Article Text

Abstract
Objectives Novel imaging methods based on specific molecular targets to detect both established neoplasms and their precursor lesions are highly desirable in cancer medicine. Previously, we identified claudin-4, an integral constituent of tight junctions, as highly expressed in various gastrointestinal tumours including pancreatic cancer. Here, we investigate the potential of targeting claudin-4 with a naturally occurring ligand to visualise pancreatic cancer and its precursor lesions in vitro and in vivo by near-infrared imaging approaches.
Design A non-toxic C-terminal fragment of the claudin-4 ligand Clostridium perfringens enterotoxin (C-CPE) was labelled with a cyanine dye (Cy5.5). Binding of the optical tracer was analysed on claudin-4 positive and negative cells in vitro, and tumour xenografts in vivo. In addition, two genetically engineered mouse models for pancreatic intraepithelial neoplasia (PanIN) and pancreatic cancer were used for in vivo validation. Optical imaging studies were conducted using 2D planar fluorescence reflectance imaging (FRI) technology and 3D fluorescence-mediated tomography (FMT).
Results In vitro, the peptide-dye conjugate showed high binding affinity to claudin-4 positive CAPAN1 cells, while claudin-4 negative HT1080 cells revealed little or no fluorescence. In vivo, claudin-4 positive tumour xenografts, endogenous pancreatic tumours, hepatic metastases, as well as preinvasive PanIN lesions, were visualised by FRI and FMT up to 48 h after injection showing a significantly higher average of fluorochrome concentration as compared with claudin-4 negative xenografts and normal pancreatic tissue.
Conclusions C-CPE-Cy5.5 combined with novel optical imaging methods enables non-invasive visualisation of claudin-4 positive murine pancreatic tumours and their precursor lesions, representing a promising modality for early diagnostic imaging.
- Clostridium perfringens enterotoxin
- fluorescence reflectance imaging
- pancreatic cancer
- PanIN
- claudin-4
- pancreatic fibrosis
- pancreatic disease
- pancreatic tumours
- imaging
- pancreatic damage
- image analysis
- adenocarcinoma
- molecular biology
- cancer genetics
- pancreas
- colorectal cancer genes
- abdominal MRI
- endoscopy
- gene expression
- pancreatitis
- cancer
- gastrointestinal cancer
- cell migration
- carcinogenesis
Statistics from Altmetric.com
- Clostridium perfringens enterotoxin
- fluorescence reflectance imaging
- pancreatic cancer
- PanIN
- claudin-4
- pancreatic fibrosis
- pancreatic disease
- pancreatic tumours
- imaging
- pancreatic damage
- image analysis
- adenocarcinoma
- molecular biology
- cancer genetics
- pancreas
- colorectal cancer genes
- abdominal MRI
- endoscopy
- gene expression
- pancreatitis
- cancer
- gastrointestinal cancer
- cell migration
- carcinogenesis
Significance of this study
What is already known on this subject?
-
Pancreatic cancer exhibits the poorest prognosis of all solid tumours. Novel imaging methods are urgently required to detect this tumour at an early and potentially curable stage.
-
Claudin-4, an integral constituent of tight junctions, has been identified as highly expressed in various gastrointestinal tumours including pancreatic cancer.
-
Clostridium perfringens enterotoxin (CPE) is a naturally occurring ligand of claudin-4. Its C-terminus (C-CPE) binds to claudin-4 but lacks the toxic effects of full-length CPE.
What are the new findings?
-
The non-toxic C-terminal fragment of CPE (C-CPE) labelled with a fluorochrome shows high binding affinity specifically to claudin-4 positive pancreatic cancer cells.
-
In vivo, claudin-4 positive tumour xenografts as well as endogenous pancreatic cancers and their metastases in genetically engineered mouse models of pancreatic cancer can be reliably detected by fluorescence imaging techniques using the C-CPE as tracer.
-
C-CPE-based imaging is also able to detect preinvasive precursor lesions of pancreatic cancer in genetic mouse models.
How might it impact on clinical practice in the foreseeable future?
-
Based on these data, C-CPE-based tracers for routine clinical diagnostic applications, such as photon emission computer tomography or positron emission tomography should be developed.
-
C-CPE/claudin-4-based molecular imaging might be a valuable tool for early diagnosis and for screening of high-risk populations for pancreatic cancer.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the fifth-leading cause of cancer-related deaths in industrialised countries, and accounts for about 30 000 deaths per year in the USA. It exhibits the poorest prognosis of all solid tumours with a 5-year survival rate <5% and a median survival of 3–6 month after diagnosis.1 The best option currently available for ameliorating pancreatic cancer survival is to diagnose the neoplasm at an early and potentially curable stage.2 Emerging optical imaging techniques have recently been proven to be powerful tools for in vivo molecular imaging of specific target structures.3–5 The combination of new quantitative fluorescence imaging methods with novel pancreatic cancer-specific fluorochromes may lead to improved diagnosis of this fatal disorder at an early stage.6
The family of claudins comprise a multigene family of at least 20 transmembrane proteins forming integral constituents of tight junctions that determine cell polarity and paracellular permeability in human epithelial cells.7 ,8 Our group provided the first evidence of claudin-4 upregulation in pancreatic cancer tissues using various expression-profiling approaches, such as representational difference analysis and DNA array technology.9 ,10 Furthermore, intense claudin-4 immunoreactivity was also reported in human pancreatic intraepithelial neoplasia (PanIN), the preinvasive precursor lesion of PDAC, suggesting that claudin-4 may serve as a promising candidate for molecular diagnostics not only for advanced pancreatic cancer but also in high-risk populations before the development of an invasive carcinoma.11
Interestingly, claudin-4 was described as receptor of Clostridium perfringens enterotoxin (CPE), a polypeptide of 35 kilodaltons.12 In C perfringens type A food poisoning, a common human food-borne illness, interaction of CPE with its receptor, claudin-4, results in watery diarrhoea caused by the destruction of the intercellular barrier of the intestinal epithelial cells.13 In pancreatic cancer, administration of full-length CPE to claudin-4-expressing pancreatic tumour cells leads to an acute dose-dependent cytotoxic effect in vitro and in vivo.14 Interestingly, cytotoxicity of CPE is mediated by its NH2-terminal half, whereas binding of CPE to claudin-4 is accomplished by its COOH-terminal half (C-CPE).15 ,16
In this study, we designed a target-specific fluorochrome (C-CPE-GST-Cy5.5) with high affinity to claudin-4 and evaluated its ability to visualise pancreatic cancer cells in vitro. In vivo, xenograft tumours, as well as two genetically engineered mouse models for both preinvasive pancreatic precursor lesions and invasive pancreatic cancer, were imaged by two-dimensional planar fluorescence reflectance imaging (FRI) technology and three-dimensional fluorescence-mediated tomography (FMT).
