Article Text

Abstract
Background: We previously reported that endogenous prostaglandin I2, generated by a mild irritant, sensitised calcitonin gene related peptide (CGRP) containing sensory nerves and facilitated the release of CGRP and gastric mucosal protection against ethanol. Administration of capsaicin also inhibited ethanol induced gastric mucosal injury through immediate release of CGRP from primary sensory neurones, which is termed the neural emergency system. In the present study, we tested whether endogenous prostaglandin I2 also modulates the cytoprotective action of capsaicin using prostaglandin I receptor knockout mice (IP−/−).
Methods: The stomachs of IP−/− or their wild-type counterparts (IP+/+), anaesthetised with urethane (1.225 g/kg), were doubly cannulated from the oesophageal and duodenal sides, and the gastric mucosa was perfused (1 ml/min) with physiological saline. Perfusate was changed to 50% ethanol alone, or 50% ethanol containing capsaicin (16∼1600 μM). The injured area was estimated at the end of each perfusion experiment. In some animals, CGRP-(8–37), a CGRP antagonist (0.3 mg/kg), or indomethacin (1 mg/kg) was intravenously injected before perfusion of 50% ethanol containing capsaicin.
Results: Capsaicin inhibited the injured area in a dose dependent manner. Fifty per cent ethanol containing capsaicin (480 μM) immediately increased intragastric levels of CGRP although 50% ethanol alone did not. The protective action of capsaicin (480 μM) against ethanol was completely abolished by intravenous injection of CGRP-(8–37). Indomethacin also inhibited the protective action of capsaicin, and this was accompanied by reduced levels of intragastric CGRP. Intragastric levels of prostaglandin E2 were not increased by capsaicin treatment but those of 6-keto-prostaglandin F1α, a metabolite of prostaglandin I2, were markedly increased. No protective action of capsaicin was observed in IP−/− which lacked the ability to increase intragastric CGRP levels in response to ethanol containing capsaicin. The CGRP content of the stomach from untreated IP−/− did not differ from those in IP+/+. Capsaicin (160 μM) together with intragastric perfusion of beraprost sodium (PGI2 analogue, 2.5 μg/ml) showed enhanced protection against ethanol induced injury. This enhanced protection was completely blocked by intravenous injection of CGRP-(8–37).
Conclusions: The present results suggest that endogenous prostaglandin I2 enhances the protective action of the capsaicin mediated neural emergency system against ethanol induced gastric mucosal injury through enhancement of CGRP release.
- neural emergency system
- capsaicin
- prostaglandin I2
- calcitonin gene related peptide
- gastric mucosal injury
- CGRP, calcitonin gene related peptide
- IP, prostaglandin I receptor
- IP−/−, prostaglandin I receptor knockout mice
- IP+/+, wild-type mice
- EP3−/−, prostaglandin E (EP3) receptor knockout mice
- GAPDH, glyceraldehyde-3-phosphate dehydrogenase
- RT-PCR, reverse transcription-polymerase chain reaction
- VR1, vanilloid receptor 1
Statistics from Altmetric.com
- neural emergency system
- capsaicin
- prostaglandin I2
- calcitonin gene related peptide
- gastric mucosal injury
- CGRP, calcitonin gene related peptide
- IP, prostaglandin I receptor
- IP−/−, prostaglandin I receptor knockout mice
- IP+/+, wild-type mice
- EP3−/−, prostaglandin E (EP3) receptor knockout mice
- GAPDH, glyceraldehyde-3-phosphate dehydrogenase
- RT-PCR, reverse transcription-polymerase chain reaction
- VR1, vanilloid receptor 1
Prevention of gastric mucosal injury is highly dependent on the rapid alarm systems which can sense the harmful chemical or mechanical stimuli exposed to the mucosa. The gastrointestinal tract is known to be rich in neural systems, among which afferent neurones of extrinsic origin are reported to operate as the protective emergency system during exposure to certain noxious chemicals, such as ethanol. This protective system was called by Holzer the “neural emergency system” of the stomach.1 The functions of these afferents sensitive to chemicals can be mimicked by capsaicin, whose binding sites (vanilloid receptors) are expressed on C and Aδ fibres of sensory nerves.
A rich plexus of nerve fibres containing sensory neuropeptides such as substance P and calcitonin gene related peptide (CGRP) is distributed throughout the gastric mucosa.2,3 CGRP was reported to be located mainly on sensory nerves although it is also found in cells other than nerves.4,5 CGRP is a peptide which protects against the gastric mucosal injury induced by ethanol and other substances.6–10 We reported previously that capsaicin released CGRP, and prevented ethanol induced gastric mucosal injury.11 In our series of intravital microscopic studies,11–14 ethanol induced rat gastric mucosal injury was initially attributed to congestion of mucosal blood flow, which was caused by constriction of the collecting venules of the gastric mucosa. Both CGRP and capsaicin prevented ethanol induced constriction of the collecting venules and inhibited mucosal injury.15
We have previously reported that increased production of prostaglandin E2, prostaglandin I2, or both, in the rat gastric wall was related to inhibition of acid secretion by tetragastrin, motility of the gastric smooth muscle, and development of ethanol induced gastric mucosal lesions.16 It is well established that prostaglandins sensitise sensory nerves to potentiate the pain sensation.17 Thus endogenous prostaglandins generated in the stomach may prevent gastric mucosal injury by means of facilitation of the neural emergency system by sensitising sensory nerves and subsequently increasing release of neuropeptides. But, in spite of intense studies, the findings that may implicate prostaglandins as mediators of sensory neurone mediated gastric mucosal protection are still controversial.18–23 Furthermore, the receptors involved in such mucosal protection are not known. Prostaglandins exert their biological actions by binding to specific receptors that contain seven transmembrane domains. Eight different prostaglandin receptors have been defined pharmacologically and cloned, including four subtypes of prostaglandin E receptor (EP1, EP2, EP3, and EP4), and prostaglandin I receptor (IP).24 Five types of these knockout mice have been produced as a result of disruption of the genes for each of these receptors.25–30 At present, using these knockout mice is the only way of identifying the prostaglandin receptors involved.
