

Abstract
Although nitrous oxide (N2O) has been used to facilitate surgery for >150 years, its molecular mechanism of action is not yet defined. Having established that N2O-induced release of norepinephrine mediates the analgesic action at α2 adrenoceptors in the spinal cord, we now investigated whether activation of noradrenergic nuclei in the brainstem is responsible for this analgesic action and which α2adrenoceptor subtype mediates this property. In rats, Fos immunoreactivity was examined in brainstem noradrenergic nuclei after exposure to nitrous oxide. After selective lesioning of noradrenergic nuclei by intracerebroventricular application of the mitochondrial toxin saporin, coupled to the antibody directed against dopamine β hydroxylase (DβH–saporin), the analgesic and sedative actions of N2O were determined. Null mice for each of the three α2 adrenoceptor subtypes (α2A, α2B, and α2C), and their wild-type cohorts, were tested for their antinociceptive and sedative response to N2O. Exposure to N2O increased expression of Fos immunoreactivity in each of the pontine noradrenergic nuclei (A5, locus coeruleus, and A7). DβH–saporin treatment eliminated nearly all of the catecholamine-containing neurons in the pons and blocked the analgesic but not the sedative effects of N2O. Null mice for the α2B adrenoceptor subtype exhibited a reduced or absent analgesic response to N2O, but their sedative response to N2O was intact. Our results support a pivotal role for noradrenergic pontine nuclei and α2B adrenoceptors in the analgesic, but not the sedative effects of N2O. Previously we demonstrated that the analgesic actions of α2 adrenoceptor agonists are mediated by the α2A subtype; taken together with these data we propose that exogenous and endogenous α2adrenoceptor ligands activate different α2 adrenoceptor subtypes to produce their analgesic action.
Nitrous oxide (N2O) has been used to provide anesthetic conditions suitable for the performance of surgery for >150 years. Because it is relatively impotent, it cannot be used as a sole anesthetic agent except when administered under hyperbaric conditions; therefore, it is most often used as an adjunctive general anesthetic agent for surgical procedures.
The state of general anesthesia encompasses a syndrome of “behaviors” including analgesia, hypnosis–sedation, amnesia, and muscle relaxation. We have investigated the mechanisms for N2O analgesic effect (it is more accurate to refer to its antinociceptive effect when studies are performed in animals who are unable to communicate the emotive experience of pain) and demonstrated that intrathecally, but not supraspinally, administered α2 antagonists could block the antinociceptive action of N2O, thereby localizing the site of noradrenergic action to components within the spinal cord (Guo et al., 1996).
It is not clear whether the spinally transduced antinociceptive action of N2O originates in supraspinally located noradrenergic neurons. Noradrenergic projections to all regions of the spinal cord arise almost entirely from the dorsolateral pontine catecholamine cell groups A5, the locus coeruleus (LC), and A7. Electrical or chemical stimulation in the dorsolateral pons produces analgesic effects mediated by spinal α2adrenoceptors that can be differentiated from cardiovascular effects, and such stimulation causes inhibition of nociceptive neurons in the deep dorsal horn (Byrum et al., 1984; Kingery et al., 1997; Willis and Westlund, 1997). We found that transection of the spinal cord eliminated the antinociceptive property of N2O, suggesting that spinal pathways were involved (Zhang et al., 1999). Furthermore, exposure to N2O provoked release of norepinephrine at the level of the dorsal horn of the spinal cord, and when this neurotransmitter was depleted, N2O was no longer able to produce antinociception (Zhang et al., 1999). Therefore, we proposed that N2O could be activating a descending noradrenergic pathway, which stimulates α2 adrenoceptors in the spinal cord through the released norepinephrine. Now, using indirect measures of neuronal activation and toxins targeted for norepinephrine-containing neurons, we elucidate the role played by the noradrenergic nuclei in the brainstem for the antinociceptive action of N2O.
There are three α2 adrenoceptor subtypes (α2A, α2B, and α2C), and each of the genes for these receptor subtypes has been cloned (Bylund et al., 1994), thereby facilitating the creation of genetically modified reagents with dysfunctional (“point mutation”) or deficient (“knock-out”) subtypes. Using a moderately selective pharmacological probe in rats, as well as the D79N mouse, which has a point mutation of the α2Aadrenoceptor gene causing its dysfunction, we showed that the α2A subtype was not responsible for the antinociceptive effect of N2O (Guo et al., 1999). Using null mice for α2A, α2B, and α2C subtypes we now reveal the precise subtype involved in mediating the antinociceptive action of N2O. This appears to differ from the mechanism involved in the antinociceptive response to exogenously administered α2 adrenergic agonists.
Surprisingly, the noradrenergic mechanisms we identify as mediators of N2O antinociception are not instrumental in the hypnotic–sedative action of N2O and therefore challenge the notion that the same anesthetic produces its “continuum of effects” at a single site of action.
MATERIALS AND METHODS
Animals
These experiments were reviewed and approved by our institute's Subcommittee on Animal Studies and were in accordance with the provisions of the Animal Welfare Act, the Public Health Service Guide for the Care and Use of Laboratory Animals, and Veterans Affairs Policy. All immunolesioning and immunohistochemistry experiments were performed in adult male Sprague Dawley rats (240–260 gm) from B & K Universal (Fremont, CA). Additional behavioral studies were performed in adult (20–30 gm) male mice. Various genetically engineered mice strains were examined, including: (1) D79N mice with a nonfunctioning α2A adrenoceptor because of a point mutation in its gene substituting aspartic acid by asparagine at amino acid residue #79, (2) α2A−/− null mice with a knock-out of the α2A adrenoceptor gene, (3) α2C−/− null mice with a knock-out of the α2C adrenoceptor gene, and (4) their wild-type (WT) controls. All these strains were on a congenic C57BL/6J background. The α2B −/− null mice had a knock-out of the α2B adrenoceptor gene on a hybrid C57BL/6J and 129SvJ background; as their controls we used generationally matched wild-type mice on the same hybrid background (C57BL/6J × 129SvJ). Production of the α2A−/−, α2B-/, α2C−/−, and D79N mice have been described previously (Link et al., 1996; MacMillan et al., 1996; Altman et al., 1999). Rats and mice were housed in a temperature- and humidity-controlled environment and were maintained on a 12 hr light/dark cycle. Food and water were available ad libitum.
