

Abstract
Along with ionotropic and metabotropic glutamate receptors, the cystine/glutamate antiporter x
 may play a critical role in CNS pathology. High levels of extracellular glutamate inhibit the import of cystine, resulting in the depletion of glutathione and a form of cell injury called oxidative glutamate toxicity. Here we show that a portion of the cell death associated with NMDA receptor-initiated excitotoxicity can be caused by oxidative glutamate toxicity. In primary mouse cortical neurons the cell death resulting from the short-term application of 10 μmglutamate can be divided into NMDA and NMDA receptor-independent phases. The NMDA receptor-independent component is associated with high extracellular glutamate and is inhibited by a variety of reagents that block oxidative glutamate toxicity. These results suggest that oxidative glutamate toxicity toward neurons lacking functional NMDA receptors can be a component of the excitotoxicity-initiated cell death pathway.
may play a critical role in CNS pathology. High levels of extracellular glutamate inhibit the import of cystine, resulting in the depletion of glutathione and a form of cell injury called oxidative glutamate toxicity. Here we show that a portion of the cell death associated with NMDA receptor-initiated excitotoxicity can be caused by oxidative glutamate toxicity. In primary mouse cortical neurons the cell death resulting from the short-term application of 10 μmglutamate can be divided into NMDA and NMDA receptor-independent phases. The NMDA receptor-independent component is associated with high extracellular glutamate and is inhibited by a variety of reagents that block oxidative glutamate toxicity. These results suggest that oxidative glutamate toxicity toward neurons lacking functional NMDA receptors can be a component of the excitotoxicity-initiated cell death pathway.
The physiological consequences of extracellular glutamate are mediated by three classes of membrane proteins within the CNS. These are ionotropic glutamate receptors, metabotropic glutamate receptors, and the cystine/glutamate antiporter. Ionotropic glutamate receptors have two known roles. They are responsible for the majority of excitatory neurotransmission and also for a great deal of CNS pathology. In cases of stroke or trauma, excessive extracellular glutamate leads to nerve cell death via the activation of NMDA receptors (Rothman and Olney, 1986). This phenomenon, which can be reproduced in cell culture (Rothman, 1985;Choi, 1987), is termed excitotoxicity (Olney, 1986). In contrast to ionotropic glutamate receptors, the metabotropic glutamate receptors (mGluRs) are G-protein-coupled membrane proteins with a wide variety of biological functions (Nakanishi, 1994). Finally, a third target for extracellular glutamate in the CNS is the inhibition of the glutamate/cystine antiporter x
 , which results in a form of oxidative stress and cell death called oxidative glutamate toxicity (Murphy et al., 1989). The glutamate/cystine antiporter couples the import of cystine to the export of glutamate (Sato et al., 1999). Concentrations of extracellular glutamate as low as 100 μm, which are well below the level of extracellular glutamate found in models of stroke and trauma (see, for example,McAdoo et al., 1999), completely inhibit the uptake of cystine (Sagara and Schubert, 1998). Cystine is required for the synthesis of the potent intracellular-reducing agent glutathione (GSH). When GSH is depleted by extracellular glutamate, cells die from a form of programmed cell death (Tan et al., 1998a,b).
, which results in a form of oxidative stress and cell death called oxidative glutamate toxicity (Murphy et al., 1989). The glutamate/cystine antiporter couples the import of cystine to the export of glutamate (Sato et al., 1999). Concentrations of extracellular glutamate as low as 100 μm, which are well below the level of extracellular glutamate found in models of stroke and trauma (see, for example,McAdoo et al., 1999), completely inhibit the uptake of cystine (Sagara and Schubert, 1998). Cystine is required for the synthesis of the potent intracellular-reducing agent glutathione (GSH). When GSH is depleted by extracellular glutamate, cells die from a form of programmed cell death (Tan et al., 1998a,b).
The potential role of oxidative glutamate toxicity in ischemia and trauma is not understood, but there have been strong indications that several cell death pathways are involved. In localized cerebral infarction the neurons in the epicenter die rapidly, whereas those more distal remain viable for several hours (Siesjo, 1992). Multiple forms of nerve cell death also have been identified in excitotoxic CNS primary culture paradigms that follow exposure to glutamate (for review, see Choi, 1992). In primary cultures of cerebellar granule cells that are exposed to glutamate, there is a rapid necrotic phase, followed by delayed apoptotic-like cell death (Ankacrona et al., 1995). During oxygen–glucose deprivation of primary mouse cortical cultures or organotypic cultures of the rat hippocampus, some cell death occurs from ionotropic receptor-independent mechanisms (Gwag et al., 1995;Newell et al., 1995). All of these observations are consistent within vivo data, which show that glutamate receptor-independent programmed cell death may occur after ischemic insults (Shigeno et al., 1990; Linnik et al., 1993; MacManus et al., 1993; Okamoto et al., 1993). In addition, animals that lack caspases undergo a form of cell death that is morphologically very similar to oxidative glutamate toxicity (Tan et al., 1998a,b; Oppenheim et al., 2001). A number of parameters change dramatically during CNS stress, leading to the observed high exogenous glutamate. These include the direct release of glutamate from cells, the enzymatic conversion of high extracellular glutamine to glutamate, and the shutdown of nerve and glial glutamate uptake systems by pro-oxidant conditions (see Discussion). It is therefore of interest to determine whether oxidative glutamate toxicity can play a significant role in nerve cell death that is associated with the excitotoxicity cascade.
MATERIALS AND METHODS
Cell culture. Primary cultures of cortical neurons that die reproducibly by excitotoxicity were prepared by combining aspects of two published protocols (Rose et al., 1993; Dugan et al., 1995). Embryonic day 14 (E14) BALB/c mouse embryo cortices were minced and treated with 0.1% trypsin for 20 min. After centrifugation the cells were resuspended in B27 Neurobasal medium (Life Technologies, Grand Island, NY) plus 10% fetal calf serum and were dissociated by repeated pipetting through a 1 ml blue Eppendorf pipette tip. Then the cells were plated at 1 × 105cells per well in 96-well poly-l-lysine and laminin-coated microtiter plates (Becton Dickinson, Bedford, MA) in B27 Neurobasal plus 10% fetal calf serum and 20% glial growth-conditioned medium prepared according to Dugan and colleagues (Dugan et al., 1995). The growth-conditioned medium improved plating efficiency by ∼30%. Then 2 d later the medium was aspirated and replaced by serum-free B27 Neurobasal medium plus 10 μg/ml cytosine arabinoside. The cultures were used without media change between 7 and 12 d after plating and were essentially free of astrocytes (Brewer et al., 1993).