In the present study, we determined the amount of CGRP release after administration of capsaicin and ethanol, and clarified the involvement of endogenous prostaglandins in the prevention of gastric mucosal injury by facilitating the release of CGRP using prostanoid receptor knockout mice. The present study addresses the controversial issue of the involvement of endogenous prostaglandins in the modulation of the neural emergency system.
MATERIALS AND METHODS
Prostanoid receptor knockout mice
A strain of male IP receptor knockout mice (IP−/−, eight weeks old) recently developed by us,25 and their wild-type counterparts (IP+/+, male, eight weeks old), were used. Prostaglandin E receptor knockout mice, EP3 knockout mice (EP3−/−), were also developed by us,28 and were used in some experiments. They were starved for 18 hours before the experiments began but had free access to water. The experiments were performed on the animals after they had been anaesthetised with urethane by intraperitoneal injection (1.225 g/kg; Aldrich Chemical Co., Milwaukee, Wisconsin, USA).14 The studies were carried out in accordance with the Guidelines for the Treatment of Experimental Animals of Kitasato University School of Medicine.
RT-PCR
Transcripts encoding IP1, EP3, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were quantified by reverse transcription-polymerase chain reaction (RT-PCR) analysis. Mouse stomach was removed and rapidly frozen in liquid nitrogen. Frozen tissue was pulverised in a stainless steel cylinder cooled with liquid nitrogen. Total RNA was extracted from the tissue with Isogen (Wako, Osaka, Japan), and cDNA was synthesised from 1 μg of total RNA with the use of an oligo-p(dT)15 primer and AMV reverse transcriptase (Boehringer Mannheim, Mannheim, Germany). cDNA (50 ng) was amplified with 1 U of Taq DNA polymerase in a 25 μl reaction mixture containing 10 mM Tris HCl (pH 8.3), 50 mM KCl, 1.5 mM MgCl2, 0.2 mM of each deoxynucleoside triphosphate, and 0.6 μM each of forward and reverse primers. The amplification protocol comprised 25 cycles (IP and EP3), and 20 cycles (GAPDH) of 45 seconds at 94°C, 60 seconds at 55°C, and 60 seconds at 72°C. The reaction mixtures were subsequently applied to a 2% agarose gel and the amplified products were stained with ethidium bromide. Primers used were as follows: 5′-AAC GGG CTG GCA TTG GGC ATC C-3′ (sense) and 5′-AGC AGG GGC AGT GAG CAG AAG AGG-3′ (antisense) for IP, 5′-GGA GAG ACT CAG TGC AGA AAT ATC-3′ (sense) and 5′-GAA CTG TTA GTG ACA CCT GGA ATG-3′ (antisense) for EP3, and 5′-CCC TTC ATT GAC CTC AAC TAC AAT GGT-3′ (sense) and 5′-GAG GGG CCA TCC ACA GTC TTC TG-3′ (antisense) for GAPDH.
Perfusion of the gastric lumen and measurement of CGRP in the perfusate
Surgical preparation for stomach perfusion
After laparotomy of the anaesthetised mice, the stomach was doubly cannulated from the oesophageal and duodenal ends. The cannulas were secured with thread at the middle of the oesophagus and at the pylorus, respectively. Physiological saline (37°C) containing 10 μg/ml pepstatin (pepstatin A; Peptide Institute, Inc., Osaka, Japan) was perfused at 1 ml/min, using a constant rate pump (Model 11; Harvard Apparatus, Harvard, Massachusetts, USA) connected to the oesophageal cannula, and perfusate was collected from the duodenal cannula. Before collection of the first sample, in order to stabilise the stomach, it was perfused with the above solution for more than 30 minutes. Body temperature was monitored with a thermometer (Model CTM-303; Terumo, Tokyo, Japan) and was maintained throughout at 37±1°C with a desk lamp and a heated table. Passage for air was kept patent by insertion of a plastic cannula (Clay Adams, Parsippany, New Jersey, USA) into the trachea.
Experimental procedure for the perfusion
All solutions for perfusion of the lumen of the stomach, including physiological saline, 50% ethanol solution, and 50% ethanol solution containing capsaicin, contained 10 μg/ml of pepstatin to prevent degradation of CGRP during perfusion and collection.
(1) Perfusion of the stomach with 50% ethanol alone
The stomach lumen was perfused with physiological saline at a rate of 1 ml/min, and 4 ml of intragastric perfusate were collected in four minutes. The perfusate was placed directly into a plastic tube kept on ice. After baseline sampling, the perfusion solution was replaced with 50% ethanol (Kanto Chemical Co., Inc., Tokyo, Japan) prepared with distilled water, for four minutes. We previously reported that 50% ethanol caused immediate development of mucosal lesions within four minutes after contact with the mucosa.11 Therefore, in the present experiment, we exposed the gastric mucosa for four minutes to this concentration of ethanol, which was then replaced with physiological saline containing 10 μg/ml pepstatin.