Drugs
The anti-dopamine β-hydroxylase–saporin (DβH–saporin) immunotoxin was produced by conjugating saporin, a ribosome-inactivating protein, with a mouse monoclonal antibody for DβH (Advanced Targeting Systems, San Diego, CA). DβH, a key enzyme for the synthesis of norepinephrine, is only present in noradrenergic and adrenergic neurons. The membrane-bound subunit of DβH is exposed during exocytotic release of norepinephrine, allowing the anti-DβH antibody to attach and to be endocytosed. When the DβH–saporin conjugate is injected intracerebroventricularly in rats, it is taken up in the axon terminals of catecholaminergic neurons, undergoing retrograde transport to the neuronal cell bodies where it arrests protein synthesis. This technique gradually destroys the noradrenergic neurons in the locus coeruleus and in the A5 and A7 brainstem nuclei, taking 14 d to complete the selective neurolytic process (Rohde and Basbaum, 1998; Martin et al., 1999). Once the neuronal cell bodies are destroyed, the lesion is permanent (Wrenn et al., 1996). The DβH–saporin immunotoxin was diluted in saline (1 μg/μl).
Behavioral testing
All behavioral testing was performed in a blinded manner, and the experimental groups were mixed together during each testing session to ensure identical gas exposure conditions. Using a heating blanket, the tail and paw temperatures were maintained within 0.5°C of 32°C. A K-type fine wire contact thermistor (5SC-66-K-30–36) and a multimeter thermometer (HHM25; Omega, Bridgeport, NJ) were used to measure the plantar paw surface and volar tail surface temperatures for each baseline and N2O exposure nociceptive threshold test. During the 30 min exposure to N2O, both rats and mice exhibited increased motor activity. This made tail-flick testing more difficult during N2O exposure, but using patience and care to calm the animals before testing we could confirm that the animals were withdrawing from the noxious heat stimulus during the tail-flick assay and not simply spontaneously moving their tails.
Tail-flick latencies were determined from the mean of three (in rats) or two (in mice) consecutive latencies using a tail-flick apparatus (Columbus Instruments, Columbus, OH). The interstimulus interval was ∼60–90 sec. A high-intensity light was focused on the ventral surface of the tail, and the time for the animal to move its tail from the light beam was recorded. A different patch of the middle (rat) or distal (mice) third of the tail was exposed to the light beam each time to minimize the risk of tissue damage. The same light stimulus intensity was used for all experiments in a given strain, having been preset at an intensity that elicited a mean latency of 2.9–3.2 sec in room air. To avoid the possibility of tissue damage, a cutoff time of 10 sec was used; if no response had occurred by this time, a value of 10 sec was assigned to the experimental subject. The tail-flick latency in both rats and mice was performed, whereas the animal was gently held under a towel. The animals were trained to stay under the towel, which was tented up under the investigator's hand like a cloth burrow. Latency measurements were taken only when the rat or mouse was calmly resting with tail protruding from underneath the towel.
Hot-plate latencies were determined from the mean of two consecutive latencies on a hot-plate device set at a constant 55°C surface temperature (Columbus Instruments). The latency was determined when the mouse first licked a hindpaw, and the cutoff time for this test was 30 sec. No mouse was used for more than one test session on the hot-plate assay, because repeated test sessions result in frequent jumping behavior, sometimes within seconds of being placed on the hot plate.
The loss of righting reflex (LORR) was measured by placing the rat on its back and determining if the animal could right itself. The calculation of the ED50 for LORR was determined for each rat, based on interpolation of the gas concentrations that bracketed the righting response (Koblin et al., 1979). LORR testing was performed inside a large (3.66-m-diameter, 4.58-m-height) clinical hyperbaric chamber, with the entire Plexiglas gas exposure chamber and the investigator inside the hyperbaric chamber.
For the measurement of the sedation response, mice were placed on a rotarod (IITC; Life Sciences Instruments, Woodland Hills, CA) turning at 10 rpm (Lakhlani et al., 1997). The mice learned to remain on the rod for 60 sec during the course of three training sessions. Drug-naive D79N, α2A−/−, α2B−/−, α2C−/− and their respective wild-type mice were indistinguishable in their ability to remain on the rod. Each mouse was tested once for its ability to remain on the rod at one of three exposure conditions [air, 35%, and 70% atmospheres absolute (ATA) N2O]. The cutoff time was 60 sec.
Normobaric gas exposures
Behavioral studies were performed in a Plexiglas chamber (91-cm-long, 48-cm-wide, and 38-cm-high) with a sliding door for insertion of the animals. The investigator's forearms could be inserted through two circular openings on the side of the chamber, which were sealed with rubber flap iris diaphragm air seals. The chamber was large enough to contain the tail-flick and hot-plate devices. Antinociceptive testing was always performed after 30 min N2O exposure because we had previously demonstrated a maximal antinociceptive effect in mice and rats at this time interval (Guo et al., 1999; Fender et al., 2000).
Fresh gas flow (rate varied between 3 and 10 l/min) was introduced into the chambers via an inflow port; a fan was used to achieve adequate mixing within the chamber, and gases were purged by vacuum set to aspirate at the same rate as the fresh gas inflow. Oxygen concentration in the chamber was maintained between 25 and 30% ATA, and the N2O concentration was maintained at 70% ATA. Control exposure was with room air. An airway gas monitor (model 254; Datex, Helsinki, Finland) was used to continuously monitor the concentrations of N2O, oxygen, and carbon dioxide in the chamber, and flow rates were adjusted to maintain the desired concentrations. Temperature in the chamber was controlled by a heating blanket, and the tail and paw temperatures were monitored before each behavioral test.
Hyperbaric gas exposures
During hyperbaric studies, the gas mix inside the gas chamber was kept at 70% v/v of N2O and 30% v/v of oxygen. The chamber pressure was increased in 0.3 atmosphere increments to generate 91, 112, and 133% ATA of N2O over a 90 min testing session. After a 15 min exposure at each N2O% ATA the LORR testing was performed. Rectal temperature was monitored and maintained within 0.5°C of 36.5°C with a heating pad.
Immunohistochemistry
After 90 min exposure to 70% ATA N2O, rats were anesthetized with pentobarbital (100 mg/kg, i.p.) and transcardially perfused with 100 ml of sodium PBS 0.1m, followed by 500 ml of paraformaldehyde 4% in sodium phosphate buffer (PB) 0.1 m. After decapitation, the whole brain was removed and submerged in the same fixative for 4 hr. Tissues were then stored in 30% sucrose solution in 0.1 m PB at 4°C overnight for cryoprotection. The brainstem was sliced into 40-μm-thick sections with a cryotome (CM1800; Leica, Heidelberg, Germany) at −15°C. Every third section of the brainstem (from caudal periaqueductal gray to rostral medulla) was retained and placed in the PB solution.