For glutamate toxicity assays, test drugs (e.g., antioxidants) were added 30 min before glutamate exposure. Then the culture medium was moved with a multichannel pipette to a new 96-well plate, and the cells were exposed to glutamate (usually 10 μm) in a HEPES-buffered salt solution [HCSS (Rose et al., 1993)] containing (in mm) 120 NaCl, 5.4 KCl, 0.8 MgCl2, 1.8 CaCl2, 15 glucose, and 20 HEPES, pH 7.4. In some cases, 1 μm glycine was included, but this had no net effect on excitotoxic death. After 10 min at room temperature the HCSS was aspirated, and the original growth medium was returned to the cells. In some cases the NMDA antagonist aminophosphonopentanoic acid (AP-5) was added at this point to inhibit the downstream activation of glutamate receptors.
MTT assay. Cell survival was determined by the MTT [3-(4,5-dimethyldiazol-2-yl)-2,5-diphenyl tetrazolium bromide] assay as described (Schubert et al., 1992), which correlates with cell death as determined by trypan blue exclusion and a colony-forming assay (Davis and Maher, 1994). At 24 hr after the addition of glutamate, 10 μl of the MTT solution (2.5 mg/ml) is added to each well and the cells are incubated for 3 hr at 37°C. Then 100 μl of solubilization solution (50% dimethylformamide and 20% SDS, pH 4.8) is added to the wells, and the next day the absorption values at 570 nm are measured. The results are expressed relative to the controls specified in each experiment. They are expressed as the mean of triplet determinations within the same experiment ± SEM; each experiment has been repeated at least three times with similar results.
Western blotting and glutamate assays. For Western blotting the cells were collected directly in Laemmli buffer (Laemmli, 1970). Cell lysates were resolved in 10% polyacrylamide gels containing SDS and transferred electrophoretically to hybridization membranes (Micron Separations, Westboro, MA). The membrane was probed first with a rabbit antiserum at a dilution of 1:2000 and then with horseradish peroxidase-conjugated goat anti-rabbit IgG secondary antibody at a dilution of 1:20,000. The antibody conjugates were detected with a chemiluminescence Western blot kit (Amersham, Buckinghamshire, UK).
Glutamate assays in growth-conditioned medium were performed by both mass spectroscopy and standard amino acid analysis with similar results (Iwabuchi et al., 1994). The standard amino acid data are presented. The removal of glutamate from growth-conditioned medium was done exactly as described by Matthews et al.(2000), except that the enzyme was added every 6 hr during the experiment. Media were treated initially for 1 hr at 37°C with 100 μg/ml glutamate pyruvate transaminase (GPT), 100 μmpyridoxal-l-phosphate and 10 mm pyruvate; these reagents were left in the culture medium.
Reagents. The mGluR agonists and antagonists were all from Tocris Cookson (Ballwin, MO), and mGluR 1 and 2/3 antisera and anti-NMDA antisera were from Chemicon (Temecula, CA). Anti-mGluR5 was a gift from Dr. R. Gereau (The Salk Institute, La Jolla, CA). The remaining reagents were obtained from Sigma (St. Louis, MO).
RESULTS
Cortical neuron cell death can be initiated by a purely NMDA receptor-dependent mechanism
As outlined above, there is some evidence for a non-ionotropic glutamate receptor component of the excitotoxicity cascade, but there have been only limited attempts to isolate and study this event. To do so, a number of criteria should be met. These include reproducibility, a pure nerve cell population to avoid confounding interactions with glia, a quantitative cytotoxic assay, and a system in which the process is initiated by the activation of a single class of ionotropic receptors, ideally NMDA receptors. By combining and modifying a number of published procedures (Rose et al., 1993) (also see, for example,Dugan et al., 1995), we devised a cell culture system that meets these criteria. Briefly, E14 mouse cortical neurons are dissociated and plated into 96-well microtiter plates in Neurobasal medium containing B27 supplements (Brewer et al., 1993) and fetal calf serum. Then 2 d later the medium is replaced with serum-free B27-supplemented medium alone containing cytosine arabinoside. The experiments are done between 7 and 14 d after plating, and cell viability usually is determined by the reduction of MTT (Liu et al., 1997) 24 hr after a 10 min exposure to glutamate. After 8 d in culture the cells are killed by glutamate with an EC50 of ∼2 μm and by NMDA with an EC50 of 20 μm. AMPA and kainate are not toxic to these cells unless concentrations in excess of 100 μm are used (Fig.1). The toxicity of 10 μmglutamate is blocked completely by the NMDA receptor antagonists AP-5, DCQX, and MK-801, but not by the kainate/AMPA antagonists CNQX, GYKI-52466, or AMOA (Table 1). These data show that the cytotoxic cascade in this culture system is initiated exclusively by the activation of NMDA receptors, therefore meeting the criteria for excitotoxicity as initially defined by Olney (1986).

Ionotropic glutamate receptor-mediated toxicity. After 8 d in culture, E14 cortical neurons were exposed to the indicated reagents for 10 min, and cell viability was measured 24 hr later by the MTT assay, as described in Materials and Methods. The results were confirmed by visual (trypan blue exclusion) assays and are the mean of triplicate determinations ± SEM. x, Glutamate; ○, NMDA; ▵, kainate; ▿, glutamate plus 100 μm AP-5; ■, AMPA.
Toxicity of 10 μm glutamate
Cell death is initiated rapidly
To determine how rapidly cells die under the experimental conditions outlined above, we exposed cultures to 10 μmglutamate for 10 min, followed by a 3 hr MTT viability assay at various times after glutamate exposure. The results were confirmed by visual assays, including propidium iodide exclusion. Figure2A shows that most of the cell death is quite rapid, with maximal levels at ∼4 hr postglutamate exposure. The duration of exposure to 10 μm glutamate that is required to elicit maximum cell death is also short. When cells are exposed to 10 μm glutamate for various lengths of time, followed by a viability assay 24 hr later, cell death is significant after 1 min and maximum with a 3–4 min exposure (Fig.2B). All cell death can be prevented by the inclusion of 100 μm AP-5 in the glutamate incubation medium. Therefore, there is a very efficient coupling between NMDA receptor activation and the initiation of the cell death pathways.