Material and methods
Materials, cell lines and human tissue
Cell lines were obtained from the following suppliers: PANC1, from the European Collection of Animal Cell Cultures, Salisbury, UK; CAPAN1, HT1080, HT29, Caco2 from the American Type Culture Collection, Manassas, Virginia, USA. Cells were maintained in Dulbecco's modified minimal essential medium (Gibco, Invitrogen Corp, Carlsbad, California, USA) supplemented with 10% fetal calf serum (Gibco), 100 μg/ml streptomycin (Sigma-Aldrich, St Louis, Missouri, USA) and 100 U/ml penicillin (Sigma). All cell lines were grown at 37°C in 5% CO2. The tissue microarray (TMA) was constructed as described before.17 Briefly, six TMAs containing 300 cores of normal ducts and PanINs (collected from 21 disease-free pancreata and 81 resected pancreata because of GI neoplasms), 30 alcoholic chronic pancreatitis and 10 autoimmune pancreatitis specimens and 48 PDACs (G1-G3) were used for analysis.
Design of C-CPE-GST protein and Cy5.5 labelling
The CPE receptor-binding activity of full-length CPE (319aa) is restricted to the 30 C-terminal amino acids.16 ,18 ,19 Recently, site-directed mutagenesis revealed three Tyr residues, located at positions 306, 310 and 312 to be critical for receptor binding to claudin-4.20 Furthermore, competitive binding experiments showed that C-terminal CPE (C-CPE) binds with 1:1 stoichiometry and submicromolar affinity to pure claudin-4.21 Therefore, we designed a COOH-terminal fragment of CPE (aa184-319) linked to a GST fusion protein, cloned into XhoI and NotI sites of pGEX-4T (GE Healthcare Lifesciences, Chalfont St Giles, UK), produced in Escherichia coli and purified as described previously.12 Cy5.5 monoreactive NHS-ester was obtained from Amersham Biosciences (Amersham, Piscataway, New Jersey, USA) and labelled on lysine according to the manufacturer's protocol. Briefly, C-CPE-GST peptide fragments (1 mg/ml) were dialysed once against 2 l of 0.15 M sodium chloride (4 h) at 4° and twice against aqueous bicarbonate buffer (0.1 M, pH 8.4, 4 h, overnight). A solution of the dye in DMSO (10 mg/ml) was added (ratio peptide/dye 1:20). The mixture was stirred for 60 min at room temperature in the dark. Purification was accomplished using a PD10 column (Amersham) diluting the peptide with phosphate buffer saline (PBS). The product fractions were pooled, dialysed against PBS and were assessed by fluorometry and photometry for determining the coupling degree (Hitachi, F-4500 fluorescence spectrometer & U-3310 UV/VIS spectrophotometer, Tokyo, Japan). The degree of labelling was between 1.5 and 2.0 (1 Mol C-CPE-GST to 1.5–2 Mol Cy5.5), and equimolar tracer was used for subsequent experiments as indicated. The size and structure of C-CPE-GST-Cy.5.5 is displayed in supplementary figure 1.
Immunoblotting
After pretreatment with C-CPE-GST (1 μg/ml), cells were washed and incubated in lysis buffer (50 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (pH 7.5), 150 mM NaCl, 1 mM ethyleneglycol-bis (aminoethylether)-tetraacetic acid, 100 mM NaF, 10 mM Na4P2O7, 10% glycerol and 1% Triton X-100) supplemented with a cocktail of protease inhibitors (Complete, Roche Applied Science). Proteins were sonicated, separated on a 10% SDS-PAGE and transferred onto Polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, Massachusetts, USA). After blocking, immunoblots were incubated with antibodies against CPE (rabbit polyclonal, AbD Serotec), GST (mouse monoclonal, Abcam, Cambridge, UK), and claudin-4 (rabbit polyclonal, Abcam), followed by incubation with peroxidase-conjugated secondary antibodies (Sigma). All blots were detected by ECL chemiluminescence (Amersham Biosciences, Freiburg, Germany).
C-CPE-GST-Cy5.5 binding assay
Cells were seeded in 24-well plates and incubated in culture medium overnight. For binding studies, cells were washed twice with PBS and blocked in PBS/BSA (0.1%). After washing twice with PBS, C-CPE-GST-Cy5.5, or free Cy5.5 (0.2 nmol/ml), was added into each well. After an incubation period of 1 h at 4°C, the cells were washed twice with PBS. The cells could be directly visualised by fluorescence microscopy (20× objective magnification, Nikon TE 2000-S, Tokyo, Japan) as previously described.22
Mouse models of pancreatic cancer
Female NMRI-nu/nu mice (6–8 weeks old), LSL-KrasG12/D+ ; Pdx-1-Cre (KC) mice (2–10 months old) with PanIN lesions and LSL-KrasG12/D+ ; LSL-Tpr53R172H/+ ; Pdx-1-Cre (KPC) mice (3–6 months old) with spontaneous pancreatic cancer23 and Pdx-1-Cre (PC) control mice were propagated and maintained in a pathogen-free environment. All experiments were carried out according to the guidelines of the local animal welfare committee. In KPC mice, tumour development was monitored from the age of 2 months by weekly palpation. In addition, MRI imaging was performed with a 9.4 Tesla small-animal MRI system (Biospec 94/20, Bruker, Rheinstetten, Germany) using standard T1- and T2-weighted imaging techniques. Only mice with clearly palpable tumours were enrolled in the imaging study. For subcutaneous xenograft experiments, CAPAN1 (5×106 in 0.1 ml DMEM) and HT1080 cells (1×106 in 0.1 ml DMEM) were subcutaneously injected in the flanks of NMRI-nu/nu mice, respectively. When tumours reached 4–6 mm in diameter, the tumour-bearing mice were anaesthetized by inhalation of isofluorane (2%) and subjected to in vivo imaging studies.