Sections were stained using antibodies for Fos and tyrosine hydroxylase (TH; a catecholamine-synthesizing enzyme) in sequence as follows. Sections were first incubated for 1 hr in blocking solution (3% normal goat serum and 0.3% Triton X-100 in PBS) and then incubated overnight with rabbit Fos antibody (1:20,000; Santa Cruz Biotechnology, Santa Cruz, CA) diluted in 1% normal goat serum and 0.3% Triton X-100 in PBS (buffer 1). After vigorous rinsing in buffer 1, sections were incubated for 1 hr in biotinylated goat anti-rabbit Ig (1:200; Chemicon, Temecula, CA) in buffer 1. Sections were vigorously rinsed with 0.3% Triton X-100 in 0.1 m PBS (buffer 2), then incubated for 1 hr in avidin–biotin–peroxidase complex (Vectra Elite ABC; Vector Laboratories, Burlingame, CA) in buffer 2. Visualization of the reaction product was achieved by incubation for 4 min with diaminobenzidine (DAB) and nickel-ammonium sulfate to which hydrogen peroxide was added (DAB kit; Vector Laboratories). Sections were rinsed in PB and subsequently reacted with rabbit TH antibody (1:50,000; Chemicon) by the same procedures as for Fos immunostaining, except that DAB reaction was performed for 2 min without nickel-ammonium sulfate. All the incubations were performed at room temperature. After these staining procedures, the sections were rinsed in water and placed on a slide glass. The sections were dehydrated in 100% ethanol, cleared in 100% xylene, and covered.
For those sections subjected only to TH staining, sections were exposed to rabbit TH antibody (1:20,000) and reacted with DAB–nickel solution for 4 min.
Quantitation
The sections were examined by light microscopy. The Fos-positive neurons were identified by dense black staining of the nucleus. The TH-positive neurons were identified by yellow–orange staining of the cytoplasm. The brainstem regions were located according to a rat brain atlas (Paxinos and Watson, 1986). The A5, LC, and A7 noradrenergic neurons were easily identified by TH staining of the cytoplasm. For each region, all the Fos-positive nuclei in TH-positive cytoplasm were counted for each section, and the four sections with the highest counts were pooled, giving a count that was the sum of all the double-stained neurons in those four sections. At the time of quantitation, the investigator performing the Fos counting was unaware of the origin of the sections.
In the DβH–saporin study, TH-positive neurons in the A5, LC, and A7 nuclei were counted and totaled for all sections for each nucleus in each rat. Again, the investigator performing the counting was blinded as to treatment.
Intracerebroventricular injections
Rats were anesthetized with intraperitoneal injection of pentobarbital (50 mg/kg). While the skull was fixed in a stereotaxic apparatus, the animal was injected with DβH–saporin (3 μg/3 μl) or saline (3 μl) into the lateral cerebral ventricle according to coordinates obtained from Paxinos and Watson (1986) (anteroposterior, −1.0 mm; lateral, −1.5 mm from bregma; dorsoventral, −4.3 mm from skull surface) (Paxinos and Watson, 1986). The location of the injection site was confirmed in a pilot study by direct visualization of Evans blue dye injected into the lateral ventricle using these coordinates. The intracerebroventricular injections were performed with a 27 gauge needle connected via PE-20 catheter (25-cm-long) to a 10 μl Hamilton syringe that was manually injected. A small air bubble (1 μl) separated the injectant from the saline-filled catheter to visualize the injection. The injection of 3 μl of immunotoxin or saline was performed over 2 min, and the needle was left in place for an additional 10 min after the injection to avoid reflux.
RT-PCR
Total RNAs were isolated from the kidney, spinal cord, and heart of the wild-type mouse (Chomczynski and Sacchi, 1987). For each sample, total RNA concentration was assessed by measuring the absorption at 260–280 nm with a spectrophotometer. The quality of total RNA for each sample was examined by fractionating 10 mg of total RNA stained with ethidium bromide in formaldehyde/1% agarose gel by electrophoresis to confirm clear bands for 28s and 18s ribosomal RNAs. Total RNA (2 mg each) from each sample was reverse-transcribed in 20 ml of solution consisting of 2.5 μm random primer, 5 mmMgCl2, 1× PCR buffer, 1 mm of dNTP, 1 U of RNase inhibitor, and 2.5 U of reverse transcriptase. The reaction conditions were 42°C for 15 min, 99°C for 5 min, and 5°C for 5 min. The products were then subjected to PCR using 15 μm backward primer (5′CTGGAAGCCAAGATGTACCAGG 3′) and 15 μm forward primer (5′TCATCCTCTTCACCATTTTCGG 3′) in 100 ml of a solution consisting of 2 mmMgCl2, 1× PCR buffer, and 2.5 U of AmpliTaq polymerase. The expected PCR product size was 466 nucleotide pairs. The PCR conditions were: 1 cycle of 95°C for 2 min, 35 cycles of 95°C for 1 min, 60°C for 1 min, and 72°C for 30 sec, terminated by 1 cycle of 72°C for 7 min. PCR products (16 ml each) were then fractionated by 1% agarose gel (with ethidium bromide) electrophoresis, and Polaroid pictures were taken on a UV transilluminator.
Statistical analysis
All data are presented as the mean ± the SEM, and differences are considered significant with a p value < 0.05. The immunohistochemistry (Fos and TH staining) quantitation data were analyzed using unpaired t tests. The tail-flick latencies were compared using paired (air vs N2O) and unpaired (saline vs DβH–saporin) t tests. The LORR ED50 values were compared between the saline and DβH–saporin-treated rats using the Mann–Whitney U test. A one-way ANOVA was performed on the hot-plate latencies when comparing groups of mice, and a t test was used to test for contrasts. Figures comparing N2O analgesic effects between groups of mice or rats show the mean latency changes as the percentage of the maximum possible effect (%MPE): %MPE = ([post-gas latency − baseline latency]/[cutoff latency − baseline latency]) × 100. The cutoff latencies were 10 sec on tail-flick and 30 sec on hot-plate.
Experimental protocols
N2O and Fos. We used Fos and TH double-staining in the brainstem to determine whether N2O exposure activated the catecholaminergic neurons in the A5, LC, and A7 nuclei. Before the Fos experiment, the rats were habituated to the experimental conditions to minimize background Fos expression induced by the stimulus of a novel environment. For 7 consecutive days the rats were taken to the laboratory and individually placed in the Plexiglas test chamber (open to room air) for 90 min. Gas exposures of N2O or air were performed on rats individually housed in an airtight cylindrical Plexiglas chamber (20 cm in diameter, 30 cm in height) for 90 min.
Noradrenergic lesioning and N2O analgesia. To determine whether the noradrenergic brainstem neurons mediated N2O analgesia, DβH–saporin was injected to destroy the noradrenergic neurons in the brain. Rats were injected with the immunotoxin DβH–saporin (3 μg/3 μl) or with saline intracerebroventricularly (n = 14 for each group). Fifteen days after the intracerebroventricular injection, the analgesic effect of N2O was evaluated by measuring the tail-flick latency before and during N2O exposure. After the tail-flick measurement, the rat was anesthetized, and brain was harvested and single-stained for TH to evaluate the efficacy of noradrenergic lesioning.
