

Abstract
Brain lesions containing filamentous and aggregated α-synuclein are hallmarks of neurodegenerative synucleinopathies. Oxidative stress has been implicated in the formation of these lesions. Using HEK 293 cells stably transfected with wild-type and mutant α-synuclein, we demonstrated that intracellular generation of nitrating agents results in the formation of α-synuclein aggregates. Cells were exposed simultaneously to nitric oxide- and superoxide-generating compounds, and the intracellular formation of peroxynitrite was demonstrated by monitoring the oxidation of dihydrorhodamine 123 and the nitration of α-synuclein. Light microscopy using antibodies against α-synuclein and electron microscopy revealed the presence of perinuclear aggregates under conditions in which peroxynitrite was generated but not when cells were exposed to nitric oxide- or superoxide-generating compounds separately. α-Synuclein aggregates were observed in 20–30% of cells expressing wild-type or A53T mutant α-synuclein and in 5% of cells expressing A30P mutant α-synuclein. No evidence of synuclein aggregation was observed in untransfected cells or cells expressing β-synuclein. In contrast, selective inhibition of the proteasome resulted in the formation of aggregates detected with antibodies to ubiquitin in the majority of the untransfected cells and cells expressing α-synuclein. However, α-synuclein did not colocalize with these aggregates, indicating that inhibition of the proteasome does not promote α-synuclein aggregation. In addition, proteasome inhibition did not alter the steady-state levels of α-synuclein, but addition of the lysosomotropic agent ammonium chloride significantly increased the amount of α-synuclein, indicating that lysosomes are involved in degradation of α-synuclein. Our data indicate that nitrative and oxidative insult may initiate pathogenesis of α-synuclein aggregates.
Abnormal accumulations of filamentous protein aggregates in neurons, glia, or the extracellular space are pathological hallmarks of many sporadic and hereditary neurodegenerative diseases (Clayton and George, 1998; Goedert et al., 1998; Duda et al., 2000a). Recently, α-synuclein (α-syn) was determined to be the major component of Lewy bodies (LBs) in sporadic Parkinson's disease (PD), dementia with LBs, and LB variant of Alzheimer's disease, as well as inclusions in multiple system atrophy and neurodegeneration with brain iron accumulation type 1 (Clayton and George, 1998; Goedert et al., 1998; Duda et al., 2000a). The biochemical and biophysical factors that induce aberrant α-syn aggregation are not clearly understood, but recent reports describing a PD-like phenotype in transgenic α-syn mice and flies, as well as rotenone-induced parkinsonism in rats and in vitrofibrillogenesis of α-syn, provide new insights into the pathogenesis of α-syn aggregation (Giasson et al., 1999; Betarbet et al., 2000;Conway et al., 2000; Feany and Bender, 2000; Masliah et al., 2000; Van der Putten et al., 2000). Collectively these studies have indicated that wild-type α-syn is capable of forming fibrils and potentially toxic aggregates.
Although α-syn polymerization has been characterized biophysically (Conway et al., 2000; Serpell et al., 2000), the intracellular conditions that favor the initiation and propagation of this process are not completely understood. One potential biochemical pathway that may lead to protein aggregation is the inhibition of proteolytic processes. Other studies not related to α-syn aggregation have shown that selective inhibition of the proteasome results in protein aggregation and formation of inclusion bodies (Johnston et al., 1998). Another biochemical pathway that may lead to protein aggregation is oxidative stress. Consistent with the oxidative stress hypothesis,in vitro oxidation and nitration of α-syn stabilize protein polymers by forming stable cross-linked α-syn aggregates (Souza et al., 2000). Similar evidence has been provided (Hashimoto et al., 1999) for a cytochrome c or hemin plus hydrogen peroxide-mediated α-syn aggregation. Moreover, immunohistochemical studies using novel monoclonal antibodies against nitrated α-syn revealed robust and abundant staining of numerous LBs, Lewy neurites, glial cell inclusions, and neuroaxonal spheroids in brains from diverse types of synucleinopathies (Giasson et al., 2000a). In addition to the specific nitration of α-syn, evidence for protein nitration in human neurodegenerative diseases as well as animal models of neurodegeneration has been provided (Smith et al., 1997; Ara et al., 1998; Good et al., 1998; Hensley et al., 1998; Liberatore et al., 1999;Duda et al., 2000b; Giasson et al., 2000a; Kowall et al., 2000). Oxidative stress in the form of relatively high concentrations of hydrogen peroxide and ferrous iron induced aggregation of α-syn in human neuroblastoma cells transfected with α-syn (Ostrerova-Golts et al., 2000). On the basis of these observations, we explored the possibility that exposure of cells to pathophysiologically reasonable fluxes of reactive oxygen and nitrogen species or selective inhibition of the proteasome may induce the formation of protein aggregates in HEK 293 cells transfected with wild-type or mutant human α-synuclein.
