for P10-P12 and P16-P18 synapses. The asterisk indicates statistical significance (p < 0.05). Error bars represent SE.
Distinctive Ca2+ cooperativity in P8-P12 and P16-P18 synapses
Under our experimental condition, the relationship between Ca2+ currents and EPSC can be simply described by a power function in the form of IEPSC α [ICa]m (where m denotes Ca2+ cooperativity and IEPSC as a readout of transmitter release). It is generally accepted that the release of synaptic vesicles often requires the cooperative action of many VGCCs by forming overlapping Ca2+ domains (Luebke et al., 1993; Takahashi and Momiyama, 1993; Wheeler et al., 1994; Wu and Saggau, 1994; Mintz et al., 1995; Iwasaki and Takahashi, 1998). If spatial coupling of VGCCs and synaptic vesicles tightens, as suggested by the EGTA loading experiments, we would predict that the Ca2+ cooperativity decreases as the release of single vesicles becomes less dependent on the cooperative action of VGCCs. To test this prediction, we compared the effect of AP waveform on Ca2+ currents and quantal output and measured the Ca2+ cooperativity in P8-P12 and P16-P18 synapses. To mimic the changes in AP waveform that takes place in the developing calyx of Held synapse, we gradually increased the width of the AP-like waveform (-70 to +60 mV; 100 μs increments in repolarization time) (Fig. 3A, top). It was thought that such changes in spike width would recruit an increasing that the amplitudes of both Ca2+ currents and EPSCs increased as the width of APL was increased. We found that the minimal Ca2+ current at which we could detect quantal events was significantly higher in P8-P12 synapses than that in P16-P18 ones (P8-P12, 470 ± 150 pA; P16-P18, 230 ± 50 pA; p < 0.008) (Fig. 3B), suggesting different Ca2+ current thresholds for triggering release in these two populations of synapses. This result is consistent with our observation that P8-P12 terminals failed to release transmitter in response to APM (Fig. 1), likely because of the fact that the narrow APM waveform usually generated Ca2+ currents that were subthreshold for triggering release from P8-P12 terminals but suprathreshold in P16-P18 calyces. When the normalized area integrals of EPSCs were plotted against that of Ca2+ currents on a log-log scale, we determined the Ca2+ cooperativity from the slope of linear fits to these input-output relationships (see Materials and Methods) (Fig. 3C). We found that the m value for P8-P12 synapses was significantly higher than that for P16-P18 synapses, being 4.8 ± 0.3 and 2.6 ± 0.5, respectively (p < 0.005) (Fig. 3C, D). When [Ca2+]o was raised from 1 to 2 mm, we observed a decrease in m values for both age groups, but a highly significant difference in this parameter was observed in P8-P12 synapses (from 4.8 ± 0.3 to 2.8 ± 0.3; p < 0.0005) (Fig. 3D). Our interpretation of these observations is that an increase in the driving force for Ca2+, through individual open channels, reduces the total number of VGCCs or Ca2+ domains required for a fusion event, leading to a decrease in m. Hence, many VGCCs are likely required to trigger release of single vesicles in young synapses, whereas tighter coupling in older synapses reduces the number of VGCCs or Ca2+ domains required for a release event.

Measurement of Ca2+ cooperativity with action potential-like waveforms. A, Examples contrasting two paired recordings of presynaptic Ca2+ currents (Pre-ICa) and EPSCs from postsynaptic neurons (Post-EPSC) from P9 (middle) and P18 synapses (bottom) in response to pseudo-action potential-like voltage ramps (-70 to + 60 mV; rise time, 0.2 ms; falling phase, from 0.2 to 0.6 ms with 0.1 ms increments). B, The mean amplitude of threshold Pre-ICa at which quantal release events were detected in P8-P12 and P16-P18 synapses. Min., Minimum. C, Area integral of Pre-ICa and EPSCs from two recordings in A are normalized (norm) and plotted on a log-log scale. Solid lines are linear regression of paired data for two synapses and m values given. Note that results from A-C were all obtained in 1 mm [Ca2+]o. D, A summary plot of cooperativity m values in 1 and 2 mm [Ca2+]o from two populations of synapses. Single or double asterisks in B and D indicate statistical significance (p < 0.05) between P8-P12 and P16-P18 groups or within the same age group, respectively. Error bars represent SE.