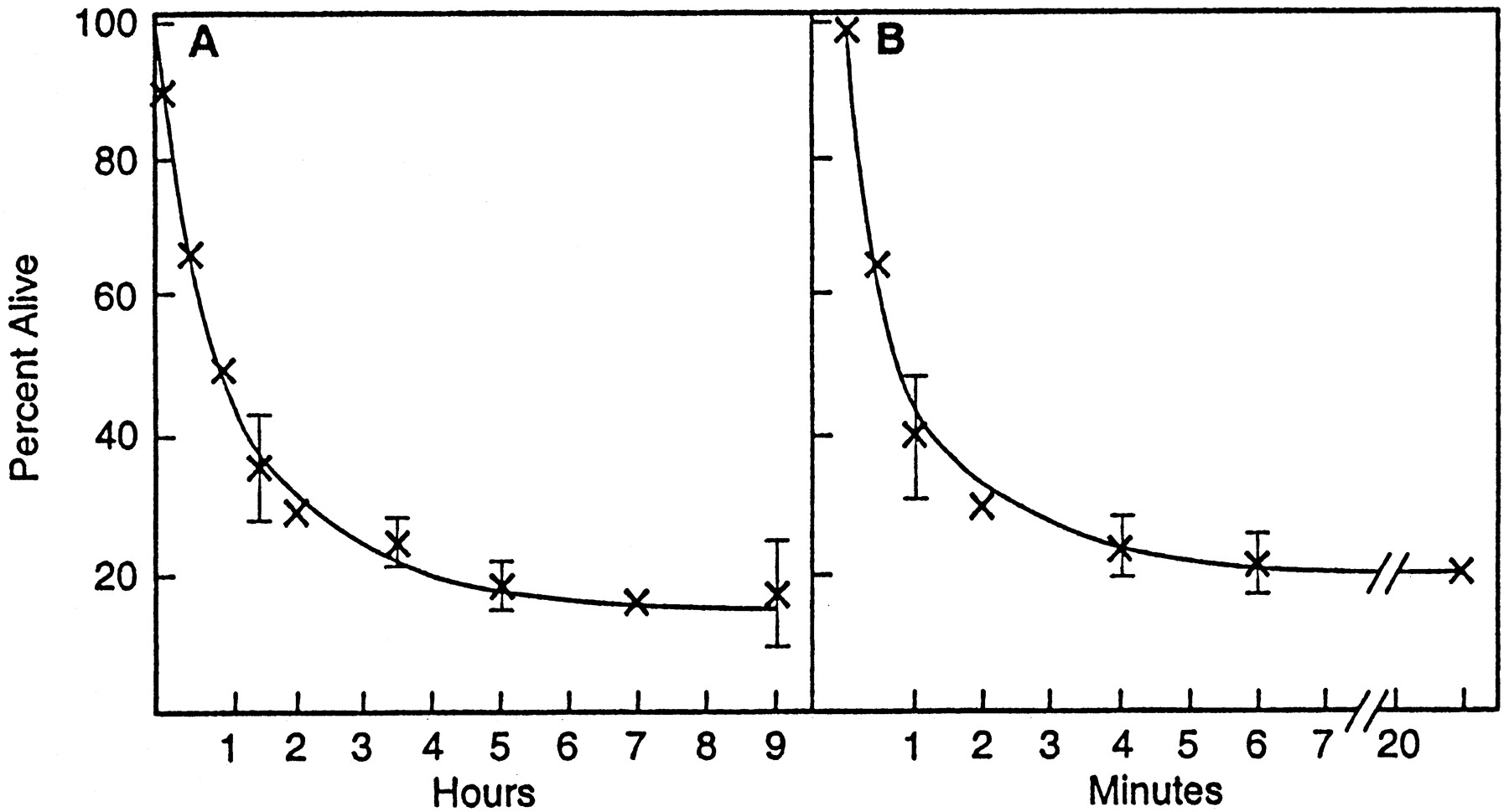
Temporal requirements for glutamate excitotoxicity. A, Cells 9 d in culture were exposed to 10 μm glutamate for 10 min, followed by a 3 hr MTT assay for viability at various times after glutamate exposure. For example, at 0 hr the cells were exposed to glutamate and assayed immediately for viability in the 3 hr MTT assay; the 5 hr point is a 5 hr incubation after glutamate, followed by a 3 hr MTT assay.B, Cells were exposed to 10 μm glutamate for 0–20 min, followed by the MTT viability assay 24 hr later. At the 30 sec time point ∼35% of the cells died during the next 24 hr. The results are the mean of triplicate determinations ± SEM.
Cell death can be divided into three components
Although the initiation of cell death is totally dependent on the activation of NMDA receptors, it is possible that other forms of cell death are hidden within the ionotropic receptor-initiated process. To isolate a possible NMDA receptor-independent component, we exposed cells to 10 μm glutamate for 10 min and then cultured them continuously in the presence or absence of AP-5, a potent NMDA antagonist that completely blocks glutamate toxicity in these cultures (Table 1). Figure 3A shows that, at 8 d in culture, three components of the excitotoxicity cascade are revealed by this procedure. Approximately 80% of the cells are killed by a 10 min exposure to 10 μmglutamate (arrow A), and none are killed when AP-5 is present with glutamate. However, if AP-5 is added immediately after the exposure to glutamate, ∼30% of the cells are rescued from cell death (arrow B). It follows that the 30% of the cells that are rescued by AP-5 require NMDA receptor activation after glutamate exposure, whereas the remaining 50% (arrow C) must be killed either by the initial exposure to glutamate via the activation of NMDA receptors or by a downstream mechanism that is independent of the NMDA receptor. If AP-5 is present during the exposure to glutamate and then removed from the cultures, there is still no cell death, for under these conditions glutamate cannot activate receptors and initiate the cascade. These observations are consistent with previous observations showing that a significant fraction of cells destined to die after glutamate exposure can be rescued by NMDA antagonists applied after the initial glutamate exposure (Rothman et al., 1987; Hartley and Choi, 1989; Manev et al., 1989). Approximately 20% of the cells never die under these conditions; the reason for this is unknown.

A portion of excitotoxic cell death is non-NMDA receptor-mediated. A, After 8 d in culture, E14 cortical neurons were exposed for 10 min to the indicated concentrations of glutamate in the presence or absence of AP-5 and then incubated for 24 hr in the presence or absence of AP-5, at which time cell viability was monitored by the MTT assay. x, Glutamate alone; ▵, glutamate plus 100 μm AP-5 during and after the 10 min glutamate exposure; ○, glutamate plus 100 μm AP-5 added immediately after glutamate exposure. A indicates total cell death in the system. B indicates the fraction of cells that die after glutamate exposure by a NMDA receptor-mediated process. C indicates the fraction of cells that die by virtue of the initial NMDA activation of the cell death pathway plus those that die independently of the NMDA receptor after the initial exposure to glutamate. B, Schematic representation of alternative cell death pathways identified above. Open circles represent cells lacking NMDA receptors andcircles enclosing an N represent cells with functional NMDA receptors. The two-headed arrow inA indicates that there may be a reciprocal interaction leading to cell death between cells with and without NMDA receptors.