(2) Exposure of the gastric mucosa to 50% ethanol containing capsaicin
After collection of a baseline sample for four minutes, 50% ethanol containing capsaicin at final concentrations of 16∼1600 μM was perfused for four minutes, followed by physiological saline for four minutes.
In some experiments, the stomach was perfused for 10 minutes with a PGI2 analogue, beraprost sodium (2.5 μg/ml), starting two minutes before perfusion of ethanol and capsaicin.
Measurement of intragastric CGRP
CGRP levels in the perfusate from anaesthetised mice were determined by the method described in our previous report.11 To obtain a high recovery of endogenous CGRP released into the gastric lumen, 10 μg/ml of pepstatin, a dose sufficient to inhibit pepsin released from the stomach, was added to the perfusate. Perfusates were collected into plastic tubes and kept on ice. Immediately after sample collection, the tubes containing perfusate were placed in boiling water for 10 minutes and centrifuged at 2000 g for 15 minutes at 15°C. The supernatant was transferred to other tubes and evaporated to dryness. Samples were dissolved in EIA buffer (500 μl) and centrifuged (6000 g for 15 minutes at 15°C).
CGRP in the sample was determined by a highly sensitive EIA for CGRP (Societe de Pharmacologie et d’Immunologie-BIO, Massy Cedex, France) after the pretreatments outlined above.31 When samples were diluted 2-, 4-, 8-, and 16-fold with EIA buffer and the measured absorbency was plotted against the dilution rates shown on the abscissa of a graph, the curves for the diluted samples were in agreement with the standard curves provided with authentic standards in the EIA kit, suggesting that there was probably no contamination by substances that could have affected the EIA. The detection limit of the EIA was 0.4 pg/4 min perfusate.
In separate recovery experiments, physiological saline containing exogenous CGRP (50 pg/2 ml) was perfused for four minutes from the oesophageal cannula in the same way as above, and 76 (11)% (n=3) of the amount of CGRP perfused was recovered in the first four minutes.
Measurement of intragastric prostaglandin E2 and 6-keto-prostaglandin F1α levels
Levels of prostaglandin E2 and 6-keto-prostaglandin F1α in the perfusate from anaesthetised mice were measured as in our previous report.32 Briefly, the perfusate for every four minutes was collected directly in ice cold absolute ethanol, and after centrifugation at 6000 g for 15 minutes at 4°C, the supernatant was evaporated at a reduced pressure. The residue was applied to a Sep-Pack C18 and HPLC, and the resulting fractions for 6-keto-prostaglandin F1α and prostaglandin E2 were determined by EIA (Cayman Chem Corp., Ann Arbor, Michigan, USA), as reported previously.32
Administration of a CGRP antagonist, CGRP-(8–37), or indomethacin to mice
A polyethylene cannula (PE10) was inserted into the femoral artery and physiological saline containing human CGRP-(8–37) (100 μg/ml) was injected (0.3 ml/kg) five minutes before the first sample collection. We previously reported that CGRP-(8–37) inhibited dilation of gastric mucosal arterioles induced by rat CGRP in a dose dependent manner in rats33 and it has been reported that this compound did not inhibit relaxation of the vascular smooth muscle induced by bradykinin, histamine, isoprenaline, substance P, or prostaglandin E1.34–37 We confirmed that rat CGRP (1 μM and 10 μM) induced arteriolar dilatation in mice in the basal part of the mucosa to 175.5 (17.5)% (n=6) and 198.9 (29.8)% (n=4) of the original diameter, respectively. Human GCRP-(8–37) (10 μM) completely inhibited CGRP (rat, 1 μM and 10 μM) induced arteriolar dilatation to 97.1 (3.5)% (n=4, p<0.01) and 96.9 (6.0)% (n=4, p<0.05) of the original diameter, respectively. Control mice were injected only with physiological saline. Some mice were injected intravenously with indomethacin (1 mg/kg) five minutes before the first sample collection.
After ethanol perfusion, physiological saline was perfused for four minutes, and the stomach was immediately excised. The reddened area was assessed as described below.
Assessment of gross lesions in the glandular stomach
Gross lesions in the glandular stomach were determined as reported previously.11 Briefly, at the end of each perfusion experiment, the stomach was excised, and the reddened areas were calculated as percentages of the glandular stomach area using Adobe Photo Shop 4.0J. software on a Macintosh computer.
Statistical analysis
Values are expressed as means (SEM). ANOVA was used to evaluate the significance of differences with a post hoc test. A p value of less than 0.05 was regarded as significant.