Because the pseudo-APL waveforms used above had different repolarization phases, it is conceivable that not only the number of VGCCs being recruited but other confounding factors may also contribute to the different efficacy of vesicular release. For example, the density and voltage dependence of these channels in P8-P12 and P16-P18 calyces may also differ. To address these issues, we next examined the properties of VGCCs with a series of command voltage-steps (10 ms steps from -70 to +60 mV in 10 mV increments). We found that the two sets of current and voltage relationships were virtually identical, in that Ca2+ currents from both age groups activated at approximately -40 mV and reversed around +40 mV with an identical maximal current at -10 mV (Fig. 4A, B). These results suggest that both the total number and voltage dependence of VGCCs are very similar for both age groups of calyces. In line with this, the maximal tail currents evoked during this protocol were not statistically different in amplitude (1.5 ± 0.1 nA for P8-P12; 1.2 ± 0.2 nA for P16-P18; p > 0.1) and occurred at approximately +20 mV for both age groups.

Voltage dependence of VGCCs in P8-P12 and P16-P18 calyces. A, Example recordings of presynaptic Ca2+ currents (Pre-ICa) in response to voltage steps (10 ms) from -70 to +60 mV in 10 mV increments (top) from a P11 (middle) and a P17 (bottom) synapse in 1 mm [Ca2+]o. B, Current-voltage relationships of Pre-ICa from P8-P12 (open diamonds; n = 8) and P16-P18 calyces (filled diamonds; n = 7). Error bars represent SE.
Knowing that VGCCs in both groups of terminals have similar properties, we used another voltage-clamp protocol (APD; -70 to +60 mV; 100 μs increments in plateau time) (Fig. 4A, top) to further explore the presynaptic coupling between VGCCs and vesicular release. Incremental increases in plateau duration were used to recruit an increasing number of VGCCs while maintaining the driving force for Ca2+ as well as the on and off kinetics of the command voltage steps. Because voltage steps to +60 mV in APD presumably maximized the open probability of VGCCs, while the driving force for Ca2+ remains constant for all open channels (Augustine et al., 1987; Roberts, 1994; Borst and Sakmann, 1998, 1999; Gentile and Stanley, 2004), the only variable at any given plateau duration would be the number of VGCCs engaged in mediating release of synaptic vesicles. Using these APD waveforms, we compared the relationships between ICa and IEPSC and determined m values for P8-P12 and P16-P18 synapses in 1 and 2 mm [Ca2+]o (Fig. 5A, B). We again found that the m values from P8-P12 synapses were significantly higher than those of P16-P18 ones (5.5 ± 0.5 for P8-P12, 3.0 ± 0.4 for P16-P18 synapses in 1 mm [Ca2+]o; p < 0.0004) and that raising [Ca2+]o from 1 to 2 mm led to a reduction in m values for both populations of synapses, producing values consistent with that reported previously under similar conditions (Borst and Sakmann, 1999) (3.5 ± 0.5 for P8-P12, 2.1 ± 0.2 for P16-P18 synapses in 2 mm [Ca2+]o; p < 0.02) (Fig. 5C). Moreover, we found no difference in the rise or decay time of Ca2+ currents evoked by the APD protocol between two populations of calyces (10-90% rise time, 0.18 ± 0.02 vs 0.15 ± 0.01 ms, p > 0.2; 10-90% decay time, 0.51 ± 0.05 vs 0.56 ± 0.04 ms, p > 0.5) (Fig. 5D), indicating that the difference in m is independent of gating kinetics of VGCCs.
The calyx of Held synapse undergoes dramatic morphological refinement over the first two postnatal weeks (Morest, 1968; von Gersdorff and Borst, 2002). The “spoon-shaped” calyx at early postnatal stages may slow glutamate removal, whereas older “finger-digit-like” terminals allow more rapid clearance of glutamate from the synaptic cleft. Because we use AMPA receptor (AMPAR) EPSC as a readout of transmitter release, different residence times of transmitter in P8-P12 and P16-P18 synapses may lead to different extents of AMPAR desensitization (Joshi and Wang, 2002; Taschenberger et al., 2002), potentially contributing to the difference in the m value between the two age groups. To test this, we repeated the experiments in 1 mm [Ca2+]o with the APD paradigm in the presence of cyclothiazide (CTZ; 50 μm), which blocks AMPAR desensitization. Despite application of CTZ, the age-dependent difference in m remained (5.5 ± 0.7 for P8-P12; 2.9 ± 0.1 for P16-P18 synapses; p < 0.01) (Fig. 5C), indicating the developmental difference in Ca2+ cooperativity is independent of postsynaptic receptor desensitization.