FRI
The optical FRI was performed using the Image Station In Vivo FX Pro Imaging System (Carestream Health Inc, Rochester, New York, USA), which is equipped to provide multi-wavelength fluorescence, luminescence, x-ray, and radioisotopic imaging. Fluorescence is generated by a 150-W halogen illuminator with selectable bandpass excitation and emission filters (Cy5.5 excitation, 665±18 nm and emission 694±17.5 nm). Light from the fluorescence screen is captured with a 4-million-pixel cooled CCD camera equipped with a 10× zoom lens. Images acquisition times were 30 s per animal. Fluorescent images were co-registered with white light background images, regions of interest were selected and analysed with the Carestream MI software, V.5.X. Signal intensity is expressed as mean intensity/pixel (arbitrary unit = AU) in the regions of interest.
For in vivo experiments, C-CPE-GST-Cy5.5 (2 nmol/100 μl PBS) was injected intravenously into the tail vein of each mouse. In order to assess probe specificity, GST-Cy5.5 was injected as control. FRI was performed 1, 3, 6, 24 or 48 h post-injection. For biodistribution studies, FRI images of isolated organs were obtained immediately after sacrificing the mice 24 or 48 h after tracer injection. (tumour tissue, normal pancreas, heart, spleen, kidney, liver, muscle and lung).
FMT
All tomographic optical imaging studies were performed with a small-animal FMT system from VisEn Medical, Inc (FMT 2500, now Perkin Elmer, Woburn, Massachusetts, USA).24 Mice were scanned in an animal-imaging chamber by a movable optical fibre. The light source is software-selectable from two high-power laser diodes at 670 nm ad 745 nm, operating in the range of 5 mW–150 nW. Detection is performed with a thermoelectrically cooled CCD camera. Tomographic reconstruction is based on the normalised Born approximation, a diffraction optical tomographic technique that uses a diffusion-type theoretical photon propagation model in biological tissue.25 The instrument is calibrated on each channel with a phantom of known Cy5.5 fluorochrome concentration.26 Calibration of the phantom was performed according to the steps described in the VisEnMedical FMT 2500 Quantitative Tomography System user guide using a calibration kit provided by VisEn Medical. Animal scan times are in the range of 2–5 min within a predefined scan field. In vivo experiments using FMT were performed as described for FRI.
Immunohistochemistry
Immunohistochemical analysis of claudin-4 was performed as previously described.27 Briefly, after antigen retrieval (microwave in antigen unmasking solution, Vector Laboratories, Burlingame, California, USA), paraffin sections were incubated with rabbit polyclonal antibody (Abcam, 1:1000), or mouse monoclonal anti-Claudin-4 antibody (Clone 3E2C1, Life Technologies, Darmstadt, Germany, 1:50) for human sections. Antibody binding was visualised using a biotinylated secondary antibody, avidin-conjugated peroxidase (ABC method; Vector Laboratories), and 3,3′diaminobenzidine tetrachloride as substrate, with haematoxylin as counterstain. For TMA analysis, the intensity of the reactions was scored as mild, moderate or strong (score 1, 2 or 3, respectively). The proportion of the positive cells in ducts and tumour areas was estimated in per cent and divided into scores (<10%, 1; 10–50%, 2; 51–80%, 3; 81–100%, 4). The final score was determined as a product of the intensity of the staining and the proportion of positive cells (minimum 0, maximum 12).
Statistical analysis
Data are presented as mean ± SEM. Statistical analysis of tumour fluorescence in vivo was conducted using the two-tailed, unpaired Mann–Whitney test. The differences between the immunohistochemical scores were analysed by means of the Mann–Whitney U and Kruskal-Wallis H tests. When multiple comparisons were performed, the p value was modified according to Bonferroni. All statistical tests were performed using SPSS V.10.1 software. p Value ≤0.05 was considered to be significant.
Results
Claudin-4 is upregulated in human PDAC and high-grade PanIN lesions
Using a large TMA, we investigated the expression of claudin-4 in a variety of healthy and diseased human tissues. Claudin-4 showed a predominantly membrane-bound, partly cytoplasmic expression. Most ducts in disease-free pancreata n=45 (mean IHC score 2.02, range 0–9), or in chronic pancreatitis n=34 (mean IHC score 1.09, range 0–6) and low-grade PanINs n=86 (mean IHC score 1.4, range 0–9), exhibited no or mild claudin-4 staining. Claudin-4 expression was more pronounced in high-grade PanINs n=36 (mean IHC score 7.64, range 1–12) and in PDACs n=46 (mean IHC score 6.72, range 1–12). Ducts in chronic pancreatitis exhibited comparable claudin-4 staining to that in normal ducts. Statistically, there were no differences in claudin-4 expression between normal ducts, ducts in chronic pancreatitis and low-grade PanINs. In contrast, single comparisons between normal ducts, chronic pancreatitis, low-grade PanINs and high-grade PanINs/PDACs showed significantly elevated claudin-4 expression in the latter entities (p<0.001) (figure 1A,B). To study the interaction of C-CPE with claudin-4 positive cell lines in vitro, we first confirmed that claudin-4 protein is highly expressed in human pancreatic adenocarcinoma cell lines, such as PANC1 and CAPAN1. Moreover, claudin-4 expression was also upregulated in adenocarcinoma cells of other origin, such as the colon carcinoma cell lines HT29 and Caco2. In contrast, claudin-4 expression was absent in the human sarcoma cell line HT1080 (figure 2A).