The interpretation of these data, and the basis for the following experiments, is that the activation of NMDA receptors during the 10 min exposure to 10 μm glutamate initiates the death of a population of cells, which is represented within the “A” and “C” components. This event triggers two additional responses caused by the initial lysis of cells and the accumulation of glutamate in the culture medium. One is the subsequent activation of NMDA receptors on additional cells, resulting in more receptor-dependent cell death (population B); the other possible outcome is the death of a population of cells that do not have functional NMDA receptors (a subset of population C). These alternatives are shown schematically in Figure3B, in which the circles on the left represent cells directly killed during the 10 min glutamate exposure and the circles on the right are cells killed after glutamate exposure via NMDA (circled N) and NMDA receptor-independent (open circles) mechanisms. The experiments below define the cell death pathway by which this latter population is killed.
Glutamate receptor expression changes with length of time in culture
It has been observed repeatedly that the efficiency of excitotoxic cell death is dependent on the length of time the cells have been maintained in culture (see, for example, Dugan et al., 1995). This is presumably attributable to the time required for the cells to express functional ionotropic receptors. To assay the distribution of NMDA receptor versus non-NMDA receptor-mediated killing as a function of time in culture, we repeated the experiment described in Figure 3 on days 7–11 of cell culture. The fraction of the total nerve cell culture that is killed by a 10 min exposure to 10 μmglutamate increases from 40% at day 7 to ∼80% on days 10 and 11 (Fig. 4). In contrast, ∼60% of the cells that die are rescued by the postglutamate addition of AP-5 at day 7. This decreases to 20% between days 10 and 11.

Changes in cell death mechanism as a function of time in culture. E14 cortical cultures were monitored for glutamate-induced cell death exactly as described in Figure 3 but as a function of time in culture. The endpoint that is plotted is the plateau of killing by 10 μm glutamate (see Fig. 3). ●, Percentage of the initial cell population killed by glutamate (10 min exposure); ○, percentage of total late cell death in the culture rescued by AP-5 (see Fig. 3B). The data are the mean ± SEM of three or four experiments.
The observation that the total number of cells killed increases with culture age suggests that either the level of NMDA receptor expression increases or its coupling to relevant second message systems is dependent on the amount of time the neurons are in culture. Because one NMDA receptor subunit, NR1, is common to most NMDA ionotropic channels (for review, see Akazawa et al., 1994), the expression of this subunit was followed by Western blotting as a function of time in culture. Figure 5A shows that the expression of the NR1 receptor dramatically increases between days 3 and 10 in culture, suggesting that NMDA receptor availability may be limited in the NMDA receptor-mediated killing. Concomitant with culture age is an increase in neurite density (data not shown). Actin is a major component of neurites, and the amount of actin in neuronal cultures correlates with neurite density. Figure 5A shows that there is an increase in actin accumulation closely paralleling that of NR1, suggesting that most of the NR1 may be associated with neurites.

Expression of glutamate receptors as a function of time in culture. Cell lysates were made from E14 cortical neurons cultured for 3–11 d. Then the lysates were run on SDS-acrylamide gels and immunoblotted with the indicated anti-receptor antibodies. The same fraction of each culture dish was loaded per lane; the amount of protein per culture increased only ∼20% from day 7 to 11. Quantitation was accomplished by scanning the negatives.A, Top, mGluR1; mGluR2/3; mGluR5; NMDA NR1; A, Bottom, Actin. The experiments were repeated at least three times with similar results. B, Quantitation: ●, mGluR1; x, mGluR2/3; ○, mGluR5; ▵, NR1; ▿, actin, shown as a percentage of maximal expression.
In addition to ionotropic receptors, glutamate activates metabotropic receptors (mGluRs). mGluR activation has been associated with a variety of physiological processes, including protection from oxidative glutamate toxicity (Sagara and Schubert, 1998). Therefore, the expression of mGluRs 1, 3 and 4, and 5 was monitored by Western blotting in the same lysates as NR1 and actin. Figure 5 shows that all of these receptors are expressed in the cortical cultures but that their expression patterns vary. The expression of mGluRs 1 and 5 increases with time in culture until day 7, after which their expression declines. In contrast, the expression of mGluRs 2 and/or 3 increases with culture age in a manner similar to that of NR1 and actin.
Oxidative glutamate toxicity is a component of excitotoxicity
Oxidative glutamate toxicity is a well studied programmed cell death pathway that is independent of ionotropic glutamate receptors (Murphy et al., 1989; Maher and Davis, 1996; Li et al., 1997a,b; Tan et al., 1998a,b). If oxidative glutamate toxicity is a component of excitotoxicity, then it should be inhibited by reagents that selectively block oxidative glutamate toxicity, but not by AP-5. If a compound blocks the NMDA-mediated component in addition to oxidative glutamate toxicity, then the whole cascade would be inhibited because its initiation is dependent on NMDA receptor activation. Therefore, to determine whether oxidative glutamate toxicity is involved in the excitotoxicity pathway, a variety of components that inhibit oxidative glutamate toxicity but do not block excitotoxicity were screened for their ability to block the C fraction of the excitotoxicity cascade (see Fig. 3).
A defining characteristic of oxidative glutamate toxicity is that it is strongly inhibited by many antioxidants, including vitamin E (Murphy et al., 1989). To determine whether part of the C component shares this trait with oxidative glutamate toxicity, we preincubated 8-d-old cultures of cortical cells for 30 min with 100 μmα-tocopherol, followed by glutamate exposure and a 24 hr incubation with α-tocopherol ± AP-5. Figure6A shows that part of the C phase of cell death is blocked by α-tocopherol, whereas the viability of the cells exposed to glutamate in the absence of AP-5 is increased by the same amount. This increase in viability is expected in the absence of AP-5, because this condition contains both the NMDA receptor-independent and NMDA receptor-mediated components of glutamate toxicity. Because one-half of the cells survive at day 8 in the presence of α-tocopherol and because α-tocopherol has no effect on excitotoxicity at days 10 and 11 (data not shown), α-tocopherol must not block the NMDA receptor-mediated excitotoxicity component. Although these results are consistent with oxidative glutamate toxicity being a component of the excitotoxicity cascade, a number of other reagents known to inhibit oxidative glutamate toxicity were examined also. These include the group I metabotropic glutamate receptor (mGluR1) agonists and a caspase 1 inhibitor that has been shown previously to block oxidative glutamate toxicity (Tan et al., 1998a,b).