Because temperature may profoundly affect synaptic efficacy at this synapse (Taschenberger and von Gersdorff, 2000; von Gersdorff and Borst, 2002), we next measured the Ca2+ cooperativity in 1 mm [Ca2+]o at near-physiological temperature (35°C). We found that an increase in temperature reduced the m value significantly in young synapses but only slightly in older ones (Fig. 5C). However, a significant difference in the m value between two age groups remains (3.3 ± 0.1 for P8-P12, 2.5 ± 0.2 for P16-P18 synapses in 1 mm [Ca2+]o; p < 0.002). These results led us to suggest that raising temperature may lead to an expansion of individual Ca2+ domains, presumably as a result of increased channel conductance and Ca2+ diffusion rate. Such a manipulation reduces the number of Ca2+ channels required to trigger single fusion events in P8-P12 synapses, similar to the effect of raising [Ca2+]o. In contrast, temperature has little effect on P16-P18 synapses, because the tight coupling of VGCCs and synaptic vesicles minimizes the dependence on cooperative action of multiple Ca2+ domains, independent of increases in Ca2+ domain size and diffusion rate.
Collectively, the results acquired with the APD paradigm reinforce our interpretation of age-dependent decreases in the Ca2+ channel/domain cooperativity, based on the APL protocol. As such, the number of VGCCs or Ca2+ domains required for release of single synaptic vesicles decreases as the calyx of Held synapse matures.
The role of Ca2+ channel types in developmental transformation of the release modality
Previous studies have shown that multiple types of VGCCs (N, P/Q, and R type) are present in young calyces and cooperatively mediate transmitter release (Wu et al., 1998, 1999); however, only P/Q-type VGCCs exclusively mediate transmitter release in older ones (Iwasaki and Takahashi, 1998). Immunofluorescence staining of young calyces with subtype-specific antibodies further revealed that N- and R-type VGCCs appear to be physically more distant from release sites than P/Q type (Wu et al., 1999), raising the possibility that subtypes of VGCCs may be differentially involved in the developmental reorganization of channels and synaptic vesicles. To test this possibility, we measured Ca2+ cooperativity in the presence of toxins that specifically block different types of Ca2+ channels in young synapses (P8-P12). This age window covered the critical period of developmental transition in VGCCs engaged in triggering vesicular fusion, being from N-, R-, and P/Q-type channels together to P/Q type alone after P10. By dividing these immature synapses into two age subgroups (P8/9 vs P11/12), we found that blocking P/Q-type Ca2+ channels with ω-agatoxin-IVA (100 nm) did not change the m value (5.6 ± 0.4 for P8-P12 in 1 mm [Ca2+]o), despite a sharp reduction in quantal output (Fig. 6). However, when N-type Ca2+ channels are blocked with ω-conotoxin-GVIA (1 μm), we found a significant difference in m value within two subpopulations of young synapses, the P11/12 subgroup being significantly lower (3.8 ± 0.2; p < 0.03) in comparison with that of the control (no toxin) group (5.5 ± 0.5 for P8-P12), whereas the m value for P8/9 subgroup was unchanged (5.3 ± 0.5) (Fig. 6 B, C). These observations suggest that N-, P/Q-, and probably R-type Ca2+ channels are spatially intermingled in the active zone (AZ) in P8/9 and that release of single vesicles relies on the cooperative action of many channels, regardless of Ca2+ channel subtype. As maturation progresses, P/Q-type VGCCs predominate over N- and R-type VGCCs (Wu et al., 1999) and become more tightly associated with vesicles, capable of triggering release with a lower m value, even as early as P11/12, when N-type channels are blocked. It should be noted that previous studies have simultaneously measured changes in the volume-averaged Ca2+ concentration and transmitter release during application of a toxin that gave an estimate of the effectiveness (also termed as m or n) for the toxin-sensitive subtype of VGCCs to induce quantal release (Wu and Saggau, 1994; Mintz et al., 1995; Wu et al., 1999). In general, these values of m or n are high (3-4) when P/Q-type VGCCs are gradually blocked with ω-agatoxin-IVA and are low (1-2) when other subtypes are blocked in a similar manner. Such experiments reveal the nature of the cooperation between different subtypes of VGCCs in triggering release. Our m values, derived by APD paradigm after a complete blockade of one subtype VGCCs by a toxin, have a distinct meaning in that the cooperative domain interactions among the remaining type(s) of VGCCs are assayed.