MATERIALS AND METHODS
Cell culture and exposure to nitric oxide and superoxide. The plasmids for the expression of α- and β-syn were constructed by inserting the human α- and β-syn cDNAs, respectively, into the mammalian expression vector pcDNA 3.1+ (Invitrogen, Carlsbad, CA). HEK 293 cells were obtained from the American Type Culture Collection (Vienna, VA) and cultured in 4.5 gm/l high-glucose DMEM supplemented with 10% FBS, 100 U/ml penicillin, 100 U/ml streptomycin, and 2 mml-glutamine. Cells were transfected with the respective plasmids using calcium phosphate precipitation buffered withN,N-bis(2-hydroxyethyl)-2-amino-ethanesulfonic acid (Chen and Okayama, 1997). The cells were replated on 10 cm dishes 1 d after transfection, and selection with 500 μg/ml geneticin (Life Technologies, Inc., Rockville, MD) was initiated 24 hr later. Individual stable clones were isolated with glass cylinders and detached from the dish with trypsin. Stable clones were replated and maintained in culture medium with geneticin. Clones expressing high levels of syn protein were screened by Western blot analysis (Fig.1).
Cells plated at a density 1 × 106per well in 35 mm tissue culture plates were exposed to the diazeniumdiolate nitric oxide donor 1-propanamine, 3-(2-hydroxy-2 nitroso-1-propylhydrazino) (PAPA/NO; Fitzhugh and Keefer, 2000; Gow et al., 2000). PAPA/NO is stable at alkaline pH and decays with first-order rate kinetics to release NO at physiological pH (Fitzhugh and Keefer, 2000). Stock solutions of PAPA/NO (Cayman Chemical, Ann Arbor, MI) were prepared in 0.01 n NaOH and stored in nitrogen-purged airtight bottles in the dark at −20°C. The concentration of the PAPA/NO working solution was determined by measuring the absorbance at 250 nm (ε250nm = 8050 m/sec) before use. The steady-state levels of intracellular superoxide were increased by the use of three different types of reagents: (1) the redox-active compounds 2,3-dimethoxy-1,4-naphthquinone (DMNQ) and 1,1′-dimethyl-4, 4′-bipyridinium dichloride (paraquat); DMNQ is a cell-permeable, nonalkylating, and nonthiol adduct-forming compound, which produces superoxide and hydrogen peroxide intracellularly through redox cycling (Liu et al., 1998); paraquat is an alkylating agent that increases the intracellular production of superoxide but also decreases the intracellular pool of reduced thiols and injures nigrostriatal dopaminergic neurons in rodents (Clejan and Cederbaum, 1989; Brooks et al., 1999; Thiruchelvam et al., 2000); (2) the neurotransmitter dopamine that auto-oxidizes to generate superoxide and hydrogen peroxide (Hastings et al., 1996); and (3) the mitochondrial complex I inhibitor rotenone, which at high concentrations results in increased oxidant production and delayed cell death (Turrens and Boveris, 1980;Lotharius and O'Malley, 2000).
The stock solution of DMNQ (Alexis Biochemicals, San Diego, CA) was prepared in dimethylsulfoxide and stored under nitrogen in the dark at −20°C. The working solutions of paraquat, rotenone, and dopamine (Sigma, St. Louis, MO) were prepared fresh before use in phosphate buffer, dimethylsulfoxide, and water, respectively. Typically, before exposure to these compounds, the cells were rinsed twice with serum-free media to remove the nonadhered cells, and 2 ml of fresh medium containing the superoxide generating compounds was added. Cells were allowed to incubate at 37°C for 30 min before PAPA/NO was added to the medium at a final concentration of 1 mm. Cells exposed to either paraquat or DMNQ were cultured for an additional 1.5 hr (total exposure time, 2 hr), whereas cells exposed to rotenone or dopamine were maintained for an additional 4.5 hr (total exposure time, 5 hr). At the end of the exposure, the cells were washed extensively and processed for the different immunological and biochemical analyses. The concentrations of the compounds (PAPA/NO, 1 mm; DMNQ, 10 μm; paraquat, 5 mm; rotenone, 100 nm; and dopamine, 5 μm) and duration of the exposure were selected to allow sufficient time for the respective compounds to generate reactive species without inducing immediate cell death. Cell death was monitored by measuring the fluorescence of YO-PRO1, a DNA-binding and membrane-impairment dye, which is used as a marker of the loss of plasma integrity (Gow et al., 2000). To study the effect of proteasome inhibition on α-syn aggregation, cells were treated with 10 μm of the specific proteasome inhibitor lactacystin-β-lactone for 1 hr and then examined by immunofluorescence.