(A) Representative immunohistochemistry of human pancreatic intraepithelial neoplasia (PanIN) and pancreatic ductal adenocarcinoma showing increasing claudin-4 expression throughout progression to pancreatic cancer. (B) Quantification of claudin-4 protein levels as immunohistochemical claudin-4 score in normal ducts, low-grade precursor lesions (PanIN-1), high-grade precursor lesions (PanIN-2/3), pancreatic ductal adenocarcinoma and ducts in chronic pancreatitis (CP ducts). Asterisks on the top indicate significant differences (Mann-Whitney U test, each p<0.001). Circles indicate outliers, stars indicate extreme values.

(A) Claudin-4 protein expression in various human cancer cell lines and in the human sarcoma cell line HT1080. (B) Binding of C-CPE-GST to claudin-4 positive CAPAN1 cells and claudin-4 negative HT1080 cells after incubation with C-CPE-GST (0.2 nmol/ml). (C) Coimmunoprecipitation of C-CPE-GST and claudin-4: Claudin-4 positive PANC1 and CAPAN1 cells were incubated with C-CPE-GST for 4 h. Immunoprecipitation was performed with anti-GST or unspecific IgG as control. (D) Fluorescence microscopy (20×): C-CPE-GST-Cy5.5 (0.2 nmol/ml) was incubated with claudin-4 positive PANC1 and CAPAN1 cells and claudin-4 negative HT1080 cells for 1 h to assess target affinity of the probe. GST-Cy5.5, Cy5.5 and untreated cells were used as controls.
Synthesis and characterisation of C-CPE-GST-Cy5.5
Based on our previous observation demonstrating CPE as a ligand of claudin-4, we aimed to investigate the suitability of the non-toxic C-CPE fragment as a tool for diagnostic imaging of claudin-4 positive pancreatic cancers.
Therefore, we first purified a C-CPE peptide as GST-fusion protein and tested its binding characteristics to claudin-4 positive and negative tumour cells. Incubation of claudin-4 positive CAPAN1 cells with C-CPE-GST resulted in retention of this fusion protein on the cell surface as revealed by immunoblotting with anti-GST antibodies. As expected, the retention of the fusion protein on the surface of claudin-4 negative HT1080 cells was much reduced (figure 2B). Furthermore, C-CPE-GST could be co-immunoprecipitated with claudin-4 in CAPAN1 cells and another claudin-4 positive pancreatic cancer cell line, PANC1 (figure 2C), confirming a stable binding of C-CPE-GST to claudin-4.
In vitro C-CPE-GST-Cy5.5 binding studies
To evaluate the feasibility of using C-CPE-GST as a diagnostic tool, we aimed to conjugate C-CPE-GST to a near-infrared (NIR) tracer for optical imaging in preclinical studies. Based on the high claudin-4 protein expression in pancreatic tumour cells, the binding properties of C-CPE-GST-Cy5.5 were evaluated by fluorescence microscopy in claudin-4 positive pancreatic cancer cells and claudin-4 negative HT1080 sarcoma cells as negative control. Negligible signals were detected when cells were preincubated with unmodified Cy5.5 dye alone, or GST-Cy5.5. Likewise, incubation of claudin-4 negative HT1080 cells with C-CPE-GST-Cy5.5 did not reveal a significant signal. However, the labelled peptide bound consistently to claudin-4 positive CAPAN1 and PANC1 cells, exhibiting a membrane-associated distribution pattern (figure 2D). The aforementioned data confirm in vitro binding of C-CPE-GST-Cy5.5 to claudin-4 positive cells. Furthermore, claudin-4-expressing HT29 colon carcinoma cells also revealed specific tracer binding (supplementary figure 2).
In vivo optical imaging of pancreatic cancer xenografts
In vivo imaging studies with tumours were performed first with subcutaneous xenografts. Six hours after the tracer injection into the tail vein, subcutaneous CAPAN1 xenograft tumours (n=7) revealed a strong fluorescence, which peaked at 24 h (figure 3A,B) and remained visible up to 48 h, as detected by NIR FRI. An in vivo time course is shown in supplementary figure 3A. In contrast, claudin-4 negative HT1080 xenograft tumours (n=8), and CAPAN1 xenografts injected with control tracer GST-Cy5.5 (n=7) exhibited a significantly weaker signal (figure 3A,B). This imaging pattern was paralleled by a strong membranous claudin-4 expression in the CAPAN1 xenografts, but an absent immunohistochemical signal for claudin-4 in HT1080 xenografts (figure 3C–F), thus supporting a claudin-4-specific signal detected by systemically injected C-CPE-GST-Cy5.5 in vivo. The biodistribution of normal murine tissues in nu/nu mice is displayed alongside xenograft tumours in supplementary figure 3B,C.