The role of subtypes of VGCCs in regulating Ca2+ cooperativity P8-P12 synapses. A, Example recordings of presynaptic Ca2+ currents (Pre-ICa) in response to voltage steps with increasing plateau duration (top; -70 to +60 mV; rise and fall time, 0.2 ms; plateau duration, from 0 to 0.5 ms in 0.1 ms increments) and EPSCs from a P10 in ω-agatoxin-IVA (w/atx; 100 nm) (top) and a P8 synapse ω-conotoxin-GVIA (w/ctx; 1 μm) (bottom). B, A log-log plot of normalized area integral of Pre-ICa and EPSCs from four recordings including those two in A. Solid lines are linear regressions of paired data for each recording and m values given. C, A summary plot of cooperativity m values in control, ω-agatoxin-IVA group, and two subgroups (P8/9 vs P11/12) in ω-conotoxin-GVIA. The single asterisk indicates statistical significance (p < 0.05) between two subgroups of young synapses. All experiments were done in 1 mm [Ca2+]o. Note low yield of transmitter release after addition of either toxin. Error bars represent SE.
Mixed populations of loosely and tightly coupled synaptic vesicles in developing calyces
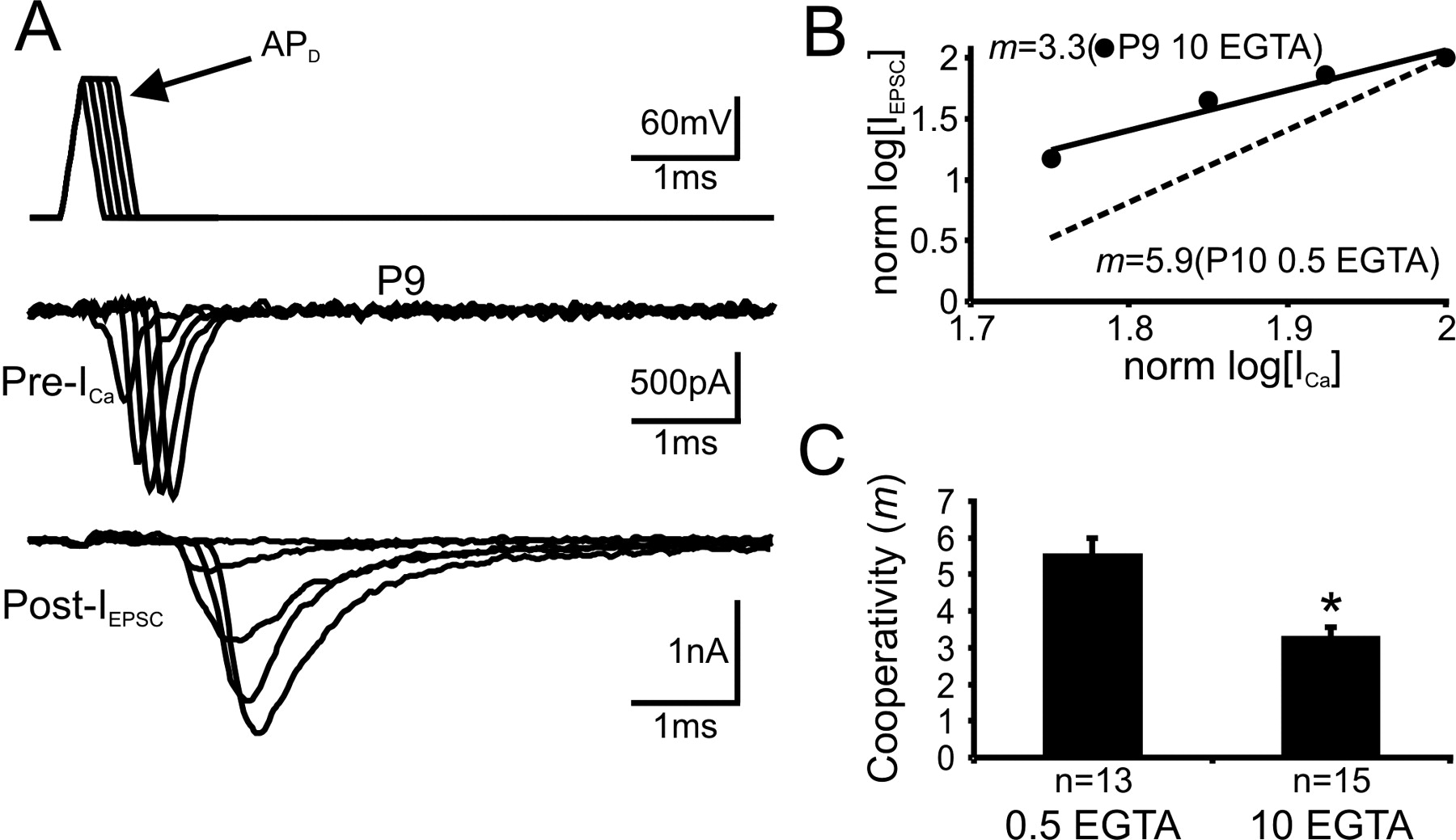
Our observations that EGTA (10 mm) did not completely attenuate transmitter release at any given age and that the extent of this attenuation was strongly correlated with synaptic maturity led us to postulate that in developing synapses, there are at least two populations of release-competent synaptic vesicles, one loosely coupled and the other tightly coupled to VGCCs. The relative proportion of the latter may be increasing with development, leading to a decrease in m. If the loosely coupled population of channel-vesicle units were functionally removed from young terminals, one would predict that the remaining population of tightly coupled vesicles be released with a similar m to that of older synapses. To test this prediction, we measured m from P8-P12 synapses after disabling loosely coupled vesicles through presynaptic loading of 10 mm EGTA (Fig. 7A, B). We found that quantal output was low under such conditions, but the m value (3.3 ± 0.3; 10 mm EGTA) decreased to that typical of P16-P18 synapses (3.0 ± 0.4; 0.5 mm EGTA) (Figs. 7B,C, 5C), in which tightly coupled vesicles presumably dominate (∼80%) as inferred from the reduction induced by 10 mm EGTA (Fig. 2C, D). This experiment appears to have similar end effects to that with application of ω-conotoxin-GVIA, suggesting, under both manipulations, release of transmitter becomes primarily dependent on tightly coupled, low-m units predominately containing VGCCs of the P/Q type. Hence, the release modality of young synapses, after loading of 10 mm EGTA or ω-conotoxin-GVIA (older than P11/12), can be functionally converted to that reminiscent of older synapses.