Evaluation of intracellular formation of peroxynitrite. The production of peroxynitrite from the reaction of nitric oxide with superoxide was confirmed by the oxidation of dihydrorhodamine 123 (DHR 123) to the fluorescent product rhodamine 123. DHR 123 is oxidized by peroxynitrite and not by superoxide, nitric oxide, or hydrogen peroxide alone (Kooy et al., 1994; Ischiropoulos et al., 1999). The cells were plated in six-well 35 mm tissue culture plates, incubated with 5 μm DHR 123 (Molecular Probes, Eugene, OR) for 2 hr at 37°C, and extensively washed to remove DHR 123 from the media. The yield of rhodamine 123 was measured continuously for 90 min starting after the addition of PAPA/NO in the cell medium using a fluorescence plate reader (Molecular Dynamics, Sunnyvale, CA) with excitation and emission wavelengths of 500 and 536 nm, respectively (Fig.2A). Data in Figure 2 show an increase in the oxidation of DHR 123 only in cells exposed simultaneously to the nitric oxide donor and a superoxide-generating agent (Fig.2A), indicating that the oxidation of DHR 123 in this model is attributable predominantly to the generation of peroxynitrite and not to hydrogen peroxide-mediated oxidation. The intracellular rate of DHR 123 oxidation resulting from the addition of 1 mm PAPA/NO and 10 μm DMNQ addition approximated 180 ± 1 pm/min, as shown in Figure 2A. The concentrations of paraquat (5 mm), dopamine (5 μm), and rotenone (100 nm) and the duration of the treatment were selected to generate similar exposure in terms of DHR 123 oxidation as well as to avoid cell death. The sequestration of rhodamine 123 into the mitochondria after cells are treated with PAPA/NO, a superoxide generator, or both indicates that these conditions do not grossly perturb the integrity of cellular organelles (Fig. 2C). Fluorescent images were obtained with an Olympus(Tokyo, Japan) 1X70 inverted microscope. Taken together with the YO-PRO1 fluorescence (data not shown), these data indicate that the cells are viable at the end of the exposure.
The percentage of cells with increased (higher than control) rhodamine 123 fluorescence was also determined by flow cytometry. Briefly, cells were loaded with 5 μm DHR 123 followed by treatment with the different compounds described above. Cells were then trypsinized, centrifuged at 1200 rpm for 3 min, and resuspended in PBS with 1 mm EDTA. Cells were evaluated for fluorescence intensity on a Beckman-Coulter (Hialeah, FL) EPICS Elite flow cytometer equipped with a 5 W argon laser operated at 488 nm and 260 mW output. Fluorescence signals were collected with a photomultiplier tube configured with 550 nm long-pass dichroic and 525 nm bandpass filters. A total of 5000 events were collected for each sample, and individual cells were selected for analysis on the basis of forward and side scatter measurements. Both mean fluorescence intensity values and percent positive cells were determined by histogram subtraction with Immuno-4 analysis software (Beckman-Coulter) using untreated cells for background fluorescence (Fig. 2B). The flow cytometry data indicated that 14 and 29% of cells showed increased rhodamine 123 fluorescence after treatment with PAPA/NO plus paraquat or PAPA/NO plus DMNQ, respectively (Fig. 2B). The percentage of cells showing increased fluorescence intensity after exposure to DMNQ (6.0%) or PAPA/NO (5.8%) alone was only modestly higher than in untreated cells (3.0%).
To further substantiate the in situ formation of peroxynitrite, cell lysates were analyzed for the presence of nitrated α-syn. α-Syn was immunoprecipitated from cells treated with PAPA/NO, paraquat, or both, but the presence of nitrated protein was detected only in cells treated with PAPA/NO and paraquat simultaneously (Fig. 2D).
Western blotting. The amount of protein was determined using the Bradford Protein Assay (Bio-Rad, Hercules, CA). Ten to 30 μg of total protein from cell lysate were resolved on 12% SDS-polyacrylamide gels and transferred electrophoretically onto a nitrocellulose membrane overnight. The blots were blocked for 1 hr with 10% powdered milk in Tris-buffered saline containing 0.05% Tween 20. The nitrocellulose membrane was washed and probed overnight at 4°C with anti-β-tubulin monoclonal antibody TUB 2.1 (Sigma) and the anti-α-syn monoclonal antibodies Syn208, Syn102, and LB509 (Giasson et al., 2000b). After several washes, the membranes were incubated with a horseradish peroxidase conjugated anti-mouse antibody, and after additional washes, the immunoreactive signals were visualized using an enhanced chemiluminescence reagent (Amersham Pharmacia Biotech, Piscataway, NJ) and exposure to film. Immunoprecipitation of α-synuclein and blotting with polyclonal anti-nitrotyrosine antibodies were performed as described in detail previously (Przedborski et al. 2001).
Immunofluorescence. Cells were washed twice with PBS and fixed in cold methanol for 30 min at −20°C. Methanol was removed, and the cells were incubated in a 1:1 mixture of cold methanol and cold acetone for 5 min at −20°C. The cells were rinsed with PBS, blocked in PBS containing 10% normal goat serum, 0.3% Triton X-100, and 5% bovine serum albumin for 30 min at room temperature, and labeled with the anti-α-syn monoclonal antibody Syn208 or Syn202 (Giasson et al., 2000b) overnight at 4°C. The following day the cells were extensively washed with PBS and 0.3% Triton X-100 and incubated with Cy3-conjugated goat anti-mouse IgG antibody (Zymed, San Francisco, CA) at a 1:100 dilution in blocking solution. In some cases, cells were double-labeled with mouse anti-α-syn antibodies and a rabbit anti-ubiquitin polyclonal antibody (Chemicon International, Temecula, CA) or with a rabbit anti-3-nitrotyrosine polyclonal antibody. An FITC-conjugated goat anti-rabbit antibody was used to detect anti-ubiquitin or anti-3-nitrotyrosine labeling.
Electron microscopy. Cells were washed with 0.1m sodium cacodylate buffer, pH 7.3, and fixed with 2.5% glutaraldehyde in 0.1 m sodium cacodylate buffer, pH 7.3, for 1 hr at 4°C. Some specimens were treated with 30% formic acid for 9 sec on the grids before imaging to improve the visualization of fibrils in the aggregates. The samples were processed for electron microscopic imaging as described previously (Gow et al., 2000).