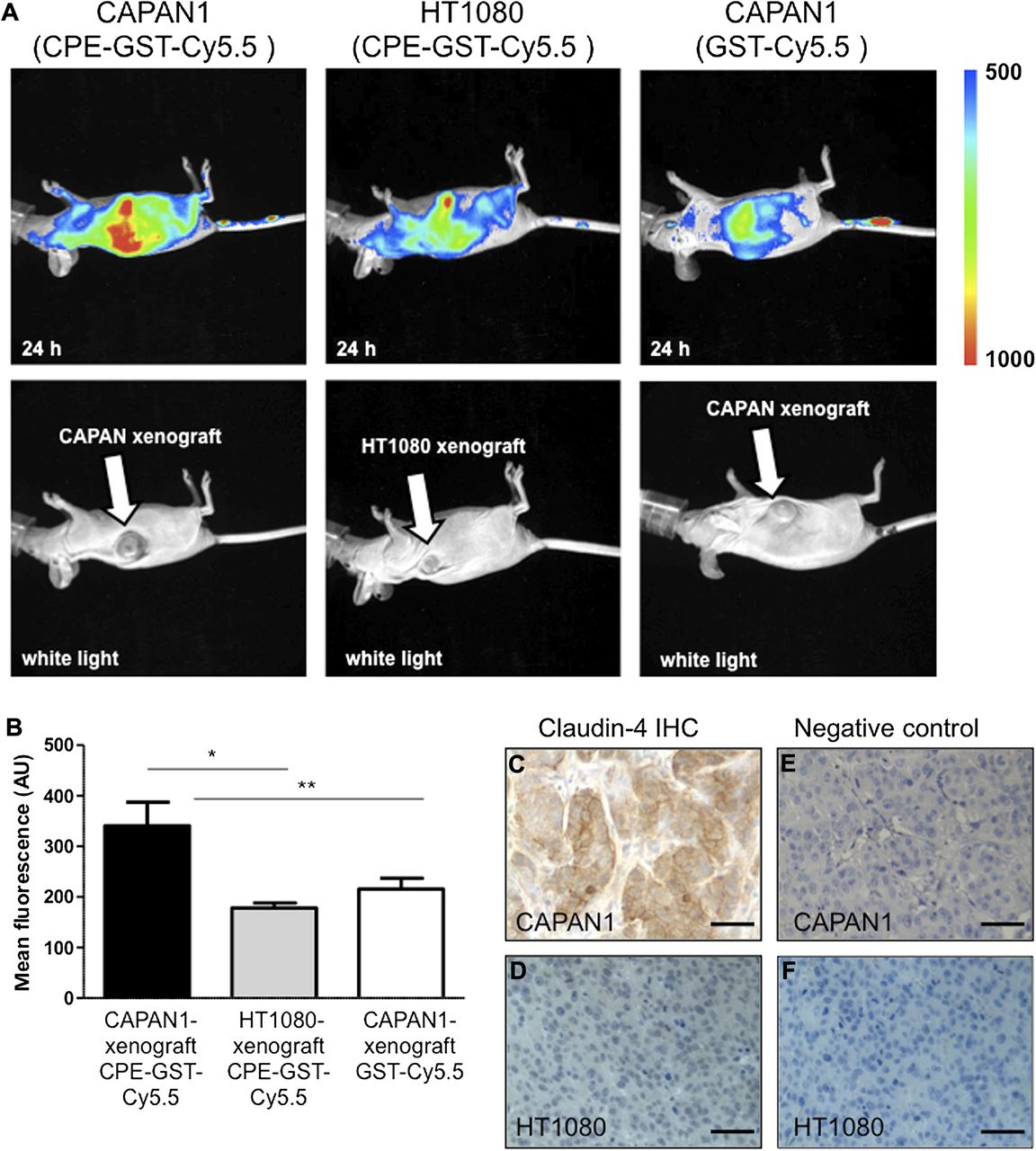
(A) In vivo fluorescence reflectance imaging images of CAPAN1 and HT1080 xenografts 24 h after injection of C-CPE-GST-Cy5.5 or GST-Cy5.5 (control) (2 nMol/100 μl). (B) Mean fluorescence of CAPAN1- (n=7) and HT1080- xenografts (n=8) after C-CPE-GST-Cy5.5 injection, and CAPAN1-xenografts (n=7) after GST-Cy5.5 injection; * p<0.001, ** p<0.01. (C) Immunohistochemistry of claudin-4 in CAPAN1 xenografts; (D) HT1080 xenografts. (E, F) Negative control without primary antibody. Bars represent 50 μm.
Claudin-4 expression in genetically engineered mice
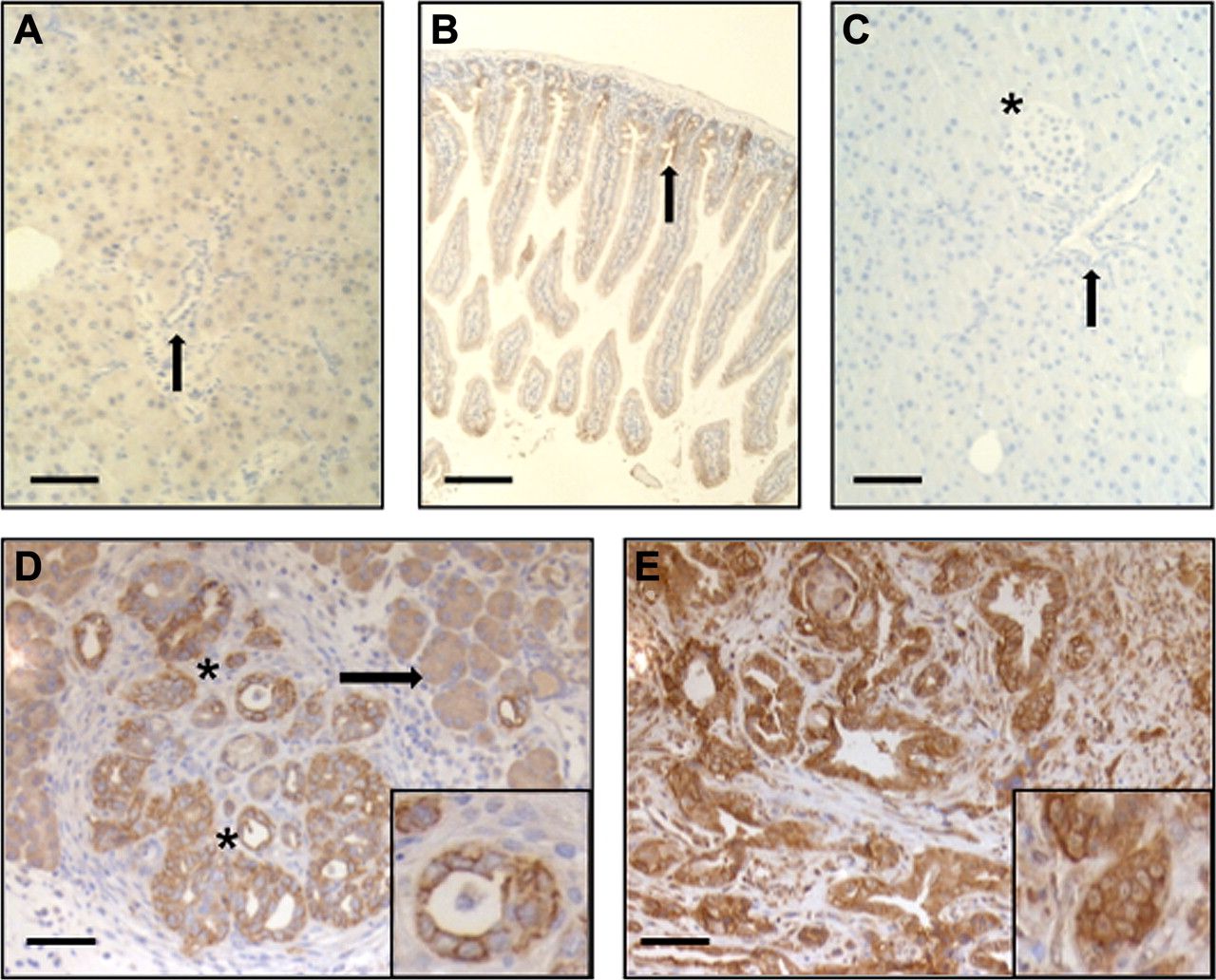
Since the histological appearance of xenograft tumours differs significantly from endogenous pancreatic cancers which are characterised by high stroma content and altered vasculature, we aimed to corroborate the in vivo imaging results in genetically engineered mouse models (GEMM). For this purpose, we chose two GEMMs, one with a conditional activating mutation of K-ras (KC), and the second with activated KC combined with a dominant negative mutation of p53 (KPC).2 ,23 KC mice develop preinvasive precursor lesions, known as PanINs, at early age, but only progress with a low penetrance to PDAC. In contrast, KPC mice exhibit invasive PDAC from 2 months of age onwards. Immunohistochemical analysis of murine PanIN and PDAC lesions showed a membranous expression pattern of claudin-4, supporting the notion that both genetically engineered models closely recapitulate the histology in humans. In analogy to our previous data,14 adjacent normal acinar tissue and acinar-to-ductal metaplasia exhibited only faint cytoplasmic claudin-4 immunoreactivity (figure 4A,C–E). In normal intestinal tissue, claudin-4 was predominantly detected in crypts and, overall, was much lower as compared with PDAC (figure 4B). Additional protein analysis by Western blot revealed much lower claudin-4 levels in murine colon and normal pancreas compared with PDAC (supplementary figure 4A). In murine cell lines, claudin-4 expression was considerably higher in tumour cells derived from KPC mice compared with PanIN cells from KC mice (supplementary figure 4B).

(A–E) Claudin-4 immunohistochemistry. (A) Murine pancreas (arrow: duct). (B) Murine colon (arrow: crypts). (C) Murine pancreas without claudin-4 antibody (arrow: duct, star: pancreatic islet). (D) Murine PanIN (star) and acinar to ductal metaplasia (arrow). (E) Murine pancreatic ductal adenocarcinoma. Insets: membraneous claudin-4 localisation. Bars equal 100 μm.