Transformation of release modality from microdomain to nanodomain coupling. A, An example recording of presynaptic Ca2+ currents (Pre-ICa) in response to APD paradigms (top; -70 to +60 mV; rise and fall time, 0.2 ms; plateau duration, from 0 to 0.5 ms in 0.1 ms increments) and EPSCs from a P9 synapse in 1 mm [Ca2+]o. Intracellular solution for presynaptic recordings contains 10 mm EGTA. B, Area integral of Pre-ICa and EPSCs from recordings in A are normalized (norm) and plotted on a log-log scale. The solid line represents the linear fit to the paired data set and m value given. Dashed line from the same experiment as in Figure 5B with low concentration of intracellular EGTA (0.5 mm) was plotted for comparison. C, A summary plot of cooperativity m values in 1 mm [Ca2+]o with 0.5 or 10 mm EGTA in presynaptic calyces from the P8-P12 synapses. The asterisk indicates statistical significance (p < 0.05) between two data sets. As a result of the low quantal output with high variance after 10 mm EGTA infusion into some of P8-P12 synapses, multiple experimental runs were taken and overlaid to derive a single m value. Error bars represent SE.
Discussion
The classical definition of Ca2+ cooperativity usually refers to the cooperative action of Ca2+ ions at the Ca2+ sensor of the release machinery (e.g., synaptotagmins), to which a minimum of three or four Ca2+ ions must bind before fusion takes place (Dodge and Rahamimoff, 1967). In this study, we measured Ca2+ cooperativity in P8-P12 and P16-P18 synapses using voltage paradigms, APD in particular, to specifically recruit different numbers of VGCCs and generate graded Ca2+ currents and quantal output. Our results imply that the m value is an index of the contribution of multiple Ca2+ channels or domains (i.e., Ca2+ channel/domain cooperativity) under this experimental condition. It is reasonable to extrapolate from channel recruitment experiments that a number of Ca2+ domains are needed to increase the likelihood of three or four Ca2+ ions binding to the sensor, when individual Ca2+ domains are small (in 1 mm [Ca2+]o) and distant from the vesicle (as in young calyces). Because the spatial distance between channel and vesicle shortens during maturation, the number of domains required for three or four Ca2+ ions to reach the sensor declines. The fact that an increase in domain size, by either raising [Ca2+]o or increasing temperature (Fig. 5), leads to a decrease in the m value further supports the interpretation that Ca2+ cooperativity in this study is likely related to the number of Ca2+ channels or domains.