Noradrenergic lesioning and N2O hypnosis. We used another group of rats to determine whether the noradrenergic brainstem neurons mediated N2O hypnosis. Rats were injected with the immunotoxin DβH–saporin (3 μg/3 μl) or with saline intracerebroventricularly (n = 14 for each group). Fifteen days after the intracerebroventricular injection, the sedative effect of N2O was evaluated by measuring the LORR ED50 in a hyperbaric chamber.
N2O antinociception in α2 adrenoceptor subtype-deficient mice. In a pilot study we were unable to demonstrate robust N2O antinociception for the tail-flick assay in the C57BL/6J WT mice used as controls for the D79N, α2A−/−, and α2C−/− mice. The α2B−/− knock-out mice were on a different genetic background (C57BL/6J × 129 SvJ hybrid); in pilot studies their genetically matched WT controls consistently exhibited significant N2O antinociception in the tail-flick assay. Therefore the antinociceptive action of N2O on tail-flick latencies was compared in the C57BL/6J × 129 SvJ WT and the α2B−/− knock-out mice. Significant N2O antinociception was evident in the hot-plate assay in both the C57BL/6J and the C57BL/6J × 129 SvJ WT controls. Hot-plate testing was performed in α2A−/−, α2B−/−, α2C−/−, and D79N mice, and in their respective WT controls. Baseline latencies were determined in the gas chamber under room air conditions, thereafter the mice were removed from the chamber, which was then equilibrated with the test gas mixture (70% ATA of N2O). After 30 min, the mice were placed back in the gas chamber and exposed to the test gas mixture for 30 min. The nociceptive testing was then repeated.
N2O sedation in α2 adrenoceptor subtype knock-out mice. To determine whether α2 adrenoceptor subtype contributes to N2O sedation, we used the rotarod latency assay. Rotarod testing was performed in α2A−/−, α2B−/−, α2C−/−, and D79N mice and in their respective WT controls. Baseline latencies were determined in the gas chamber under room air conditions; thereafter the mice were removed from the chamber, which was then equilibrated with the test gas mixture (either 35 or 70% ATA of N2O). After 30 min, the mice were placed back in the gas chamber and exposed to the test gas mixture for 30 min. The rotarod testing was then repeated.
RESULTS
N2O-activated catecholaminergic neurons in the A5, LC, and A7 nuclei
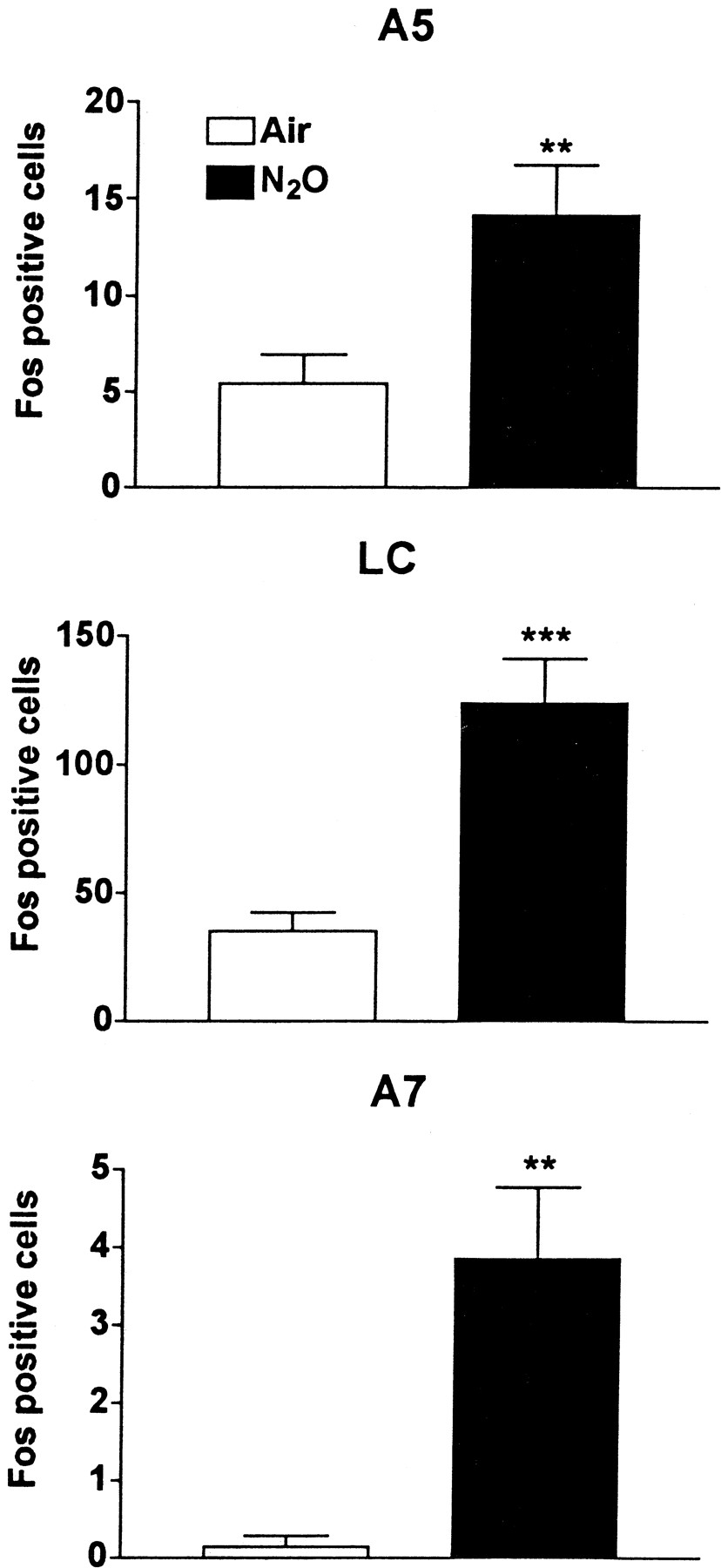
Rats exposed to 70% ATA of N2O exhibited a robust increase in Fos immunoreactivity in the TH-stained neurons in the A5, LC, and A7 brainstem nuclei (Fig.1). Representative Fos staining in each noradrenergic nucleus is shown after room air or N2O exposure (Figs.2, 3,4, 5). These data demonstrate that N2O can activate the noradrenergic pontine cell groups that send ascending and descending projections into the brain and spinal cord, respectively (Figs. 3,5).