RESULTS
RT-PCR analysis of IP and EP3 in mouse stomach
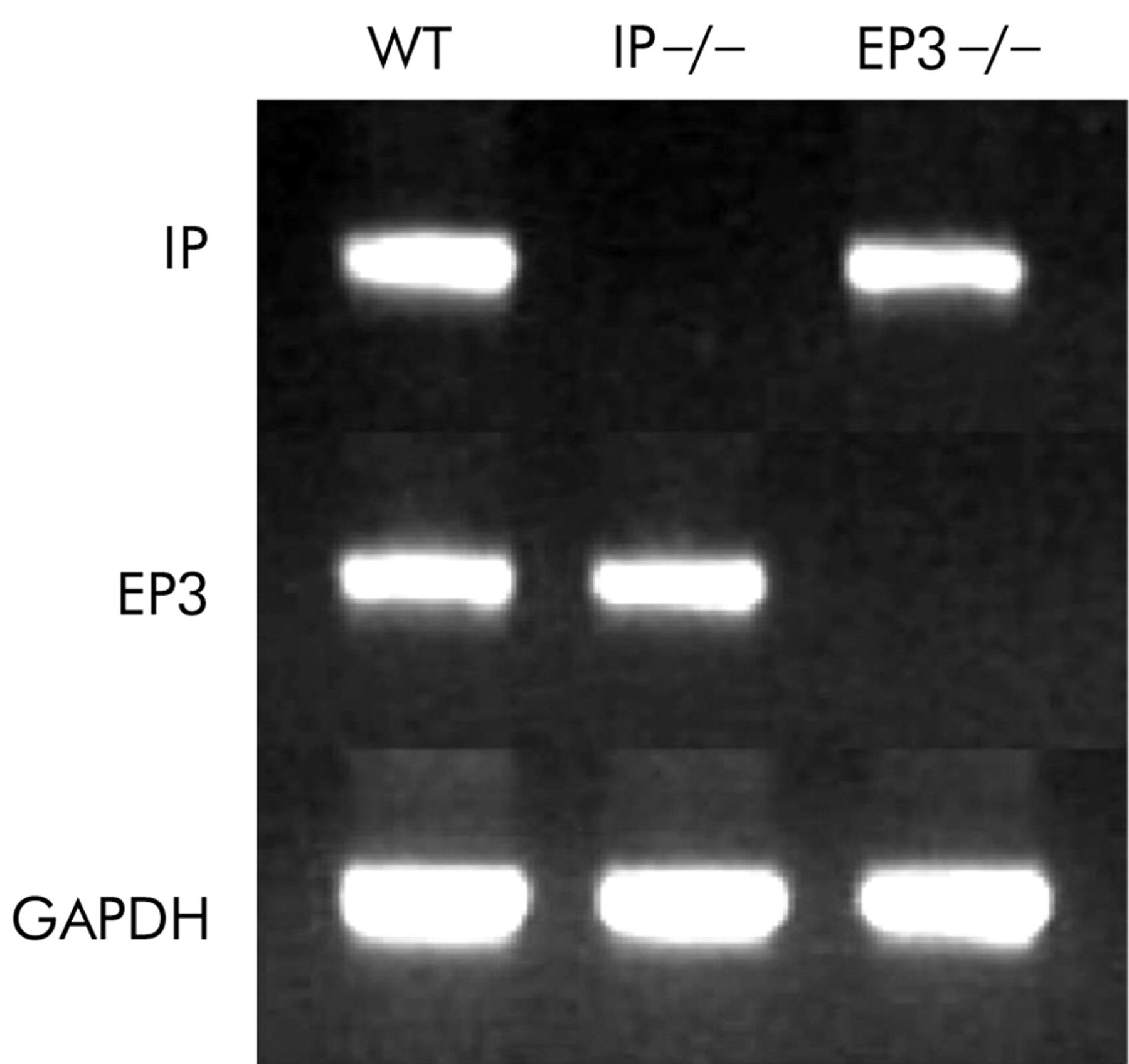
Expression of IP mRNA and EP3 mRNA were tested in whole stomach samples excised from IP−/−, PE3−/−, and their wild-type counterparts. RT-PCR analysis confirmed the lack of expression of IP in IP−/−, and that of EP3 in EP3−/− (fig 1).

Reverse transcription-polymerase chain reaction analysis of prostaglandin I receptor (IP) mRNA and prostaglandin E receptor (EP3) mRNA in the stomachs of IP knockout mice (IP−/−), EP3 knockout mice (EP3−/−), and their wild-type counterparts (WT).
Effect of capsaicin on ethanol induced gastric mucosal lesions in mice
Perfusion with 50% ethanol after preperfusion with physiological saline resulted in gastric mucosal lesions that affected approximately 20% of the area of the glandular stomach in wild-type mice (fig 2). Simultaneous application of capsaicin to ethanol reduced gastric lesions in a dose dependent manner, and the effect of more than 480 μM of capsaicin was statistically significant (fig 2).

Effect of capsaicin (Cap) on 50% ethanol (EtOH) induced mucosal injury in wild-type mice. Perfusion was performed in wild-type mice (C57BL/6) according to the protocol described in materials and methods. Capsaicin blocked mucosal injury in a dose dependent manner. Values are expressed as means (SEM) from four to six mice. ANOVA was used for statistical analysis: **p<0.01; ***p<0.001 compared with 50% ethanol only.
Changes in intragastric levels of CGRP released and effect of a CGRP antagonist on gastric mucosal lesions in mice
Intragastric CGRP levels during perfusion of physiological saline in wild-type mice were kept fairly low (1.4 (0.3) pg/stomach/4 minutes, n=5), and exposure of the gastric mucosa to 50% ethanol after perfusion with physiological saline did not raise these levels significantly (fig 3A). Nor were levels elevated when ethanol solution was returned to physiological saline. However, intragastric exposure to capsaicin (480 μM) dissolved in 50% ethanol markedly increased intragastric CGRP levels, and their elevation persisted even after the perfusate was changed to physiological saline (fig 3A).

Effect of intravenous injection of a calcitonin gene related peptide (CGRP) antagonist, CGRP(8–37), on the area of ethanol (EtOH) induced gastric lesions reduced by capsaicin (Cap), and changes in CGRP levels released during perfusion. Perfusion was performed in wild-type mice (C57BL/6) according to the protocol described in materials and methods. The reddened area reduced by simultaneous perfusion of capsaicin was restored by intravenous injection of CGRP(8–37). Intragastric CGRP levels were increased with ethanol+capsaicin perfusion, but not with ethanol alone. Values are expressed as means (SEM) from four to six mice. ANOVA was used for statistical analysis: **p<0.01; ***p<0.001.