Our results demonstrate that a majority of vesicles in young terminals are loosely coupled to VGCCs, showing high cooperativity values [i.e., m = 4.8 (APL) or 5.5 (APD) in 1 mm [Ca2+]o] and a high sensitivity to EGTA. Like other synapses (Luebke et al., 1993; Takahashi and Momiyama, 1993; Wheeler et al., 1994; Wu and Saggau, 1994; Mintz et al., 1995), the release of single vesicles at the young calyx of Held synapse depends on the cooperative action of overlapping Ca2+ domains from many channels (Borst and Sakmann, 1996, 1999; Meinrenken et al., 2002) and likely different subtypes of VGCCs such as N, R, and P/Q types (Iwasaki and Takahashi, 1998; Wu et al., 1999). R-type VGCCs mainly localized on the uninnervated side of the calyx, and therefore it is unlikely that they contribute significantly to phasic release when other two types are present (Wu et al., 1999). Our Ca2+ channel/domain cooperativity values are consistent with those from previous studies performed in the rat calyx of Held synapses at the same developmental stage (P8-P10) under similar experimental conditions (i.e., m values of ∼3-4 in 2 mm [Ca2+]o) (Borst and Sakmann, 1999). However, our m values in 1 mm [Ca2+]o (4.8-5.5) are higher than those obtained by photolysis uncaging of Ca2+ (4.2-4.4) (Bollmann et al., 2000; Schneggenburger and Neher, 2000), which may represent a theoretical ceiling for molecular cooperativity (Gentile and Stanley, 2004), because the latter measurements are independent of [Ca2+]o and the number of VGCCs. Perhaps different spatiotemporal profiles of Ca2+ ions released from the cage and inflow through VGCCs underlies this discrepancy. Alternatively, this theoretical ceiling may be an underestimate of true molecular cooperativity. In fact, five binding sites for Ca2+ ions on the sensor were used to fit the data in modeling based on photolysis uncaging experiments (Bollmann et al., 2000; Schneggenburger and Neher, 2000).
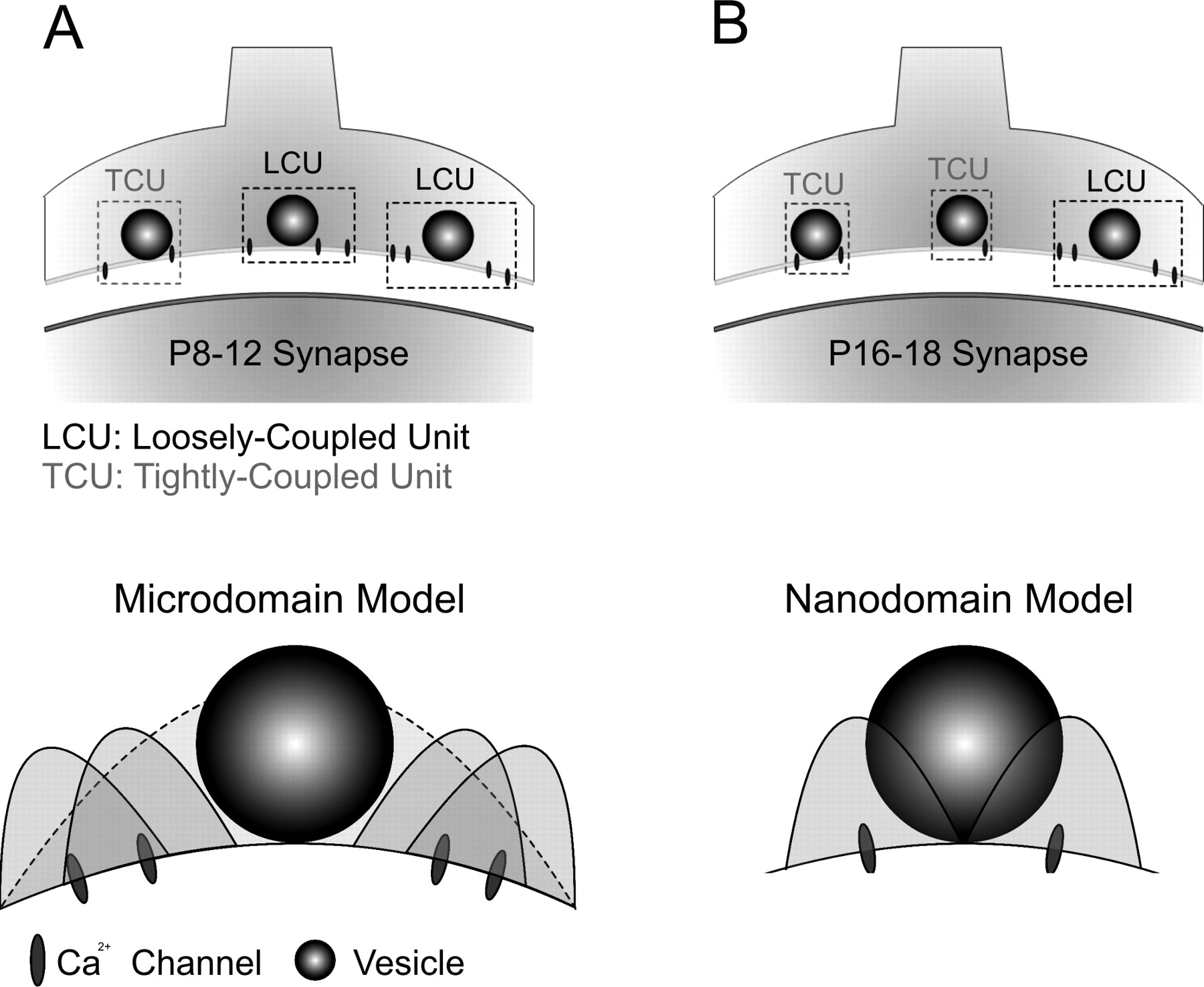
Surprisingly, we found that as the synapse matures, the physical distance between VGCCs and single vesicles appears to shorten, as reflected by a decline in the effectiveness of EGTA on attenuating release. The low m values found in older synapses (P16-P18; m = 2.1-2.6 in 2 mm [Ca2+]o) and the marked reduction in sensitivity to EGTA further suggest that the release modality between VGCCs and single vesicles shifts from primarily overlapping microdomain to nanodomain in which very few channels are required for release of a single vesicle (Fig. 8A,B), similar to those from the squid giant synapse, frog neuromuscular junction, and the chick calyx in the ciliary ganglion (Llinas et al., 1982; Yoshikami et al., 1989; Augustine, 1990; Stanley, 1993; Gentile and Stanley, 2004). However, we would emphasize that use of the terms microdomain and nanodomain in our study specifically refers to differences in the spatial distance between the source of Ca2+ entry and a SV but does not differentiate whether a nanodomain contains one Ca2+ channel or more than one Ca2+ channel. It is conceivable that Ca2+ domains from the simultaneous opening of two neighboring channels, in a cluster of VGCCs, may pool to form a single domain. Unlike other synapses that contain highly organized arrays of VGCCs and SVs, the distribution pattern of VGCCs on the release face of the calyceal terminal is not known at the calyx of Held synapse. Modeling studies suggest that VGCCs, when placed as a cluster or scattered, may produce entirely different profiles of Ca2+ transients and transmitter release (Roberts, 1994; Meinrenken et al., 2002). It is possible that the spatial distribution of VGCCs transforms from scattered in immature synapses to highly organized arrays in mature ones. As a consequence of such a developmental transformation, the relative portion of tightly coupled versus loosely coupled channel vesicle units increases (Fig. 8), so that the slow buffer EGTA becomes less effective in attenuating transmitter release as synapses mature.