In vivo optical imaging of spontaneous pancreatic tumours and precursor lesions
Similar to the xenograft experiments, the fluorescence signal in the upper abdomen peaked 24 h after injection of C-CPE-GST-Cy5.5 tracer into the tail vein. The signal could be anatomically located to the pancreas and was significantly elevated in KPC mice with PDAC compared with control mice without evidence of a pancreatic mass. KC mice with PanIN lesions also showed a significantly elevated signal by FRI when compared with control mice with normal pancreas. Subsequent ex vivo analysis and fluorescence quantification of PDAC, PanIN and normal pancreas is shown in figure 5A–D. Interestingly, sub-analysis revealed that younger KC mice with PanIN burden <80% (n=7) showed a lower mean fluorescence compared with KC mice with an extensive PanIN burden >80% (n=10), suggesting that the detected fluorescence signal increases during PanIN progression (supplementary figure 5A,B). These findings corroborate our human data showing upregulation of claudin-4 mostly in high-grade PanINs and PDAC (figure 1A,B). Altogether, our findings strongly support the feasibility of using C-CPE-GST-Cy5.5 as tracer to detect claudin-4 positive endogenous pancreatic cancer and its precursor lesions.

(A) Representative microscopic photographs of the abdominal cavity following laparotomy after injection of C-CPE-GST-Cy5.5 showing pancreatic ductal adenocarcinoma (PDAC) (upper panel), pancreatic intraepithelial neoplasia (PanIN)-containing pancreas (middle panel) and normal pancreas (NP, lower panel) which are indicated by black circles. (B) Representative fluorescence reflectance imaging (FRI) images of PDAC, PanIN and NP after laparotomy following injection of C-CPE-GST-Cy5.5. Black circles indicate the area of the pancreas. (C) Ex vivo FRI images after C-CPE-GST-Cy5.5 injection: 1: liver; 2: kidneys; 3: spleen; 4: intestine; 5: heart; 6: muscle; 7: lung. Fluorescence intensity scales indicate range of detection. (D) Ex vivo quantification of fluorescence after C-CPE-GST-Cy5.5 injection in NP, PanIN and PDAC; * p<0.02 compared with PDAC, ** p<0.002 compared with NP. (E) 24 h ex vivo biodistribution of C-CPE-GST-Cy5.5.