To evaluate the contribution of released CGRP to the prevention of 50% ethanol induced mucosal injury, the effect of intravenous injection of a CGRP antagonist on the protective effect of capsaicin against ethanol was tested (fig 3B). Prior intravenous injection of CGRP-(8–37) completely abolished the reduction of the area of gastric injury attributable to 480 μM of capsaicin (fig 3B).
Effect of indomethacin on gastric mucosal lesions and intragastric CGRP levels in mice
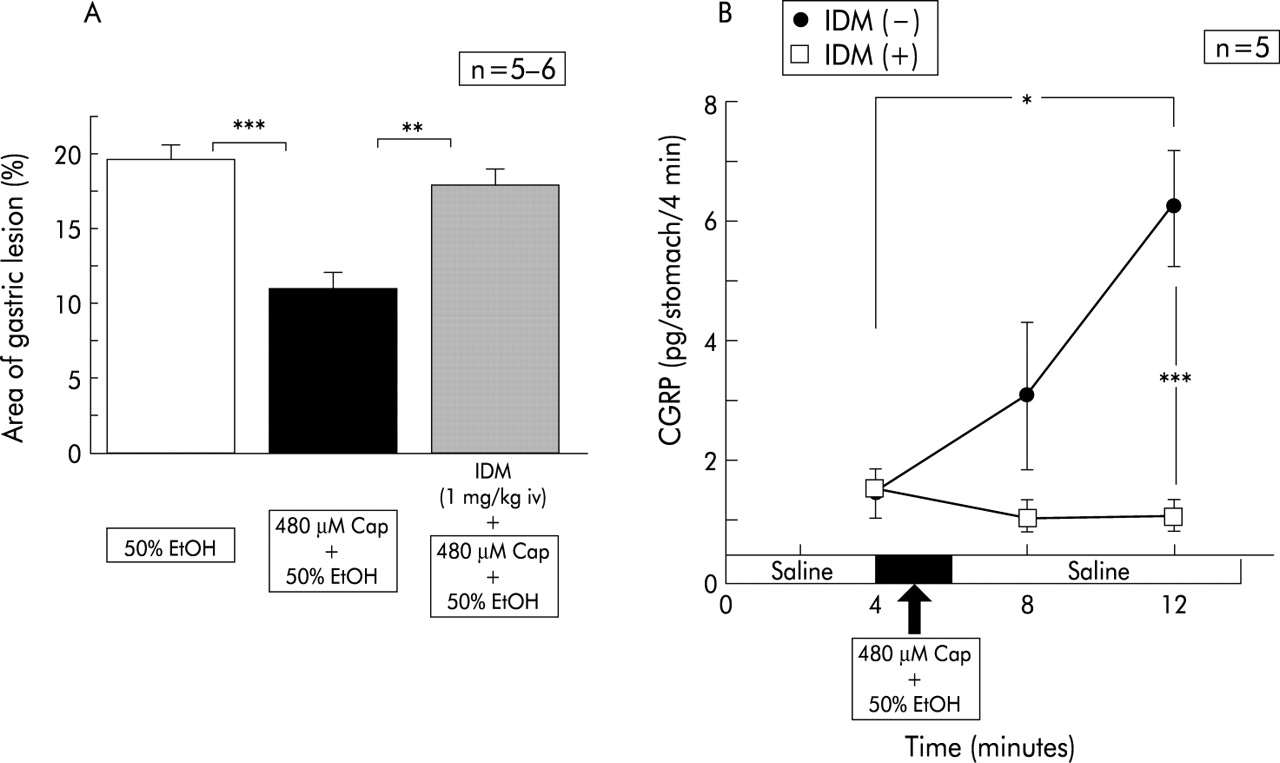
To test the involvement of endogenous prostaglandins in the preventive effects of capsaicin, we administered indomethacin prior to perfusion of ethanol containing capsaicin. As fig 4B indicates, indomethacin treatment restored the protective action of capsaicin almost completely.

Effect of intravenous injection of indomethacin (IDM) on the area of ethanol (EtOH) induced gastric lesions reduced by capsaicin (Cap), and changes in calcitonin gene related peptide (CGRP) levels released during perfusion. Perfusion was performed in wild-type mice (C57BL/6) according to the protocol described in materials and methods. The reddened area reduced by simultaneous perfusion of capsaicin was restored by intravenous injection of indomethacin. Indomethacin reduced levels of CGRP released by ethanol+capsaicin to basal control values. Values are expressed as means (SEM) from five to six mice. ANOVA was used for statistical analysis: *p<0.05; **p<0.01; ***p<0.001.
Intravenous injection of indomethacin itself five minutes before the first sample collection did not change basal values of intragastric CGRP (fig 4A). In contrast, without indomethacin, the subsequent exposure to 50% ethanol containing capsaicin did not increase intragastric CGRP levels (fig 4A).
Changes in endogenous intragastric prostaglandin levels
Resting intragastric 6-keto-prostaglandin F1α levels during perfusion of physiological saline were approximately 20 pg/stomach/4 min, and exposure to 50% ethanol containing capsaicin significantly increased intragastric 6-keto-prostaglandin F1α levels by more than fourfold in the first perfusate obtained after exposure (fig 5A). However, this increase was transient because when the perfusate was replaced with physiological saline, intragastric 6-keto-prostaglandin F1α levels were immediately reduced to their basal resting levels.