Effect of N2O on Fos induction in the neurons of the A5, LC, and A7 noradrenergic nuclei. Rats were exposed for 90 min to 70% ATA N2O (n = 7) or room air (n = 7), then transcardially perfused, and the brain was removed. Pontine sections were stained for Fos and TH. The double-stained neurons in each region were counted section by section, and the four sections with the highest counts were summed for each rat. Nitrous oxide exposure dramatically increased Fos expression in the A5 (14 ± 3 vs 5 ± 1 in air), LC (124 ± 17 vs 35 ± 7 in air), and A7 (5 ± 1 vs 0.1 ± 0.1 in air) noradrenergic neurons. **p < 0.01; ***p < 0.001 versus air.

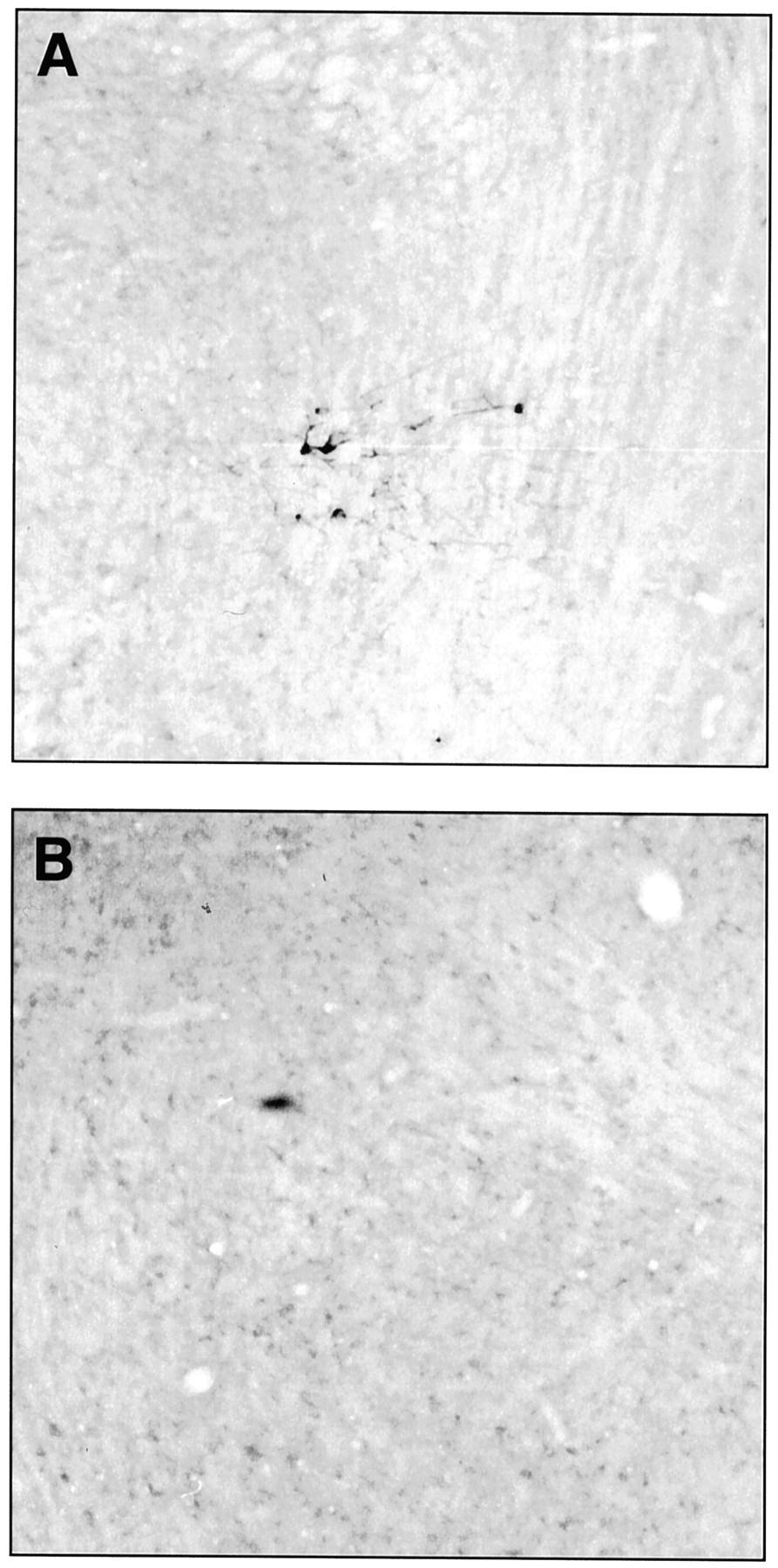
TH (light brown) and Fos (blue) staining in A5 neurons. A,Immunohistochemistry after exposure to air. B,Arrow points to double-stained neuron in A5 cell group of rat exposed to N2O. Note the increase in double-labeled neurons with N2O exposure.

TH (light brown) and Fos (blue) staining in LC noradrenergic neurons.A, Representative section in rat exposed to air.B, Marked increase in double-stained neurons after N2O exposure. Also note the numerous Fos-positive nuclei in the LC region in TH-negative cells.

TH (light brown) and Fos (blue) staining in LC noradrenergic neurons. A Fos-positive nucleus is clearly seen in a TH-positive LC neuron (arrow).

TH (light brown) and Fos (blue) staining in A7 sections. A,Representative section in rat exposed to air. B, Marked increase in double-stained neurons after N2O exposure. Also note the numerous Fos-positive nuclei in TH-negative cells.
DβH–saporin treatment destroyed pontine noradrenergic neurons and blocked the analgesic but not the sedative effects of N2O
Using TH to label catecholaminergic neurons in the brainstem, we examined the effect of DβH–saporin immunolesioning on the pontine noradrenergic cell groups. After DβH–saporin treatment, the TH-positive noradrenergic neurons in locus coeruleus completely disappeared; however, some residual staining was observed in the A5 and the A7 noradrenergic neurons (Figs. 6,7, 8,9).

Immunolesioning of brainstem noradrenergic neurons with intracerebroventricular DβH–saporin. Rats were injected with saline (3 μl) or DβH–saporin (3 μg/3 μl; n= 14 for each group). Two weeks after injection the rats underwent behavioral testing and then were transcardially perfused, and the brain was removed. The brainstem sections were stained for TH, and the number of TH-positive neurons in A5 and A7 were totaled for all sections. Data were standardized by the mean of the saline group. Approximately 30% of noradrenergic neurons survived in A5 (31 ± 6%) and A7 (28 ± 6%) after lesioning. Counting was not performed for the LC region because TH-positive neurons are densely packed and are difficult to count in the LC. Furthermore, not a single neuron survived in LC region after DβH–saporin treatment. ***p < 0.001 versus saline-treated.