Conditions that block oxidative glutamate toxicity partially protect from excitotoxic-initiated damage.A, α-Tocopherol protects from cell death. Cells cultured for 8 d were pretreated for 30 min with 100 μm α-tocopherol (natural), exposed to 10 μm glutamate for 10 min, and then returned to the original medium ± AP-5, ± α-tocopherol. x, Glutamate alone; ○, glutamate plus α-tocopherol; ▵, glutamate plus 100 μm AP-5 after glutamate exposure; ■, glutamate plus α-tocopherol plus AP-5 after glutamate; ▿, cell viability at 24 hr after continuous exposure to glutamate plus 100 μm AP-5, 100 μm GYKI-25466, and 500 μm CNQX. Multiply glutamate concentration by 1000 (e.g., complete killing at 500 μm glutamate). B, C, Group I mGluR activation is protective. Cells were pretreated for 30 min with 100 μm mGluR agonists DHPG (B) or ACPD (C), followed by a 10 min exposure to the indicated concentrations of glutamate. Then the original culture medium was returned to the cells along with the mGluR reagents and, in some cases, 100 μm AP-5 to block downstream NMDA receptor activation. Cell viability was determined 24 hr later by the MTT assay. x, Glutamate alone; ▵, glutamate plus agonist; ○, glutamate plus AP-5 after glutamate exposure; ■, glutamate plus AP-5 after the added agonist. D, A caspase inhibitor Ac-YVAD-cmk protects cells. Cells were exposed to 30 μm Ac-YVAD-cmk for 30 min before exposure to 10 μm glutamate. In some cases 100 μm AP-5 was present throughout. x, Glutamate alone; ▵, glutamate plus caspase inhibitor; ○, glutamate plus AP-5; ■, glutamate plus AP-5 after the added caspase inhibitor.
The activation of group I mGluRs protects cells from oxidative glutamate toxicity via the activation of the inositol triphosphate pathway (Sagara and Schubert, 1998). If we use the same logic applied to the experiments with vitamin E, if oxidative glutamate toxicity is a component of excitotoxicity, then mGluR1 agonists should inhibit part of component C of the cascade. Figure 6, B and C, shows that two mGluR agonists, (R,S)-3,5-dihydroxyphenylglycine (DHPG) andtrans-1-amino-1S,3R-cyclopentane dicarboxylic acid (ACPD), both protect from excitotoxic-initiated glutamate damage in 8 d cultures. It also has been shown elsewhere that ACPD has a partial protective effect on NMDA-mediated excitotoxicity (Koh et al., 1991). Another agent that protects cortical neurons from oxidative glutamate toxicity is Ac-YVAD-cmk, a potent caspase inhibitor (Tan et al., 1998a,b). Figure 6Dshows that this inhibitor protects cells in the presence of AP-5 by ∼20%. These data again substantiate the involvement of oxidative glutamate toxicity as the cause of between 20 and 30% of the cell death in the excitotoxicity cascade.
The vitamin E, the mGluR agonist, and the caspase inhibitor data show that under certain conditions excitotoxicity can be divided into three components, one of which has the characteristics of oxidative glutamate toxicity. In older cultures (10–11 d) only 20% of the cell death is blocked by the late application of AP-5, and no cell death is blocked by the oxidative glutamate toxicity antagonists described above (see Fig. 4; data not shown). These data show that the oxidative glutamate toxicity component of excitotoxicity is transient in these cultures and strongly support the argument that vitamin E, DHPG, ACPD, and Ac-YVAD-cmk do not inhibit the NMDA receptor-mediated response. The transient nature of the oxidative glutamate toxicity response may be attributable to the fact that the NMDA receptor-mediated response is more efficient in older cultures because of higher receptor density (see Fig. 5) or the loss of cells that do not express NMDA receptors from the older cultures. This also would result in a larger fraction of the cells being killed by initial glutamate exposure.
Soluble glutamate mediates late cell death
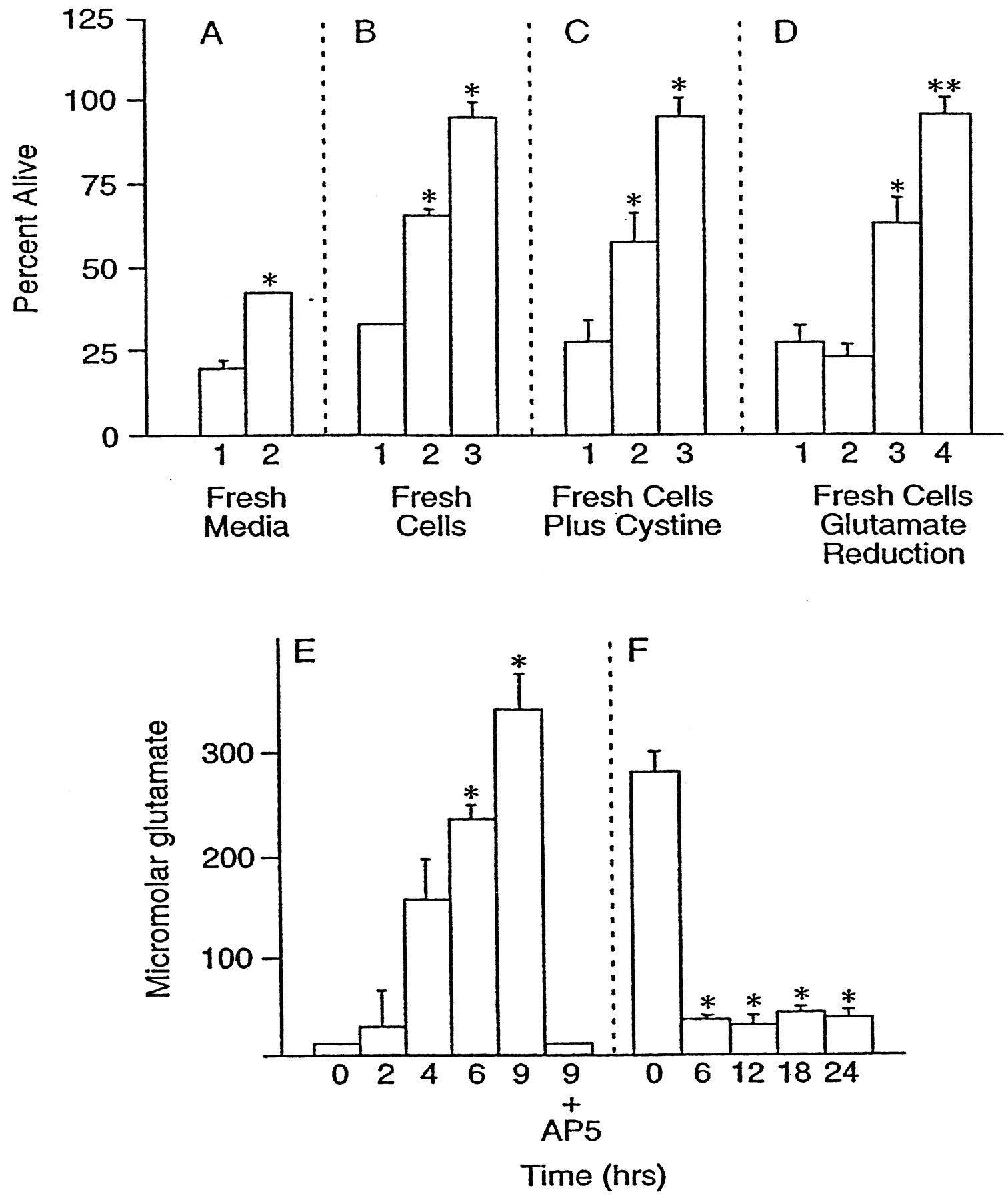