Intragastric prostaglandin (PG) levels during perfusion experiments. Perfusion was performed in wild-type mice (C57BL/6) according to the protocol described in materials and methods. Levels of 6-keto-prostaglandin F1α and prostaglandin E2 in the perfusates were determined in each four minute sample. Values are expressed as means (SEM) from five mice. ANOVA was used for statistical analysis. Values during perfusion of capsaicin (Cap)+ethanol (EtOH) were compared with basal control values (saline) (***p<0.001). Values after perfusion of capsaicin+ethanol were compared with those during perfusion of capsaicin+ethanol (***p<0.001).
Basal resting levels of intragastric prostaglandin E2 during perfusion of physiological saline were slightly lower than those of 6-keto-prostaglandin F1α, although the difference between the two prostanoids was not statistically significant. In contrast, exposure of the gastric mucosa to 50% ethanol containing capsaicin did not elevate prostaglandin E2 levels significantly (fig 5B).
Effects of capsaicin on ethanol induced gastric mucosal lesions in prostanoid receptor knockout mice
The effect of capsaicin on gastric mucosal lesions induced by 50% ethanol was tested in IP receptor knockout mice (IP−/−), EP3 receptor knockout mice (EP3−/−), and their wild-type counterparts.
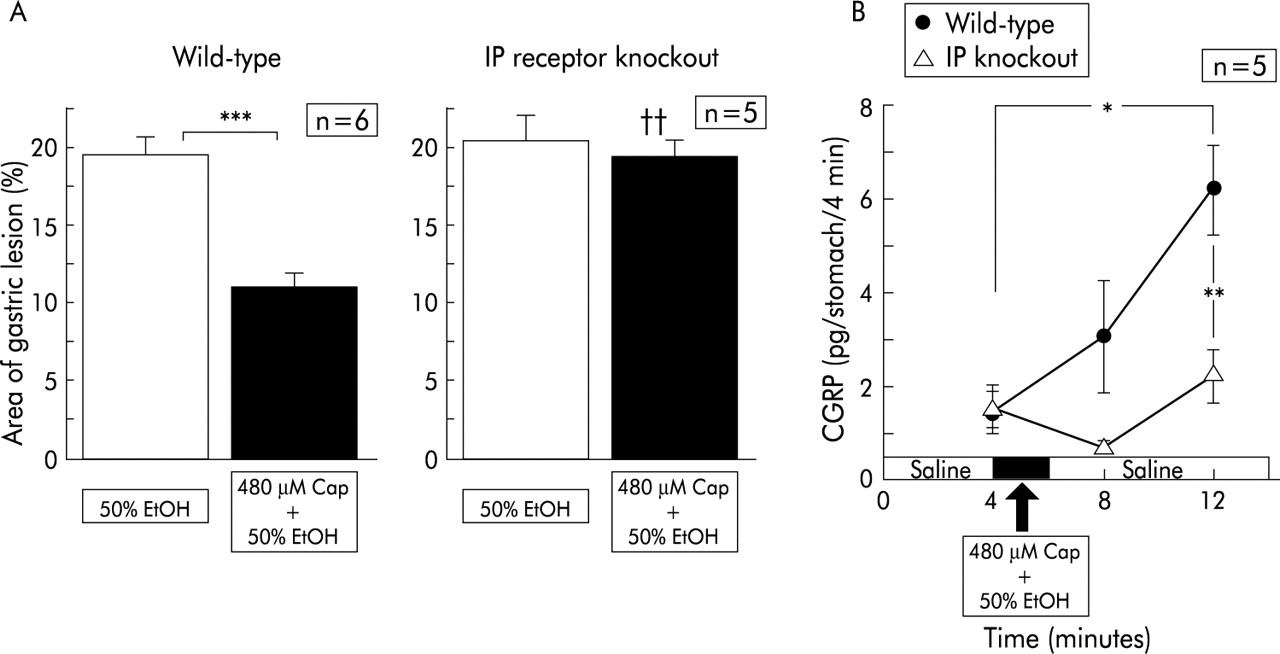
In wild-type mice, pre-exposure of the gastric mucosa to 50% ethanol after perfusion of physiological saline induced gastric mucosal lesions with a reddened area covering 19.7 (0.9)% (n=6) of the area of the glandular stomach (fig 6B). Intragastric exposure to 50% ethanol containing capsaicin significantly reduced the size of the mucosal lesions (fig 6B). When 50% ethanol containing capsaicin was perfused, CGRP levels released from the gastric mucosa were markedly increased (fig 6A).