Immunohistochemistry of noradrenergic neurons in the A5 cell group. A, TH of a saline-treated rat.B, Profound reduction in TH-stained neurons in the immunolesioned rat.

Immunohistochemistry of noradrenergic neurons in the LC. A, Very dense TH staining is observed in the LC region of the saline-treated rat. B, No immunoreactivity is observed after DβH–saporin treatment.

Immunohistochemistry of noradrenergic neurons in the A7 cell group. A, TH immunohistochemistry in the A7 cell group of a saline-treated rat. B, Note the absence of TH-stained neurons in the immunolesioned rat.
Whereas there was no significant difference in baseline tail-flick latencies between control and immunolesioned rats (Fig.10A), the tail-flick latency was significantly prolonged only in control rats after N2O exposure; DβH–saporin-treated rats exhibited no antinociceptive response to N2O. These data support the hypothesis that activation of noradrenergic pontine neurons mediates the antinociceptive response to N2O.

Effect of noradrenergic lesioning on the antinociceptive and sedative effects of nitrous oxide.A, Tail-flick latency was measured before (air) and after N2O exposure. There was no significant difference in baseline latencies of the saline (n = 11)- and DβH–saporin (n = 12)-treated rats, using the same intensity setting for the radiant heat source. The N2O antinociceptive effect was observed in the saline treatment group (27.7 ± 6.1%MPE), but not in the DβH–saporin-treated rats (1.0 ± 6.7%MPE). Three rats in the control group and two rats in the LC lesion group were excluded from the tail-flick data because of an excessive change in tail temperature after N2O exposure.B, There was no difference between saline (n = 10; 112 ± 3%)- and DβH–saporin (n = 10; 116 ± 3%)-treated rats in the sedative effect of N2O, as measured by the concentration (ATA), which prevented a righting reflex in half the rats (ED50 LORR). *p < 0.05 versus air; ###p < 0.001 versus saline.
Loss of righting reflex was used to evaluate the sedative properties of N2O in the control and immunolesioned rats. No difference was observed in the LORR ED50 values of the control and the DβH–saporin-treated rats, indicating that activation of these noradrenergic neurons do not mediate the sedative properties of N2O (Fig.10B).
The α2B adrenoceptor subtype was required for N2O analgesia but not for N2O-evoked sedation
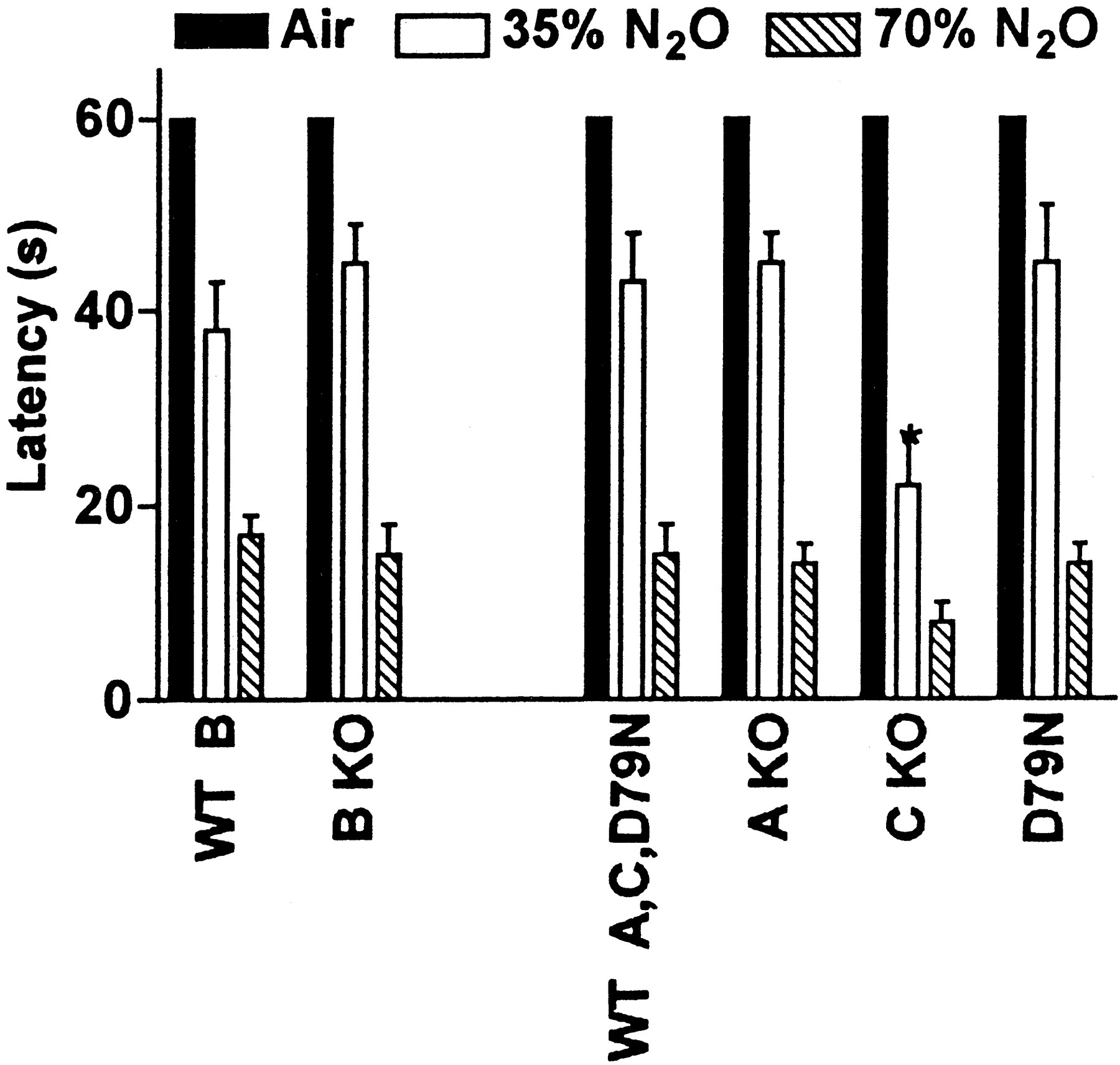
The antinociceptive effects of N2O with the hot-plate assay in the α2A−/−, α2B−/−, α2C−/−, and D79N mice, and in their respective WT controls are shown in Figure11A. The N2O response for the hot-plate assay was reduced by 65% in the α2B−/− mice (vs WT controls), suggesting that the α2B adrenoceptor subtype mediates N2O antinociception for this behaviorally complex hindpaw-licking response to nocifensive heat. Furthermore, N2O had no antinociceptive effect on the tail-flick assay in the α2B−/− mice, indicating that the α2B adrenoceptor subtype also mediates N2O analgesia for this nociceptive spinal reflex (Fig. 11B).