Turnover of α-syn in HEK 293 cells. HEK 293 cells were plated on six-well plates and maintained in complete medium (DMEM, 10% FBS, 1 mml-glutamine, 10 mm sodium pyruvate, penicillin, and streptomycin) overnight. Cells were methionine-deprived for 15 min by incubation in methionine-free DMEM (Life Technologies) before adding 100 μCi [35S]methionine (NEN, Boston, MA) per milliliter of DMEM with 10% dialyzed FBS (Life Technologies) for 45 min. Cells were rinsed with PBS, and complete medium was added. At the indicated time points, the cells were rinsed in PBS and harvested in 200 μl of immunoprecipitation buffer (50 mm Tris, pH 7.5, 100 mm NaCl, 2 mm EGTA, and 1% Triton X-100 containing a mixture of protease inhibitors). Cells were completely lysed by trituration and vortexing. Cell debris was removed by sedimentation at 13,000 × g for 20 min, and α-syn was immunoprecipitated with anti-α-syn antibody Syn211 (Giasson et al., 2000b) preincubated with anti-IgG mouse antibody conjugated to agarose beads (Sigma). The beads were extensively washed with immunoprecipitation buffer, and the protein complex was eluted by boiling in sample buffer for 10 min. The beads were removed by centrifugation, and the samples were loaded onto 12% polyacrylamide gels. After electrophoresis, gels were fixed with 50% methanol and 5% glycerol, dried, and exposed to a PhosphorImager plate. The signal was quantified using ImageQuant software (Molecular Dynamics).
RESULTS
Formation of α-syn aggregates in vivo
Exposure of cells transfected with α-syn (HEK 293/α-syn) to PAPA/NO plus intracellular superoxide-generating compounds, under the conditions described in detail in Materials and Methods and in Figures 1 and2, induced the formation of intracellular α-syn inclusions (Fig.3A–C). Exposure of HEK 293/wild type α-syn cells to PAPA/NO plus DMNQ or PAPA/NO plus paraquat resulted in the formation of α-syn inclusions in 12 ± 6 and 7 ± 4% of the cells (mean ± SD; n = 3–6 independent observations), respectively. Exposure of HEK 293 cells transfected with wild-type α-syn to PAPA/NO plus dopamine (Fig.3A) or PAPA/NO plus rotenone (Fig. 3B) resulted in 32 ± 11 and 25 ± 6% of cells with syn aggregates, respectively, whereas exposure to HEK 293 cells transfected with A30P α-syn exposed to the same conditions resulted in 4 ± 4 and 6 ± 1% of cells with syn aggregates (Fig.4B; data not shown). Finally, exposure of A53T α-syn stable transfectants to these conditions resulted in 22 ± 9 and 18 ± 7% of cells with syn aggregates, respectively (Fig. 4C; data not shown). These percentages were determined by random inspection of at least 5 different fields per well for each independent experiment (n = 3–6 independent observations). Exposure to the nitric oxide donor alone did not result in the formation of intracellular α-syn inclusions, whereas exposure to the superoxide-generating compounds alone occasionally resulted in α-syn aggregation in <1% of the cells (see Fig. 7D). Exposure of untransfected HEK 293 cells and HEK 293 cells stably expressing human β-syn (Fig. 1) to PAPA/NO plus paraquat did not result in protein aggregates, as revealed by staining with anti-α-syn (Fig.3D,E).

Expression of synucleins in stably transfected HEK 293 cell lines. Expression of human wild-type α-syn (lane 4), A53T mutant α-syn (lane 5), A30P mutant α-syn (lane 6), and human β-synuclein (lane 7) in stably transfected HEK 293 cells is shown, as demonstrated by Western blot analysis using the anti-synuclein antibody Syn102, which reacts equally to both syn proteins (Giasson et al., 2000b). Expression of α- and β-syn is not detected in untransfected HEK 293 cells (lane 3).Lanes 1 and 2 were loaded with 10 ng of recombinant human α- and β-syn, respectively. Lanes 3–7 were loaded with 10 μg of total cell lysates.

Formation of rhodamine 123 from the oxidation of DHR 123 in HEK 293/α-syn. A, HEK 293/α-syn cells were incubated for 2 hr at 37°C with 5 μm DHR 123. After extensive washing, rhodamine 123 fluorescence was monitored over time in untreated cells (triangles) and cells treated with 1 mm PAPA/NO alone (diamonds), DMNQ alone (circles), or 1 mm PAPA/NO plus 10 μm DMNQ (squares). B, Composite fluorescence histograms obtained by flow cytometric evaluation of HEK 293/α-syn cells after exposure to PAPA/NO plus DMNQ (broken trace) and control cells (solid trace). The mean fluorescence intensity value for each histogram, indicating the percentage of cells positive for rhodamine 123 fluorescence, was 6% for control and 29% for treated cells.C, Representative epifluorescence images (magnification, 17×) of cells after exposure to nitric oxide- and superoxide-generating compounds. D, Immunoprecipitation of α-syn using the anti-α-syn monoclonal antibody SYN-1 followed by Western blotting with SYN-1 or a polyclonal anti-3-nitrotyrosine antibody (Anti-3-NT). Cells were exposed to 1 mm PAPA/NO (first lane), 5 mm paraquat (second lane), or both 1 mm PAPA/NO and 5 mm paraquat (third lane).