Model diagrams illustrating developmental differences in the presynaptic arrangement of VGCCs and synaptic vesicles in young and older synapses. A, Most vesicles in young (left) synapses are loosely coupled to VGCCs to form larger functional units (LCU) than that of older synapses (right), in which a majority of vesicles are tightly coupled to VGCCs (TCU). B, Microdomain (left) and nanodomain (right) release modalities in young and older synapses. The former requires cooperative actions of multiple Ca2+ channels to trigger release of a single vesicle, whereas the latter implies that VGCCs in older synapses are physically close to synaptic vesicles, and activation of few VGCCs enables release of a single vesicle.
Our results do not exclude the possibility that other developmental changes, such as the expression level and makeup of Ca2+ sensor proteins for release (e.g., synaptotagmins), may increase the sensitivity of the release machinery to Ca2+ and also contribute to differences in release efficacy (Stewart et al., 2000; Sugita et al., 2001). Transmitter release in young calyces is already highly sensitive to intracellular Ca2+ (∼10 μm) (Bollmann et al., 2000; Schneggenburger and Neher, 2000). However, it is unknown whether there is a concurrent change in the sensitivity of the release machinery in the developing calyx of Held synapse. Direct measurement of the Ca2+ sensitivity in the future, through photolysis of caged Ca2+ compounds independent of VGCCs, may provide compelling answers to this question. The evidence in this study, and that from the literature, favors our view that developmental changes in spatial coupling is likely one of the main elements in determining the difference in Ca2+ cooperativity between young and older synapses. The strong correlation between EGTA-induced attenuation of transmitter release and maturity of synapses suggests a spatial/physical tightening of channels and vesicles. Pharmacological transformation of young synapses to older ones, in terms of Ca2+ cooperativity, through presynaptic loading of 10 mm EGTA (Fig. 7), reinforces this idea. Experiments with subtype-specific toxins suggest that P/Q-type channels appear to drive the development of the nanodomain modality (Fig. 6), implying developmental changes in Ca2+ cooperativity is likely a “channel phenomenon.” This is also in line with the notion that VGCCs physically interact with synaptic proteins (i.e., soluble N-ethylmaleimide-sensitive factor attachment protein receptors) in the active zone of nerve terminals, and these two elements can reciprocally regulate each other once in a complex (Augustine et al., 2003). Emerging evidence from other synapses demonstrates that developmental evolution of release modality involves a switch in the subtype of VGCCs, being from N type in young synapses to predominantly P/Q type in older synapses (Reid et al., 2003). In fact, N-type VGCCs fail to fully compensate for synaptic deficits, in P/Q-channel knock-out mice, in central synapses including the calyx of Held synapse (Jun et al., 1999; Inchauspe et al., 2004). This is in line with the view that the P/Q-type Ca2+ channels may occupy preferred “slots” for mediating vesicular fusion in the active zone (Cao et al., 2004).