A, N2O antinociceptive effect on hot-plate assay was reduced in α2BKO. This figure illustrates the antinociceptive effects of N2O on the hot-plate assay in the α2A−/−, α2C−/− knock-out mice (α2AKO and α2CKO), the D79N mutant mice with nonfunctional α2A adrenoceptors, and in their genetically matched WT controls (on a C57BL/6J congenic background). The α2B−/− (α2BKO) mice were on a different genetic background (C57BL/6J × 129SvJ hybrid), so they required their own genetically matched WT controls (WT for α2B). There were no differences in baseline hot-plate latencies between the various mouse strains (data not shown). Only the α2BKO mice had reduced N2O antinociception on the hot-plate assay (reduced 65%, 24 ± 9 vs 69 ± 9%MPE in WT for α2B), indicating that the α2B adrenoceptor subtype mediates N2O antinociception for this supraspinal response. The N2O antinociceptive responses (%MPE) in the other knock-out and mutant mouse strains were: WT, 62 ± 10; α2AKO, 62 ± 11; α2CKO, 49 ± 18; and D79N, 72 ± 9 (n = 16 for each group). B,N2O antinociceptive effect on tail-flick assay was lost in α2BKO. There were no differences in baseline tail-flick latencies of the α2BKO mice and their WT controls. The WT for α2B mice had a significant N2O antinociceptive effect on the tail-flick assay (14.7 ± 6.2%MPE), but the α2BKO mice had no N2O antinociceptive response on the tail-flick assay (1.3 ± 2.1%MPE), indicating that the α2B adrenoceptor subtype mediates N2O antinociception (n = 10 for each group). N2O had no effect on tail-flick latencies in the C57BL/6J WT controls for the D79N, α2AKO and α2CKO mice, so these strains were not tested for N2O tail-flick effects. *p < 0.05; **p < 0.01 versus air; #p < 0.05; ###p < 0.001 versus genetically matched WT controls.
In no instance was the sedative action of N2O diminished when one of the three α2adrenoceptor subtypes was knocked out or rendered dysfunctional (Fig.12). Therefore, although no antinociceptive action can be elicited in α2B−/− mice, their response in the rotarod test is identical to that of their wild-type control mice. At the lower concentration of N2O, the sedative effect of N2O appeared to be modestly enhanced in α2C−/− mice.

N2O sedative effect was intact in the α2B−/− knock-out mice. The sedative effects of N2O (35 and 70% ATA) in the α2A−/−, α2B−/−, and α2C−/− knock-out mice (A KO, B KO, and C KO, respectively), the D79N mice, and in their respective WT controls (n = 8 for each group). There were no differences in baseline rotarod latencies among the various mouse strains. The sedative effect was observed on the rotarod assay in all the mouse strains including the B KO mice. An enhanced sedative effect was observed in the C KO mice with the lower concentration of N2O. *p < 0.05 versus air.
α2B adrenoceptor mRNA was present in the spinal cord of wild-type mice
RT-PCR assays were repeated twice with kidney, spinal cord, and heart tissues from wild-type mice, and Figure13 illustrates representative results from these assays. The RT-PCR methods used allow relative quantitation; a large α2B adrenoceptor mRNA signal was detected from the kidney, whereas almost none could be detected in the heart (top panel). An intermediate amount of α2B adrenoceptor mRNA signal was detected in the spinal cord. The bottom panel demonstrates that total RNA from each of these tissues was of similar quality and yielded the expected ribosomal bands.

Detection of mRNA for α2Badrenoceptor in various tissues from wild-type mice (n = 2). RT-PCR analysis of total RNA prepared from kidney, spinal cord, and heart tissue revealed detectable levels of message for this receptor were found in the kidney and spinal cord but not in heart tissue. The bottom panel demonstrates that total RNA from each of these tissues was of similar quality and yielded the expected ribosomal bands after ethidium staining.
DISCUSSION
Exposure to N2O increased expression of Fos in TH-positive neurons in the pontine noradrenergic nuclei (A5, LC, and A7) of rats. After eliminating all of the TH-positive neurons in the LC and >70% in each of A5 and A7, the antinociceptive response to N2O was blocked. In contrast, the sedative response to N2O is maintained under these conditions. Null mice for the α2B adrenoceptor subtype had no N2O antinociceptive effect on tail flick and a diminished effect for the hot plate assay, whereas their WT cohorts and null mice for the other subtypes exhibited a normal antinociceptive response to N2O. Normal sedative responses to N2O were evident in all the genetically modified mice.
The use of the immediate early gene product, Fos protein, as a biochemical marker of sustained neuronal activation in the brainstem catecholaminergic neurons has been validated using a wide range of stimuli (Monnikes et al., 1997; Jordan, 1998; Hahn and Bannon, 1999; Li et al., 1999). The time course of its appearance (i.e., within 60 min of its provocation) ideally suits its application in these experiments (Presley et al., 1990). Other investigators have demonstrated that a wide variety of anesthetic agents (although no studies involved nitrous oxide) can evoke Fos protein in discrete sites of the nervous system, including the noradrenergic (A1 and A2) and adrenergic (C1) medullary nuclei (Krukoff et al., 1992; Miura et al., 1994; Takayama et al., 1994; Clement et al., 1998). Significantly, none of these previous studies have observed anesthetic evoked Fos expression in the pontine noradrenergic nuclei (A5, LC, and A7). Whereas we have shown that N2O activates pontine noradrenergic neurons, the mechanism by which this occurs has not been established. At the ion channel level, N2O is known to block NMDA channels (Jevtovic-Todorovic et al., 1998), which would tend to reduce cell excitability; however, its possible role in the activation of noradrenergic neurons has not been addressed.
Saporin is a ribosome inactivating protein that ultimately results in cell death and destruction. By coupling this toxin to an antibody directed against a key synthetic enzyme, we are able to selectively destroy only those neurons containing this enzyme (Wrenn et al., 1996). Because dopamine-β-hydroxylase only exists in noradrenergic or adrenergic neurons, only these cells will be selectively destroyed (Martin et al., 1999). In this manner we have isolated the role of activation of noradrenergic pathways in the behavioral responses to N2O. Whereas activation of noradrenergic pathways is causally linked to the antinociceptive action of N2O, it does not appear to play any role in the sedative response to N2O.