Formation of α-syn intracellular aggregates on exposure to nitric oxide- and superoxide-generating compounds. HEK 293/α-syn cells were fixed and stained with monoclonal anti-α-syn antibodies Syn208 and Syn202. A–C, α-Syn inclusions were readily visible in cells exposed to 1 mm PAPA/NO plus 5 μm dopamine (A), 1 mmPAPA/NO plus 100 nm rotenone (B), and 1 mm PAPA/NO plus 5 mm paraquat (C). Only diffuse background staining was noted in untransfected (D) and β-syn-transfected (E) cells exposed to 1 mm PAPA/NO plus 5 mm paraquat. Magnification: A, B, 125×; C–E, 65×.

Nitration of α-syn intracellular aggregates on exposure to nitric oxide- and superoxide-generating compounds.A, HEK 293/α-syn exposed to 1 mm PAPA/NO plus 100 nm rotenone. B, HEK 293/A30P mutant α-syn exposed to 1 mm PAPA/NO plus 5 μmdopamine. C, HEK 293/A53T mutant α-syn exposed to 1 mm PAPA/NO plus 100 nm rotenone. Column 1, Cells were fixed and stained with monoclonal anti-α-syn antibody Syn 208. Column 2, Stained with a polyclonal anti-3-nitrotyrosine antibody. Column 3, Superimposed image. Magnification, 125×.
Significantly, the α-syn aggregates in HEK 293 cells transfected with either wild-type or mutant α-syn stained positive with anti-3-nitrotyrosine antibodies (Fig. 4A–C,column 2), consistent with the nitration of α-syn in this paradigm (Fig. 2D). In addition, monoclonal antibodies specific for nitrated α-syn (Giasson et al., 2000a) at Tyr125 and Tyr136 (nSyn12) and at Tyr39 (nSyn14) immunostained the α-syn aggregates in HEK 293/α-syn cells exposed to PAPA/NO and rotenone or dopamine (Fig. 5). There was no staining with either antibody in untreated control cells and cells treated with PAPA/NO only (data not shown).

Intracellular aggregates of α-syn on exposure to nitric oxide- and superoxide-generating compounds are nitrated at Tyr39 as well as Tyr125 and Tyr136. A, HEK 293/α-syn exposed to 1 mm PAPA/NO plus 100 nm rotenone.B, HEK 293/α-syn exposed to 1 mm PAPA/NO plus 5 μm dopamine. Column 1, Cells were fixed and stained with monoclonal anti-nitrated α-syn antibody nSyn14, which recognizes nitrated tyrosine residue Tyr39 in the N terminus of α-syn. Column 2, Stained with monoclonal anti-nitrated α-syn antibody nSyn12, which recognizes nitrated tyrosine residues Tyr125 and Tyr136 in the C terminus of α-syn (Giasson et al., 2000a). Magnification, 98×.
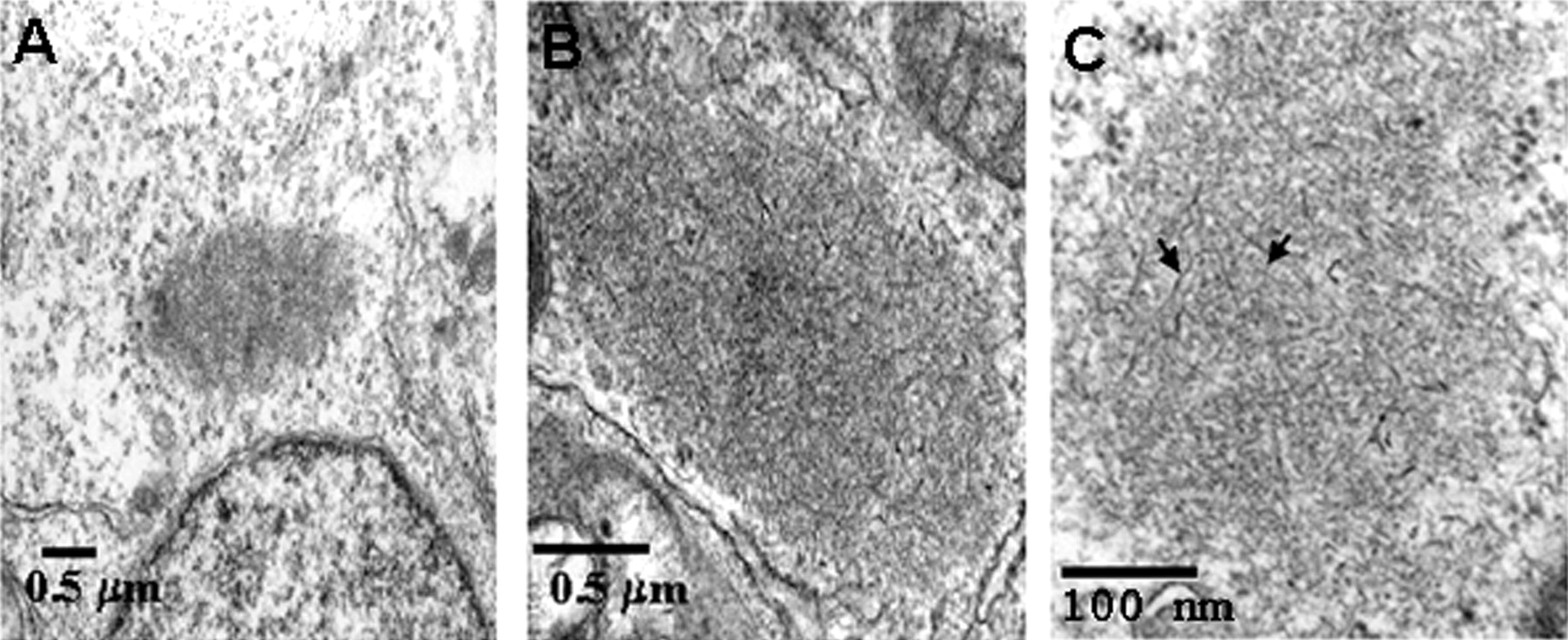
The ultrastructure of intracellular inclusions in cells exposed to nitric oxide and superoxide was examined by transmission electron microscopy (Fig. 6). Figure 6 depicts a typical perinuclear inclusion (Fig. 6A), which on higher magnification revealed the presence of fibril-like structures within the inclusion (Fig. 6B). Examination of the aggregates at higher magnification after a brief treatment with 30% formic acid confirmed that most of the inclusions contained fibril-like material (Fig. 6C). Cytoplasmic inclusions were seen only in cells treated with nitric oxide- and superoxide-generating compounds but not in untreated cells or cells treated with nitric oxide. Occasionally, inclusions are seen after exposure to superoxide-generating compounds alone (Fig.7D).