In vivo optical imaging of liver metastasis in genetically engineered mice
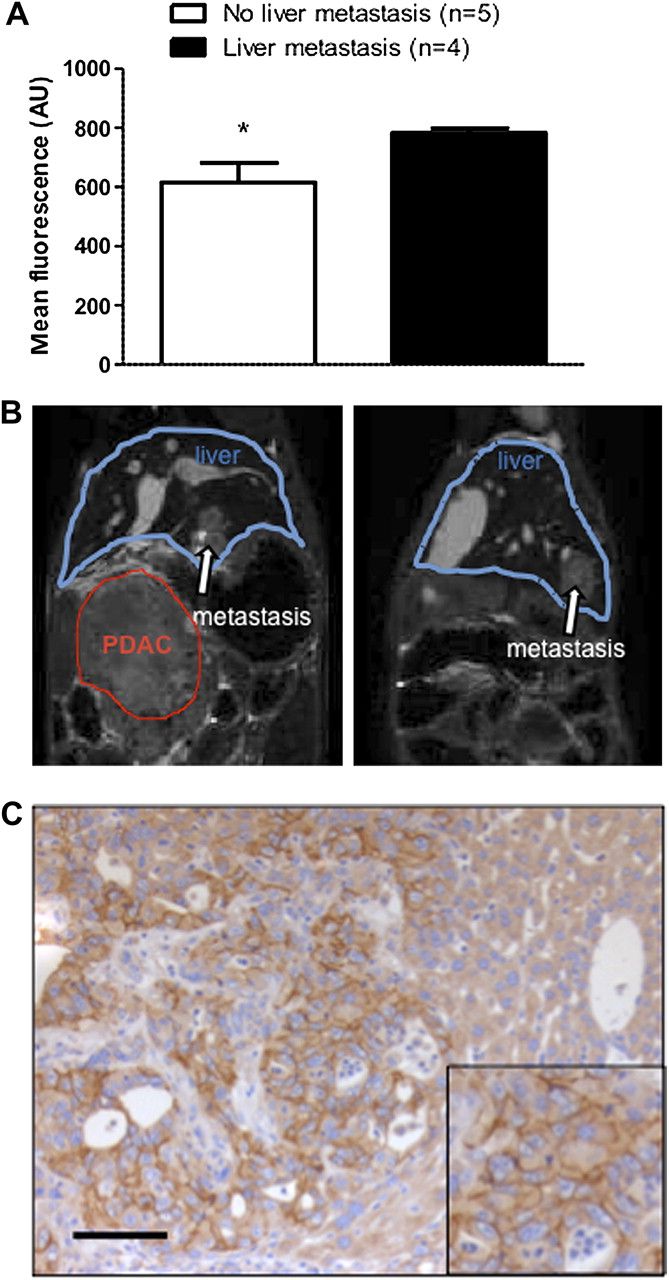
To evaluate the biodistribution of C-CPE-GST-Cy5.5, fluorescence signals were measured ex vivo in normal pancreas, muscle, heart, lung, spleen, kidney, intestine and liver tissues from all three cohorts (figure 5E). As the tracer is eliminated mainly via the kidney and liver, these two organs showed the highest tracer concentration. Strikingly, we noticed higher signals in livers from PDAC mice that almost reached the levels seen in tumours. Histological subgroup analysis of nine mice revealed liver metastases in n=4 livers exhibiting significantly higher tracer fluorescence as compared with livers without metastases (n=5); p<0.04 (figure 6A). Representative pictures of liver metastases detected by MRI, and strong claudin-4 expression in liver metastasis are depicted in figure 6B,C. These findings suggest that imaging with C-CPE-GST-Cy5.5 may also be able to detect claudin-4 positive metastatic disease in this murine PDAC model.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Ex vivo mean fluorescence of explanted livers with histologically confirmed liver metastases and without liver metastases. *p<0.04. (B) Representative magnetic resonance image of hepatic metastases (white arrow). (C) Claudin-4 immunohistochemistry in hepatic metastasis. Bars equal 100 μm.
In vivo optical imaging by FMT
In contrast with FRI technology, which detects two-dimensional planar fluorescence, FMT offers the possibility of better anatomical localisation of the emitted signal, and three-dimensional reconstruction similar to cross-sectional imaging techniques used in clinical applications, such as PET-CT. Therefore, we aimed to corroborate our data obtained in the genetic mouse models of pancreatic cancer by FMT technology. Imaging of KC and KPC mice in comparison with PC control mice by FMT confirmed consistent strong signals elicited by the pancreatic tumours of KPC mice, and by the pancreata of KC mice containing preinvasive PanIN lesions, as compared with PC control mice with normal pancreata (supplementary figure 6).
Discussion
Tumour-specific non-invasive imaging tools are urgently required to detect pancreatic cancer at early stages in order to tackle the disastrous prognosis of this disease. Recently, Eser and colleagues described an interesting approach using a cathepsin-activatable near-infrared probe in combination with flexible confocal fluorescence lasermicroscopy in a mouse model of pancreatic cancer.6
However, there is still a paucity of experimental data on suitable novel imaging tracers specifically targeting neoplastic cells of the pancreas.
Our group provided the first evidence that claudin-4 is upregulated in pancreatic cancer using various expression-profiling approaches such as representational difference analysis and DNA array technology.9 ,10 Following our report, claudin-4 has been found to be overexpressed in a number of different solid tumours such as breast, ovarian, prostate, lung and others.11 ,28–31 Claudin-4 is membrane-bound and, thus, represents an attractive imaging target for pancreatic cancer. In this study, we designed, for the first time, a target-specific fluorochrome (C-CPE-GST-Cy 5.5) using a non-toxic fragment of a naturally occurring bacterial protein as ligand with high affinity to claudin-4. By employing this tracer, we were able to visualise pancreatic cancer cells in vitro and in several mouse models of pancreatic cancer in vivo by two-dimensional planar FRI technology and three-dimensional FMT.