Because it is likely that the late cell death outlined above is attributable to glutamate, the amount of free glutamate in the culture medium was assayed as a function of time after the addition of 10 μm glutamate for 10 min. The amount of free glutamate increased from undetectable levels (<10 μm) to ∼300 μm over a period of 9 hr (Fig.7E). Glutamate (300 μm) is sufficient to inhibit extracellular cystine uptake completely, deplete intracellular GSH in clonal nerve cells (Sagara and Schubert, 1998), and kill >50% of the cells in this culture system via oxidative glutamate toxicity, as determined by the long-term exposure to glutamate in the presence of high concentrations of NMDA, AMPA, and kainate antagonists (see Fig. 6A, inverted triangles). These data clearly show that extracellular glutamate in these cultures can reach concentrations sufficient to cause damage via the oxidative glutamate toxicity pathway. The inclusion of 100 μm AP-5 during glutamate exposure completely blocked extracellular glutamate accumulation (Fig.7E). The glutamate concentrations are higher than previously reported in some cell culture systems (Strijbos et al., 1996), most probably because of the absence of astrocytes to remove free glutamate, but are very similar to those found in the culture media of lysed neurons (Newcomb et al., 1997).
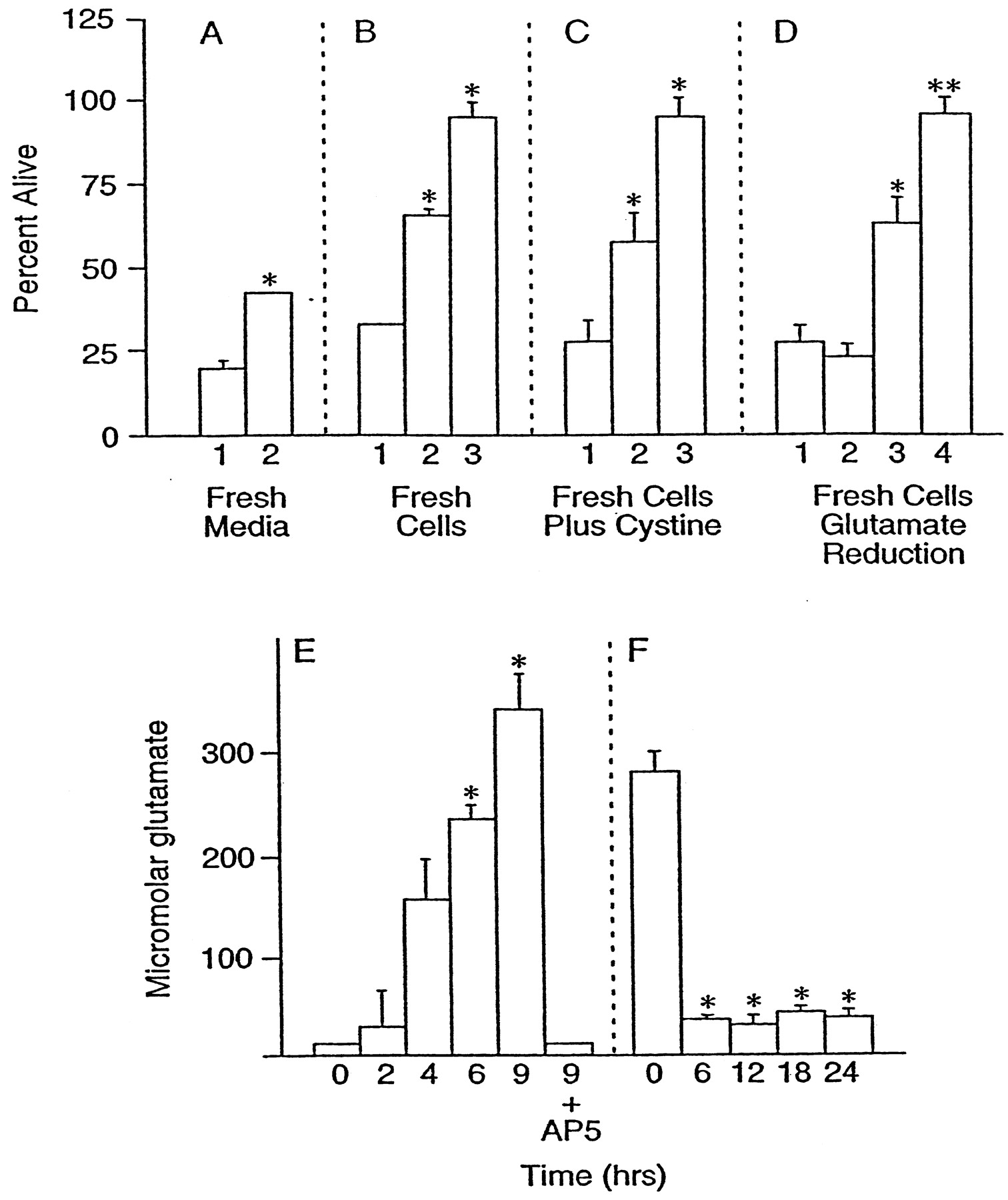
Toxicity is transferred by conditioned medium. Cell viability was measured after 24 hr in all cultures.A, Cells cultured for 8 d were exposed to 10 μm glutamate for 10 min, washed once, and returned to their original growth medium. After 11 hr either the cells were given fresh culture medium (2) or the medium was left undisturbed (1). B, In another experiment the medium was transferred to new cells of identical age in the absence (1) or presence (2) of 100 μm AP-5 or in the presence of AP-5 and 30 μm Ac-YVAD-cmk (3). C, Cells were exposed to growth-conditioned medium alone (1) or in the presence of 2 mm cystine (2) or in the presence of 2 mm cystine and 100 μm AP-5 (3). Because of the relative insolubility of cystine below pH 8, the experiments with cystine and all controls were performed at pH 8 by the reduction of incubator CO2.D, After 11 hr of glutamate exposure, growth-conditioned media were in some cases (2, 4) pretreated for 2 hr with GPT to reduce glutamate and then were transferred to fresh cells. 1, Growth-conditioned medium alone; 2, medium treated with GPT; 3, untreated medium plus AP-5; 4, glutamate-depleted medium plus AP-5. **Bar4 is significantly different from bar3,p < 0.01; n = 3.E, Concentration of glutamate in the growth-conditioned medium as a function of time after the addition of 10 μmglutamate to cultures. In one set of cultures 100 μm AP-5 was added before the addition of 10 μm glutamate (9 hr plus AP-5). F, Reduction of glutamate in the medium by GPT. GPT and cofactors were added to the growth-conditioned medium before application to the cells initially for 2 hr; then GPT was added repeatedly every 6 hr during the experiment to keep extracellular glutamate below 50 μm. *Significantly different from control (conditioned medium alone), p < 0.05;n = 3.
The above data show that there can be an ionotropic receptor-independent component of excitotoxicity, that the latter can be accounted for by the oxidative glutamate pathway, and that there is a high concentration of glutamate in media from lysed cells. If these conclusions are valid, then four additional criteria should be met. (1) Cells previously exposed to glutamate should be protected by replacing their conditioned medium with fresh medium. (2) It should be possible to transfer the late toxicity via the growth-conditioned medium. (3) Elevated exogenous cystine should reverse the inhibition by glutamate and protect cells. (4) The removal of glutamate from the conditioned medium should block downstream oxidative toxicity. Figure 7Ashows that conditioned medium replacement with fresh medium after 11 hr reduces subsequent cell death by ∼20% (Fig. 7A, bar 2). Figure 7B shows that ∼30% of the cell death caused by the transfer of 11 hr conditioned medium from cells treated for 10 min with 10 μm glutamate to fresh cells of the same age is blocked by AP-5 (Fig. 7B, bar 2) and that an additional 30% is blocked by the caspase inhibitor YVAD (Fig. 7B, bar 3).