Ultrastructure of inclusions induced by nitrative damage in HEK 293/α-syn cells. Electron microscopic examination of cytoplasmic inclusions in HEK 293/α-syn cells exposed to nitric oxide- and superoxide-generating compounds was performed. InC, The sample was treated with formic acid to enhance the appearance of fibrils (arrows) in the inclusion.

Paucity of ubiquitin immunoreactivity in α-syn aggregates. Cells were stained with the mouse anti-α-syn monoclonal antibodies Syn202 and Syn208 (column 1) or a rabbit anti-ubiquitin polyclonal antibody (column 2), and inclusions were visualized by immunofluorescence. Column 3, Overlay composite of images. Cells expressing wild-type α-syn (A, B, D) or A53T mutant α-syn (C) were exposed to PAPA/NO and paraquat (A), PAPA/NO and rotenone (B, C), or paraquat alone (D). Magnification, 125×.
α-Syn aggregation and ubiquitination
Most α-syn aggregates generated in cells transfected with wild-type or mutant α-syn after exposure to PAPA/NO and superoxide generators did not stain with anti-ubiquitin antibodies (Fig. 7). Only rare inclusions, such as the one shown after paraquat treatment, were double-labeled with anti-α-syn and anti-ubiquitin antibodies (Fig.7D). Staining of untransfected cells exposed to PAPA/NO plus superoxide-generating agents with either anti-α-syn or anti-ubiquitin antibodies failed to reveal the presence of protein aggregation (data not shown). In contrast, inhibition of the proteasome by lactacystin-β-lactone (Fenteany et al., 1994) resulted in abundant protein aggregates stained with anti-ubiquitin antibodies in both transfected and untransfected cells (Fig.8). These protein aggregates did not stain with several different anti-α-syn antibodies (Fig.9), suggesting that α-syn is not sequestered into these inclusions and is not targeted for proteolysis by the proteasome in this cell model. The presence of lactacystin did not significantly increase the percentage of cells with α-syn aggregates in α-syn transfected cells exposed to nitric oxide- and superoxide-generating compounds (data not shown).

Formation of protein aggregates after selective inhibition of the proteasome. Untransfected HEK 293 cells (A, B) or HEK 293/α-syn cells (C, D) were treated with 10 μm lactacystin-β-lactone for 1 hr and stained with anti-α-syn monoclonal antibody Syn202 (A, C) or a rabbit anti-ubiquitin antibody (B, D). Magnification, 40×.

Formation of protein aggregates after inhibition of the proteasome. HEK 293/α-syn cells were treated with 10 μm lactacystin-β-lactone for 1 hr and stained with the mouse anti-α-syn monoclonal antibody Syn202 (A) or with a rabbit anti-ubiquitin antibody (B). InC, the fluorescence fields in A andB were merged. Note that inhibition of the proteasome with lactacystin-β-lactone results in ubiquitin aggregates but not α-syn aggregates. Magnification, 65×.
Moreover, α-syn is a relatively stable protein in HEK 293 cells, with a half-life of >48 hr (Fig.10A). The relatively long half-life of α-syn prevents the use of protease inhibitors over long periods (because of cell toxicity) to measure changes in turnover rates. However, analysis of steady-state levels after 24-hr treatments with various inhibitors indicates that α-syn may be degraded by lysosomes and not by the proteasome (Fig. 10B), because lactacystin-β-lactone had no effect, whereas ammonium chloride and MG132 resulted in a significant and reproducible increase of α-syn steady-state levels.