Our experiments revealed clear visualisation of pancreatic cancer cell lines and xenografts, which was restricted to claudin-4 expression. However, xenograft studies are limited by their non-physiological tumour tissue architecture, in particular, their lack of tumour stroma, which is a predominant feature of pancreatic cancer. Genetically engineered KPC mice carrying Kras and p53 mutations develop advanced PDAC with 100% penetrance at an early age, and recapitulate human PDAC with striking histopathological similarities including a strong desmoplastic reaction, occurrence and site of metastasis, as well as comorbidities such as cachexia, activation of distinct biochemical pathways, and evidence for genomic instability.23 Interestingly, our data show, for the first time, that claudin-4 is overexpressed in KPC tumours, thus providing an ideal platform for preclinical imaging studies using claudin-4 as diagnostic target in pancreatic cancer. Our results in the KPC model provide evidence that pancreatic tumours are clearly delineated from the surrounding tissue by the claudin-4-targeted C-CPE tracer. Notably, toxic effects of the fluorochrome were not observed in any animals tested.
Importantly, as in humans, we also detected elevated claudin-4 expression in murine high-grade PanIN lesions. Paralleled by these high-expression levels in PanIN3, we observed significantly enhanced imaging signals in the pancreata of KC mice, most pronounced in older mice with advanced PanIN grades. Although claudin-4 expression was found to be very similar in human and mouse PanIN progression, accurate differentiation between low- and high-grade murine PanIN lesions proved to be challenging by optical imaging, and might have been complicated by the fact that widespread PanIN2 lesions occur early in KC mice throughout the whole pancreas increasing the overall levels of claudin-4 expression.
In contrast with genetically engineered mice with simultaneous embryonic activation of mutant Kras, in high-risk patients, we would anticipate focally distributed high-grade PanIN3 lesions surrounded by healthy pancreatic tissue potentially facilitating their detection. Recently published data indicate that PanIN progression from early preneoplastic lesions to invasive carcinoma occurs over many years opening a broad diagnostic window of about 10 years to detect curable pancreatic cancer.32 Based on these findings, claudin-4-targeted imaging techniques have to be evaluated as a diagnostic tool to detect preinvasive tumours in high-risk populations, such as patients with chronic pancreatitis or familial pancreatic cancer.33 Of note, we found claudin-4 overexpression only in preneoplastic and neoplastic pancreatic tissue, but not in inflammatory conditions such as chronic pancreatitis, further adding to the diagnostic power of this target. Interestingly, although claudin-4 is well known as the CPE receptor causing gastroenteritis in the intestinal tract, we found only faint signals originating from the small and large intestines. This is in line with our previous observations that claudin-4 expression is markedly higher in PDA than in other normal tissues including the small intestine.9 ,14 The fact that claudin-4 was also found to be overexpressed in a number of different solid tumours such as breast, ovarian, prostate and lung cancer possibly extends the applicability of claudin-4-targeted imaging approaches. Moreover, our data demonstrate that claudin-4 positivity is also retained in hepatic metastases of the murine pancreatic cancer model, and imaging might thus be employed to monitor recurrence of disease following curative surgery.
The direct clinical translation of this NIR imaging approach is limited by the tissue volume in humans, which leads to subsequent scattering and attenuation of the signal. Based on our data, further studies are warranted and are ongoing in our laboratory to evaluate the feasibility of linking other tracers such as 99mTc, 68Ga or 18F to C-CPE that can be detected by scanners used for routine clinical diagnostic applications such as single-photon emission computer tomography or positron emission tomography. Interestingly, Kelly and colleagues recently demonstrated, for the first time, that plectin-1 could be successfully employed to detect primary tumours and metastatic foci by single-photon emission computer tomography using an orthotopic mouse model of PDAC.34 ,35
To conclude, we show, for the first time, in a preclinical model the feasibility of claudin-4-targeted, non-invasive imaging of pancreatic cancer and its preinvasive precursor lesions. Extensive in vitro and in vivo validation provides evidence that claudin-4-based imaging holds great promise to improve early detection of this appalling disease, and may have the potential to be employed in surveillance strategies in high-risk populations, such as individuals from families with familial pancreatic cancer. Furthermore, GEMM represent a valid and effective experimental system for additional claudin-4-targeted imaging approaches in pancreatic cancer patients.
Acknowledgments
We thank Eva Bug and Jan Riedel, Department of Gastroenterology, University of Marburg, for expert technical assistance. Furthermore, we thank Boris Zlatopolskiy and Agnieszka Morgenroth, Department of Nuclear Medicine, University of Ulm, for their help with the in vitro binding studies; Gustavo Barreton, University of Dresden for the establishment of the TMA; and Frances Connor and Kristopher Frese from the Tuveson Lab for assistance with GEMM and murine cell lines.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data Supplement 1 - Online Figure 1
- Data Supplement 2 - Online Figure 2
- Data Supplement 3 - Online Figure 3
- Data Supplement 4 - Online Figure 4
- Data Supplement 5 - Online Figure 5
- Data Supplement 6 - Online Figure 6
- Data Supplement 7 - Online Figure description
Footnotes
-
AN and AH contributed equally to the manuscript.
-
Funding This work was supported in part by the Deutsche Krebshilfe (to PM and TG), Mildred Scheel Postdoctoral Fellowship by the Deutsche Krebshilfe (to AN), the Deutsche Forschungsgemeinschaft (DFG, KFO210 to PM and SFB656 to C.B.) and the German Ministry of Education and Research (BMBF, NGFN program of medical genome research ‘PaCa-Net’; project ID PKB-01GS08) to MB, TG, BS and SH), the European Union (6th framework program MolDiag-PaCa to TG. and the 7th framework program EPC-TM net to TG, PM, MB, DT), the state of Hessen (LOEWE, to PM, MB, TG) and the IZKF Core unit of the University of Muenster. This research was also supported by Cancer Research UK, The Li Ka Shing Foundation and Hutchison Whampoa Limited (DT).
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement All material and protocols published in this manuscript including also unpublished material and protocols will be made available to all other interested scientists upon request via email or surface mail.