In addition to the release modality transformation, it is also important to note that the morphological structure of the calyx of Held undergoes a significant transformation from spoon shape to finger digit-like (Morest, 1968; von Gersdorff and Borst, 2002). By P14, the morphological structure of calyces is considered to be fully mature. In the rat calyx of Held, the number of active zones doubles (from ∼300 to ∼600), whereas the size of each AZ decreases, during early postnatal stages (less than P14), as shown by analysis of presynaptic structures using electron microscopy (Taschenberger et al., 2002). However, without precise knowledge of the number of AZs, single-channel conductance of P/Q-type channels at physiological concentrations of [Ca2+]o and the spatial distribution patterns of VGCCs, it is perhaps premature to assign a specific number of channels to each AZ. Assuming a similar increase in the number of AZs at the mouse calyx of Held synapse during development, it is conceivable that each AZ contains fewer VGCCs in later developmental stages, given that the maximal Ca2+ current appears to be the same between the two age groups. In parallel with dramatic changes in morphology after the onset of sensory input (P11/12), similar total numbers of VGCCs spread among an increasing number of AZs may underlie the transformation to a nanodomain release modality in older synapses. Because the relationship between Ca2+ influx and quantal output (IEPSC) is highly nonlinear at this synapse (IEPSC α [ICa]m), small changes in either ICa, m, or both may have a profound effect on quantal output. At the calyx of Held synapse, developmental shortening of AP width would reduce the amount of Ca2+ influx, whereas transformation of the release modality from microdomain to nanodomain decreases the m value. These two effects would ultimately converge to reduce the release probability of the synapse, as has been reported previously (Iwasaki and Takahashi, 2001; Taschenberger et al., 2002).
The calyx of Held synapse, at maturity, is capable of phase-locking postsynaptic firing to presynaptic input at high rates, as a result of multiple adaptations (Wang and Kaczmarek, 1998; Trussell 1999; Joshi and Wang, 2002; von Gersdorff and Borst, 2002; Joshi et al., 2004). In line with previous evidence showing a developmental increase in the efficiency of exocytosis (Taschenberger et al., 2002), we suggest that tight coupling between VGCCs and vesicles may represent a critical adaptation that ensures a strong synaptic drive for high-fidelity neurotransmission with minimal jitter in synaptic delay. The low m value associated with nanodomain release modality may also constrain the effect of residual Ca2+ build-up on quantal output during repetitive synaptic activity and thus prevent depletion of synaptic vesicles. Hence, tightening the presynaptic coupling between VGCCs and single synaptic vesicles may have a profound impact on synaptic strength and fidelity during the critical period of development in this and other central synapses.
Footnotes
This work was supported by the Burroughs Wellcome Fund and EJLB Foundation and by an individual operating grant and “The Synapse” group grant from the Canadian Institutes of Health Research to L.-Y.W. L.-Y.W. currently holds the Canada Research Chair (Tier II). We thank Drs. Milton Charlton, Elise Stanley, Harold Atwood, and Mike Salter for valuable critiques and input. We thank Dr. Yimei Yang for assistance and other members of the Wang laboratory for discussions.
Correspondence should be addressed to Dr. L.-Y. Wang, Division of Neurology, The Hospital for Sick Children, 555 University Avenue, Toronto, Ontario, Canada M5G 1X8. E-mail: luyang.wang{at}utoronto.ca.
Copyright © 2005 Society for Neuroscience 0270-6474/05/254131-10$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}