Degradation of α-syn in HEK 293 cells.A, Pulse–chase analysis of wild-type (diamonds), A30P (triangles), and A53T (squares) α-syn turnover in HEK 293 stable transfectants. The residual [35S]Met is after chasing over 48 hr (n = 3). A, inset, Representative chase profile of [35S]Met-labeled wild-type α-syn.UT, Immunoprecipitation from untransfected HEK 293 cells. B, Western blot analysis using the anti-α-syn antibody LB509 showing three independent experiments. HEK 293/α-syn cells were untreated (Ct) or challenged with 25 mm NH4Cl, 10 μmlactacystin-β-lactone (Lact), or 2 μmMG132 for 24 hr. Equal amounts of protein (5 μg) were loaded in each separate lane of the gels as confirmed by the levels of β-tubulin (β-tub).
DISCUSSION
The recently reported evidence (Duda et al., 2000b; Giasson et al., 2000a) for nitration of α-syn in pathological inclusions of neurodegenerative diseases known as synucleinopathies, which include sporadic PD, dementia with LBs, a subtype of Alzheimer's disease known as the LB variant of Alzheimer's disease, multiple system atrophy, and neurodegeneration with brain iron accumulation type 1, raises the possibility that nitrative and oxidative damage may have a direct role in disease initiation, progression, or both. Nitrating agents such as peroxynitrite and nitrogen dioxide are strong oxidants capable of promoting not only nitration but also oxidation of tyrosine and other amino acids (Ischiropoulos, 1998; Alvarez et al., 1999; Pennathur et al., 1999). To study the effect of nitrating species on the biophysical properties of α-syn in intact cells, stably α-syn-expressing HEK 293 cells (Fig. 1) were challenged with reagents that when used individually result in the intracellular formation of nitric oxide and superoxide. The data indicated that simultaneous exposure to both nitric oxide and superoxide was required for the formation of the α-syn aggregates in transfected cells (Figs. 3-6). These findings in conjunction with the specific oxidation of DHR 123 and nitration of α-syn when cells are treated with nitric oxide- plus superoxide-generating compounds indicate that generation of peroxynitrite is associated with the aggregation of α-syn. In addition, the α-syn in the aggregates consists of nitrated protein (Figs. 4 and 5), which raises the possibility that this modification of α-syn directly induces the formation of aggregates, but this suggestion requires further investigation. It is unclear what cellular mechanism is triggered or disrupted by nitrative injury leading to the aggregation of α-syn. We did not detect the presence of high molecular mass species on Western blot analysis, which would be suggestive of dityrosine cross-linking that could stabilize oligomers (Souza et al., 2000). Nevertheless, our results demonstrate that nitrative injury and, to a much lesser extent, oxidative injury can directly result in the formation of intracellular fibrillar aggregations of α-syn, which may be the nidus of pathological lesions.
Exposure of cells to kinetically and biochemically defined low fluxes of nitric oxide and superoxide resulting in the formation of peroxynitrite (Fig. 2) may reflect situations encountered in vivo. Neurons can be sensitized to nitric oxide-mediated injury (Dawson et al., 1996), and mice lacking neuronal nitric oxide synthase are protected from ischemic injury, excitotoxic injury, and neurotoxins (Huang et al., 1994; Ayata et al., 1997; Eliasson et al., 1999). We have argued that nitric oxide-mediated neuronal injury is selective and pronounced in neuronal populations with higher intracellular steady-state levels of superoxide (Ara et al., 1998). For example, in dopaminergic neurons, the steady-state levels of superoxide can be increased from oxidation-reduction recycling of dopamine or impairment of mitochondrial electron transfer after a challenge with rotenone, as has been reported recently in rats (Betarbet et al., 2000). Significantly, rotenone-treated rats develop clinical and pathological phenotypic changes, including the formation of α-syn aggregates that closely resemble those seen in Parkinson's disease patients (Betarbet et al., 2000). The importance of superoxide is also highlighted by the observations that augmentation of superoxide dismutases protects neurons from neurotoxicity (Chan et al., 1991;Przedborski et al., 1992), whereas a decline in the superoxide dismutases leads to cell death that is mediated in part by nitric oxide (Rothstein et al., 1994; Przedborski et al., 1996; Troy et al., 1996;Estevez et al., 1998). The data presented herein are consistent with the importance of superoxide and nitric oxide in the process that leads to intracellular α-syn aggregation.
Although a number of studies revealed that cells expressing α-syn are sensitive to oxidative stress (Hsu et al., 2000; Kanda et al., 2000; Ko et al., 2000; Lee et al. 2001), only two other cellular models have reproduced the formation of α-syn aggregates (Ostrerova-Golts et al., 2000; Tabrizi et al. 2000). Ostrerova-Golts et al. (2000) observed formation of α-syn aggregates in ∼20% of BE-M17 neuroblastoma cells transfected with A53T α-syn after exposure to relatively high levels of hydrogen peroxide (100 μm) and ferrous iron (0.3 mm) for prolonged periods (96 hr). This observation is similar to data reported here, because 10–30% of the exposed cells developed α-syn aggregates, although the majority of the cells expressed similar levels of the protein. At this time, it is unclear why only a subset of the cells develops α-syn aggregates. It is possible that the intensity of exposure is different among the cells in culture, consistent with the observations in Figure 2, which indicated that the magnitude of peroxynitrite formation inside the cells was significant only in the same percentage of the cells that developed α-syn aggregates. Alternatively, some cells may be more resilient to oxidative stress than others by preventing oxidative modification and aggregation of α-syn. In addition, the abundance of synuclein aggregates is significantly less in cells expressing the A30P mutant compared with cells expressing similar levels of the A53T mutant or wild-type α-syn. This occurrence may be attributable to or related to the greater propensity of the A30P mutant to form “spheroid-like” intermediates rather than long fibrils in vitro (Conway et al., 2000).