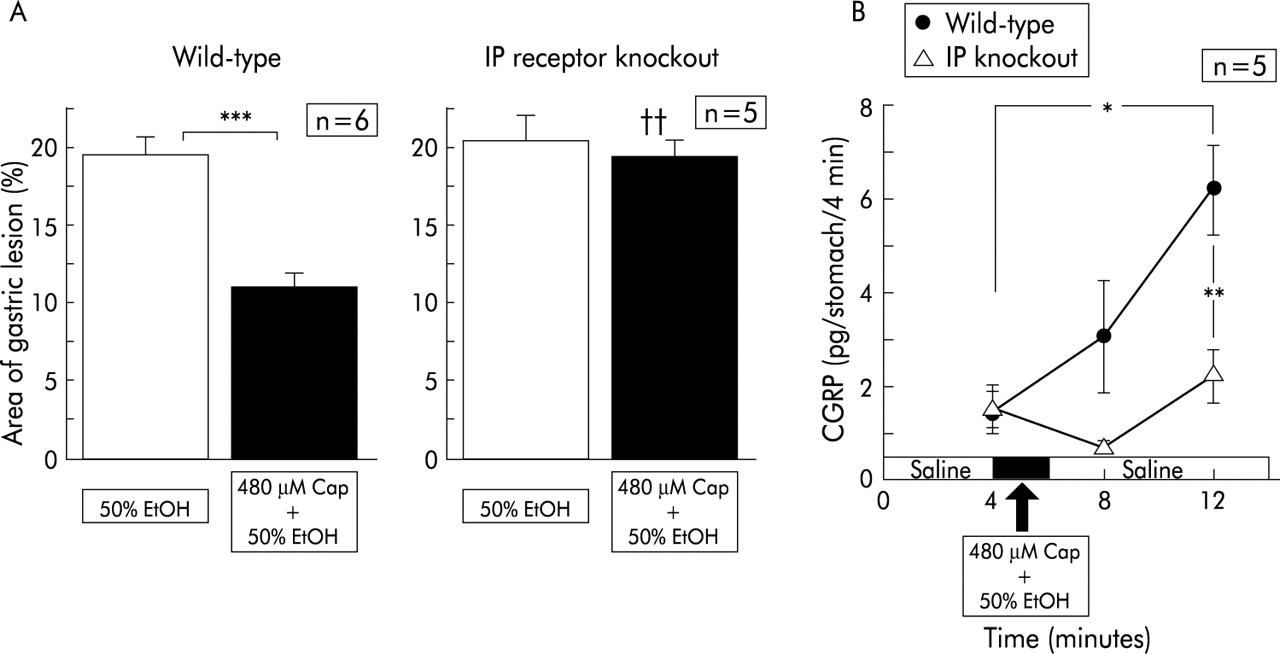
Effect of capsaicin (Cap) on gastric mucosal lesions induced by 50% ethanol (EtOH) in prostaglandin I receptor (IP) knockout mice and their wild-type counterparts, and changes in calcitonin gene related peptide (CGRP) levels released during perfusion. Perfusion was performed in IP receptor knockout mice and wild-type mice according to the protocol described in materials and methods. Values are expressed as means (SEM) from five to six mice. ANOVA was used for statistical analysis. (A) Comparisons were made between the results from ethanol treated mice and capsaicin+ethanol treated mice (***p<0.001). Comparison between IP receptor knockout mice and wild-type mice was also made (††p<0.01).
In IP−/−, exposure to 50% ethanol alone after physiological saline resulted in mucosal lesions with a reddened area extending over 20% of the glandular stomach (fig 6B). The injury that was developed in IP−/− was not substantially different from that seen in wild-type mice. In contrast, treatment with capsaicin did not elicit any protective effect against ethanol induced mucosal injury in IP−/− (fig 6B). In IP−/−, the increase in CGRP levels in response to ethanol exposure was absent even after preperfusion with ethanol containing capsaicin (fig 6A).
When EP3−/− were perfused with 50% ethanol after saline perfusion, the relative area of the reddened lesions was 20.0 (2.5)% (n=5), which was not different from that seen in wild-type counterparts. Perfusion with capsaicin reduced ethanol induced lesions significantly in EP3−/−. The marked increase in CGRP released from the stomach was seen in EP3−/− during exposure of 50% ethanol containing capsaicin. These increased levels were not different from those observed in wild-type counterparts.
Facilitation of the preventive effect of capsaicin by intragastric perfusion of a prostaglandin I2 analogue, beraprost sodium
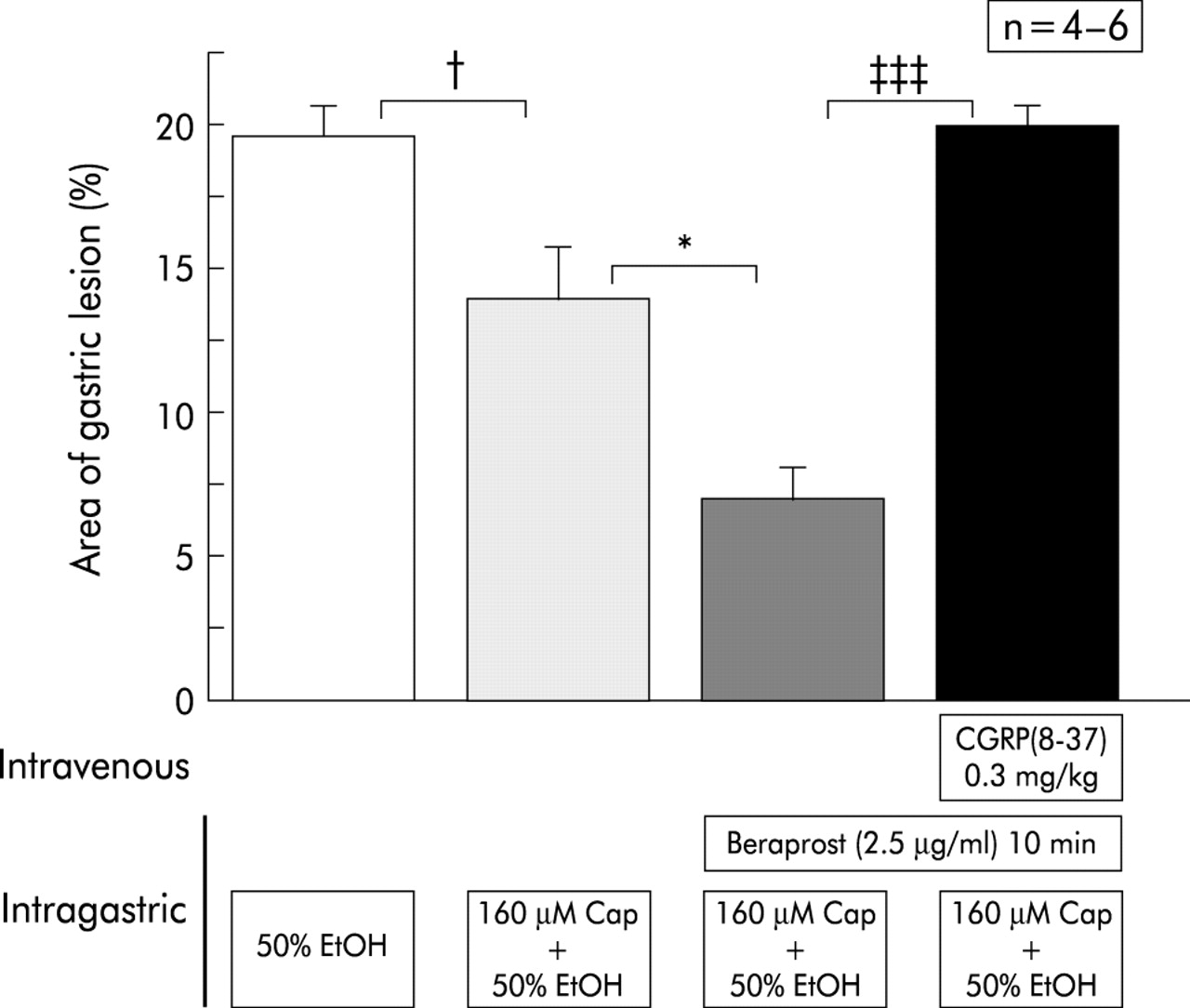
As shown in fig 7, the preventive effect of a low dose of capsaicin (160 μM) in wild-type mice was enhanced by continuous intragastric perfusion of a prostaglandin I2 analogue, beraprost sodium. The enhanced preventive effect was equivalent to that of a high concentration of capsaicin (480 μM). This enhancement in prevention was completely inhibited by intravenous injection of CGRP-(8–37) (fig 7).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of beraprost sodium on the protective action of a low concentration of capsaicin (Cap) against mucosal lesions induced by 50% ethanol (EtOH). Beraprost sodium was perfused together with 50% ethanol and enhanced the protective action of a low concentration of capsaicin. The enhancement was completely blocked by intravenous injection of the calcitonin gene related peptide (CGRP) antagonist CGRP-(8–37). Perfusion was performed in wild-type mice. Values are expressed as means (SEM) from four to six mice. ANOVA was used for statistical analysis: †p<0.05,*p<0.05, ‡‡‡p<0.001.
DISCUSSION