Because the x
 antiporter is inhibited by glutamate in a competitive manner (Sato et al., 1999), it should be possible to reverse the effect of exogenous glutamate with cystine. By increasing the concentration of cystine in the culture medium (normally 260 μm) ∼10-fold, we have shown that cells were protected from conditioned medium by ∼30% (Fig. 7C, bar 2), with almost complete protection by a combination of cystine and AP-5 (Fig. 7C, bar 3). These data, in conjunction with those presented in Figure 6, strongly suggest that the oxidative glutamate toxicity pathway can kill some of the cells in excitotoxic pathways.
antiporter is inhibited by glutamate in a competitive manner (Sato et al., 1999), it should be possible to reverse the effect of exogenous glutamate with cystine. By increasing the concentration of cystine in the culture medium (normally 260 μm) ∼10-fold, we have shown that cells were protected from conditioned medium by ∼30% (Fig. 7C, bar 2), with almost complete protection by a combination of cystine and AP-5 (Fig. 7C, bar 3). These data, in conjunction with those presented in Figure 6, strongly suggest that the oxidative glutamate toxicity pathway can kill some of the cells in excitotoxic pathways.
Finally, exogenous glutamate was reduced in the growth-conditioned medium by treatment of the medium with 100 μg/ml GPT, 100 μm pyridoxal-l-phosphate, and 10 mm pyruvate (Matthews et al., 2000). Figure 7Fshows that GPT treatment reduced glutamate from 300 to ∼40 μm. In the absence of AP-5 there should be a level of cell death caused by reduced glutamate medium similar to conditioned medium alone, because the residual glutamate is sufficient to activate NMDA receptors and all downstream cell death pathways (Fig.7D, bars 1, 2). In the presence of AP-5, which blocks all NMDA receptor-mediated events, oxidative glutamate toxicity still should occur in the high glutamate medium. However, when extracellular glutamate is reduced to a level at which it can initiate NMDA receptor-mediated toxicity but not oxidative glutamate toxicity, all of the toxicity should be eliminated in the presence of AP-5. The data in Figure 7D again support a role for oxidative glutamate toxicity, for AP-5 only partially inhibits the cell death in high glutamate medium (Fig. 7D, bar 3), whereas there is 100% survival in glutamate-depleted medium plus AP-5 (Fig. 7D, bar 4).
DISCUSSION
The above data show that the excitotoxicity cascade can be divided experimentally into three discrete components, two requiring the activation of NMDA receptors. The initiation of the cell death pathway requires NMDA receptor activation, and a second NMDA receptor-dependent phase takes place after a brief exposure to low concentrations of glutamate. In contrast, a distinct form of cell death can occur after glutamate exposure that is independent of ionotropic glutamate receptors. This pathway, which constitutes 20–30% of the total cell death in 8–9 d cultures, has characteristics of oxidative glutamate toxicity, for it is inhibited specifically by vitamin E, by group I metabotropic receptor agonists, by a caspase inhibitor, by elevated extracellular cystine, and by the removal of extracellular glutamate. These data explain earlier observations showing that there is significant cell death in excitatory amino acid toxicity, ischemia, and CNS trauma, which is independent of ionotropic glutamate receptors (Meldrum and Garthwaite, 1990) (also see, for example, Choi, 1992).