In contrast to the nitric oxide- and superoxide-mediated aggregation of α-syn, selective blockade of protein degradation through the proteasome results in protein aggregation (Johnston et al., 1998), but these inclusions did not contain α-syn (Fig. 9). Only occasional α-syn aggregates stained with anti-ubiquitin antibodies (Fig. 7D). This observation is consistent with the recent reports in two transgenic mouse lines in which ubiquitin staining was evident in some but not all α-syn aggregates (Van der Putten et al., 2000), and in wild-type α-syn aggregates in ECR293 cells (Tabrizi et al., 2000). However, these observations are in contrast with reports indicating that the majority of the α-syn aggregates induced by exposure to hydrogen peroxide and ferrous iron stained with anti-ubiquitin antibodies (Ostrerova-Golts et al., 2000). It is possible that the degree of oxidative stress in the hydrogen peroxide and ferrous iron model induced coaggregation of other proteins that were ubiquitin positive. Alternatively, because both hydrogen peroxide and ferrous iron will be consumed rather fast (within 1 hr) after exposure and will induce a rather intense stress to the cells, it is likely that the formation of ubiquitin aggregates in that model reflects inhibition of proteasome activity as well. Interestingly, inducible expression of A30P α-syn but not wild-type α-syn in PC12 cells inhibits proteasome activity, and in the presence of lactacystin, the A30P α-syn induces cell death (Tanaka et al., 2001). This occurrence suggests that at least under some conditions, the mutant protein may induce cellular accumulation of unfolded proteins by blocking the activity of the proteasome.
It has been reported that α-syn has a short half-life (1.84 hr) in SH-SY5Y cells and that it is degraded by the proteasome. We determined the half-live of wild-type, A53T, and A30P α-syn in HEK 293 cells to be >48 hr, consistent with the half-life of Flag-tagged α-syn expressed in PC12 cells (t½, >54 hr; Okochi et al., 2000). In addition, in both HEK 293 and TSM1 cells, it is unlikely that the proteasome is involved in the degradation of α-syn, because the proteasome-specific inhibitor lactacystin-β-lactone (Fenteany et al., 1994) did not affect the steady-state levels of the protein (Fig. 10B) (Ancolio et al., 2000). Thus, it is likely that the results by Bennett et al. (1999) were confounded by the expression of a His-tagged protein, although it is possible that proteasome-mediated degradation of α-syn is cell-specific. Our results indicate that α-syn can be degraded at least in part by lysosomes, as demonstrated by the increase in steady-state levels when cells are treated with ammonium chloride (Fig. 10B). Treatment with MG132 also resulted in a similar increase, but the broad range of thiol proteases inhibited by these types of peptide-aldehyde compounds renders it difficult to precisely define the specific enzyme involved (Hiwasa et al., 1990; Sasaki et al., 1990;Rock et al., 1994). However, consistent with the ammonium chloride data, these compounds can also inhibit the lysosomal enzymes cathepsin B and L (Hiwasa et al., 1990; Sasaki et al., 1990). Impairment of α-syn turnover may be an important factor leading to an overabundance of this protein, resulting in a sufficient critical concentration to facilitate aggregation into pathological inclusions.
Taken together, the studies reported here demonstrate that α-syn is sequestered from the cytoplasmic milieu into filamentous aggregates under defined conditions (e.g., in which intracellular peroxynitrite is generated). This redistribution is mostly specific to the generation of peroxynitrite and to a lesser extent to superoxide generation, because increasing the cellular levels of nitric oxide is not sufficient to generate α-syn aggregates. Thus, it is possible that the aberrant production of peroxynitrite and perhaps other reactive species triggers or facilitates the formation of pathological α-syn inclusions, because aggregation of α-syn is limited to specific cellular adversities such as oxidative stress but not to the inhibition of proteasome.
Footnotes
This work was supported by grants from the National Institute on Aging (J.Q.T., V.M.-Y.L., H.I.), by an established investigator award from the American Heart Association (H.I.), and by a pioneer award from the Alzheimer's Association (J.Q.T., V.M.-Y.L.). B.I.G. was the recipient of a fellowship from Canadian Institutes of Health Research. We thank the Biochemical Imaging Core Facility of the University of Pennsylvania for assistance with electron microscopy.
Correspondence should be addressed to Harry Ischiropoulos, Stokes Research Institute, Children's Hospital of Philadelphia, 416D Abramson Research Center, 34th Street and Civic Center Boulevard, Philadelphia, PA 19104-4318. E-mail:ischirop{at}mail.med.upenn.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}