The processes whereby activation of noradrenergic pathways produce antinociception in rodents has been investigated by our laboratory. Previously, we have shown that descending noradrenergic pathways are a pivotal component in the antinociceptive response to N2O because both spinal cord transection as well as depletion of norepinephrine in the spinal cord will prevent this behavioral response (Zhang et al., 1999). Furthermore, N2O provokes release of spinal norepinephrine over the same time course as it produces its antinociceptive action and intrathecal administration of an α2 antagonist blocks N2O evoked antinociception (Guo et al., 1996). Collectively, these data indicate that N2O can stimulate noradrenergic bulbospinal neurons, resulting in spinal norepinephrine release and activation of dorsal horn α2 adrenoceptors, thus inducing antinociception (Kingery et al., 1997).
Norepinephrine is a nonselective agonist at adrenergic receptors. Intrathecal administration of a selective α2antagonist (atipamezole) blocked the N2O antinociceptive response (Guo et al., 1996), suggesting this class of spinal adrenoceptors mediates the action. Earlier studies had revealed that spinal α2 adrenoceptors are capable of transducing antinociception from supraspinal activation (Jones, 1991;West et al., 1993; Willis and Westlund, 1997). Yet prazosin, a prototypic α1 antagonist, also blocked the antinociceptive response to N2O (Guo et al., 1999). This effect may be because of its ability to block α2B and α2Cadrenoceptors, although it has no activity at the α2A subtype (MacDonald and Scheinin, 1995). Further corroboration that the α2A adrenoceptor subtype is not involved was provided by data from studies in which the D79N transgenic animals, which have dysfunctional α2A adrenoceptor subtypes, exhibit dose-dependent antinociception in response to N2O exposure (Guo et al., 1999). Now we report that mice deficient in either the α2A or the α2C adrenoceptor subtype respond normally to N2O. However, mice that are deficient in the α2B adrenoceptor subtype exhibit little (Fig.11A) or no (Fig. 11B) antinociceptive effect. The quantitative difference in these two assays may relate to the fact that the tail-flick assay measures pain processing in the spinal cord, exclusively; supraspinal events may impact on the hot-plate test. Therefore, these data suggest that the α2B adrenoceptor mediates the antinociceptive response to the endogenously released norepinephrine in the dorsal horn of the spinal cord, which we have shown to contain message for α2B subtype.
Much of the earlier pharmacological evidence pointed to activation of α2A subtype for α2adrenoceptor-mediated antinociception (Millan, 1992; Millan et al., 1994). However, a prazosin-sensitive α2adrenoceptor subtype was shown to inhibit release of substance P in a spinal cord preparations, suggesting a role for the α2B or α2C adrenoceptor subtypes in antinociception (Ono et al., 1991). Furthermore, ST-91, a non-α2A subtype-preferring α2 agonist, was shown to induce antinociception in rats when it was administered intrathecally, and again this effect was blocked by prazosin (Graham et al., 1997, 2000; Takano et al., 1992a; Takano and Yaksh, 1992b). Inability to reconcile these disparate findings highlights the difficulties involved in using pharmacological approaches, especially when these probes lack subtype selectivity. The development of genetically modified reagents has facilitated subtype-specific approaches. Studies using D79N mice, in which the gene for α2A adrenoceptor subtype has been mutated rendering the expressed protein dysfunctional, established a role for the α2A subtype for the thermal analgesic response to the potent α2 adrenoceptor agonist dexmedetomidine (Hunter et al., 1997; Lakhlani et al., 1997). However, the D79N mice do exhibit some residual α2adrenoceptor agonist-induced effects on spinal analgesia in the intrathecal substance P-induced pain model (Stone et al., 1997) and for tail-flick analgesia after the administration of the α2 agonist moxonidine (Fairbanks and Wilcox, 1999). Furthermore, intrathecally administered α2C antisense oligodeoxynucleotides decreased mechanical antinociception induced by clonidine and other antinociceptive agents in the rat, suggesting that the α2C subtype is also involved in antinociception; however, confirmatory evidence that expression of only the α2C subtype was decreased was not provided (Aley and Levine, 1997). It should be acknowledged that studies on genetically modified reagents may provide misleading information. In the case of the knock-out models, these animals not only lack the protein of interest but the organisms have had an opportunity to adapt to this deficiency during their development. These putative adaptive responses to the knock-out remain largely undocumented.
Whereas the α2B adrenergic receptor subtype has been found to mediate some physiological responses (Link et al., 1996), its role in antinociception has not been previously established using transgenic animals. In the rat spinal cord and dorsal root ganglion, mRNA for the α2A, α2Band α2C subtypes have been identified; in the spinal cord α2A mRNA is abundant in lamina I, II, and V, whereas the α2C exhibit a weaker and less distinct signal throughout the dorsal horn, and the α2B are occasionally seen in lamina II (Zeng and Lynch, 1991; Nicholas et al., 1993; Shi et al., 1999). In the dorsal root ganglia, mRNA for the α2C subtype predominates (80% of neuron profiles), whereas α2A are seen in ∼20% of neurons, and the α2B mRNA is found only in small numbers of neurons (Nicholas et al., 1993; Cho et al., 1997; Gold et al., 1997;Shi et al., 2000). However, the low prevalence of α2B mRNA in the rat spinal cord and dorsal root ganglion may not necessarily reflect a lack of functional importance (Nicholas et al., 1996).
We have demonstrated that there is a robust signal for α2B mRNA in the spinal cord of mice. One possible site at which this receptor subtype may play a role in antinociception may be at the level of the GABAergic interneurons in lamina III; these interneurons have long been known to have a role in descending inhibition (Willis et al., 1997). It is notable that a strong signal for α2B mRNA has been detected in lamina I-V in the spinal cord of humans (Smith et al., 1995) and that α2 adrenoceptors in this region are upregulated after spinal cord transection (Giroux et al., 1999), suggesting that these may be coupled to descending inhibitory pathways.
Finally, the data from these studies indirectly refute the “unitary hypothesis for anesthetic action.” In this study we provide clear evidence that the sedative and the antinociceptive actions of N2O appear to be mediated by different molecular mechanisms. Whereas the antinociceptive response clearly requires the participation of both noradrenergic pathways and α2B adrenoceptors, neither mechanism is needed for the hypnotic–sedative action of N2O. Therefore, we posit that not only are different anesthetic agents not acting via the same mechanism, but the same anesthetic compound transduces different aspects of the anesthetic state at different sites.
Footnotes
This work was supported by National Institutes of Health Grant GM30232, a Veteran Affairs Merit Review, and the Medical Research Council. We thank the Clinical Investigation Facility of the 60th Medical Group at Travis Air Force Base and especially Master Sergeant Jonathan Gorum for assisting with the hyperbaric chamber study. We also thank Prof. Lee E. Limbird for providing us with the D79N transgenic mice.
Correspondence should be addressed to Prof. Mervyn Maze, Magill Department of Anaesthetics, Chelsea and Westminster Hospital, 369 Fulham Road, London SW10 9NH, UK. E-mail: m.maze{at}ic.ac.uk.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}