In cultures of hippocampal neurons, approximately one-half of the cells can be rescued by applying NMDA antagonists after glutamate exposure (Rothman et al., 1987; Hartley and Choi, 1989; Manev et al., 1989). These data and those presented above show that there is an initial population of cells that is killed by glutamate exposure directly and another population that dies later because of the activation of NMDA receptors. The late receptor-mediated cell death could be attributable to either the requirement for a subset of NMDA receptors that respond to the higher concentrations of extracellular glutamate derived from cell lysis or have a requirement for more prolonged exposure to cell-derived glutamate. In our experiments, of the cells that cannot be rescued by the late application of AP-5, approximately one-half die by a process with the characteristics of oxidative glutamate toxicity. The other one-half die because of the initial exposure to glutamate and require NMDA receptor activation.
Previous studies have shown that the activation of different classes of ionotropic glutamate receptors is dependent on glutamate concentration, exposure time, and probably on the cell population. For example, unlike for NMDA, a brief exposure of cortical cells to AMPA and kainate produces little cell death, but exposure of the cells to these receptor agonists for hours produces extensive cell death (Choi et al., 1989;Frandsen et al., 1989). This may be because most AMPA/kainate receptors are relatively impermeable to Ca2+, requiring the activation of voltage-dependent Ca2+ channels for toxicity. In addition to exposure duration, AMPA/kainate receptor-mediated cell death is much slower, requiring many hours for cell lysis to occur (Choi, 1992;Carriedo et al., 1998), and these later forms of cell death have some characteristics of apoptosis (Choi and Rothman, 1990; Kure et al., 1991). However, because AMPA/kainate receptor antagonists have no effect in this culture system (see Table 1), even when added after glutamate exposure (data not shown), it is unlikely that these receptors play a role in the cell death that occurs after transient glutamate exposure. However, consistent with most of the published literature is the observation that some downstream cell death occurs by a mechanism that has many characteristics of programmed cell death, such as caspase activation (Tan et al., 1998a,b). This cell death pathway is oxidative glutamate toxicity.
Oxidative glutamate toxicity requires higher concentrations of glutamate than are necessary for NMDA receptor activation (Murphy et al., 1989). Figure 7 shows that concentrations of extracellular glutamate in the 200–300 μm range are present in cultured cells after initial excitotoxic cell lysis; these concentrations are sufficient to cause oxidative glutamate toxicity (see Fig. 6A). Similar concentrations of extracellular glutamate have been reported in culture media of lysed neurons (Newcomb et al., 1997) and in CNS trauma models (McAdoo et al., 1999). Because the culture medium contains 2 mmglutamine and nerve cells possess a very active enzyme, glutaminase, which converts glutamine to glutamate, initial nerve cell lysis releases this enzyme that, in the presence of abundant substrate, leads to an accumulation of glutamate in the culture medium (Newcomb et al., 1997). The brain also contains concentrations of glutamine between 2 and 4 mm, with 0.5 mm found in CSF (Matsumoto et al., 1996). Because this culture system lacks glial cells and many of the nerve cells are damaged rapidly, there is no effective way of removing glutamate. During ischemia, trauma, and other pro-oxidant conditions there is also likely to be a loss of high-affinity glutamate transporter function because these molecules are exquisitely sensitive to biological oxidants (for review, seeTrotti et al., 1998).
In oxidative glutamate toxicity, glutamate blocks the cystine/glutamate exchange system x
 , resulting in glutathione depletion and cell death (Murphy et al., 1989). The molecular basis of x
, resulting in glutathione depletion and cell death (Murphy et al., 1989). The molecular basis of x
 function has been described recently (Sato et al., 1999). The exchange systems consist of two proteins, the heavy chain of 4F2 (4F2hc) that is involved in several amino acid transport systems and a 502 amino acid protein called XCT. Both XCT and 4F2hc are highly expressed in the brain (Kanai et al., 1998; Sato et al., 1999). Because the cells of the CNS contain sequestered concentrations of free glutamate in the millimolar range (Coyle et al., 1981), as well as the ability to convert glutamine to glutamate, it is probable that any cellular dysfunction, such as loss of energy metabolism or cell lysis, would create local concentrations of glutamate sufficient to inhibit glutamate uptake and subsequent glutathione synthesis in nearby cells. The EC50 glutamate concentration for inhibiting cystine uptake is <100 μm (Sagara and Schubert, 1998), and ∼200 μm extracellular glutamate kills 50% of the cortical neurons used in the above experiments via oxidative glutamate toxicity (see Fig. 6A). This sequence of events could lead to cell injury or death in an autocatalytic manner, resulting in a gradient of injury radiating from the site of the initial event. In addition, oxidative glutamate toxicity can generate even greater damage than excitotoxicity, because neurons lacking ionotropic glutamate receptors are killed also. It is therefore of importance to understand how x
function has been described recently (Sato et al., 1999). The exchange systems consist of two proteins, the heavy chain of 4F2 (4F2hc) that is involved in several amino acid transport systems and a 502 amino acid protein called XCT. Both XCT and 4F2hc are highly expressed in the brain (Kanai et al., 1998; Sato et al., 1999). Because the cells of the CNS contain sequestered concentrations of free glutamate in the millimolar range (Coyle et al., 1981), as well as the ability to convert glutamine to glutamate, it is probable that any cellular dysfunction, such as loss of energy metabolism or cell lysis, would create local concentrations of glutamate sufficient to inhibit glutamate uptake and subsequent glutathione synthesis in nearby cells. The EC50 glutamate concentration for inhibiting cystine uptake is <100 μm (Sagara and Schubert, 1998), and ∼200 μm extracellular glutamate kills 50% of the cortical neurons used in the above experiments via oxidative glutamate toxicity (see Fig. 6A). This sequence of events could lead to cell injury or death in an autocatalytic manner, resulting in a gradient of injury radiating from the site of the initial event. In addition, oxidative glutamate toxicity can generate even greater damage than excitotoxicity, because neurons lacking ionotropic glutamate receptors are killed also. It is therefore of importance to understand how x
 is regulated in the brain as well as how oxidative glutamate toxicity kills neurons.
is regulated in the brain as well as how oxidative glutamate toxicity kills neurons.
Footnotes
This work was supported by grants from the National Institutes of Health and Department of Defense Grant DAMD17-99-1-9562. We thank Dr. Pamela Maher for her critical review of this manuscript, Drs. John Donello and Steve Smith for the glutamate assays, and Dr. Rona Giffard for the helpful discussions on cell culture.
Correspondence should be addressed to Dr. David Schubert, The Salk Institute for Biological Studies, 10010 North Torrey Pines Road, La Jolla, CA 92037. E-mail: schubert{at}salk.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}