Maintenance of the gastric mucosal barrier is important in terms of protection from harmful substances such as hydrogen ions that are secreted from the mucosa and are capable of producing cell injury.38 Little is known of whether or not gastric neurones contribute towards gastric mucosal protection from injury, although the vagus nerve has long been regarded as a permissive factor in peptic ulcer disease. Recently, extrinsic afferent neurones were found to operate as a neural emergency system in the digestive tract.1 Released neuropeptides from their peripheral endings are known to regulate a variety of physiological functions, including an increase in resistance of the tissue to injury and enhancement of repair of damaged tissues. To study the roles of these sensory neurones, capsaicin, which can act on certain primary afferent neurones and can release neuropeptides selectively, is widely used.39
The pathophysiological involvement of prostaglandins in the nervous systems, including the peripheral sensory nervous system, has been intensively studied, and recent findings regarding constitutive expression of COX-2, an inducible isozyme of cyclooxygenase, in the nervous systems can facilitate the study of the roles of prostaglandins in the central nervous system. It has been frequently reported that prostaglandins can intensify pain sensation, and that the analgesic effects of NSAIDs can be attributed to inhibition of COX activity and consequently prostaglandin generation. However, which prostaglandins are involved in the neural emergency system is still controversial in spite of intensive study of this system. Intragastric capsaicin does not affect in vitro formation of prostaglandin E2, 6-oxo-prostaglandin F1α, or leukotriene C4, and indomethacin fails to alter the protective effect of capsaicin in rats.18 Further evidence that exogenous CGRP had a protective action against gastric mucosal injury, a process which was not affected by indomethacin, indicated that prostaglandins may not be involved in the signalling pathway of CGRP in rats.19 Some reports stated that the gastrointestinal protective action of capsaicin was not blocked by indomethacin although in these studies the authors did not measure endogenous prostaglandin levels or their inhibition by indomethacin.20–23 In the present study, we first determined levels of CGRP in the gastric perfusate and found them to be increased immediately after exposure to capsaicin. Ethanol itself did not increase CGRP levels, which was consistent with our published results.40 An increased level of CGRP exhibits a protective action, based on the fact that capsaicin induced prevention of ethanol injury was completely eliminated by pretreatment with CGRP-(8–37). We further demonstrated the inhibition of capsaicin induced release of CGRP by indomethacin, which was accompanied by complete inhibition of the protective action. These results confirm the involvement of prostaglandins in the protective action of capsaicin, and this action can be explained by increased release of CGRP. Previous results of other investigators confirmed prostaglandin involvement but all failed to determine the level of CGRP, which can strengthen the prostaglandin involvement. As capsaicin and indomethacin reversed completely the intensity of injury, the direct harmful actions of these agents may be excluded in the present experiment. Furthermore, the effect of capsaicin was reversed by CGRP antagonist. This also supports the lack of direct actions of these agents.
The major arachidonate metabolites in the stomach in rabbits were reported to be prostaglandin I2 and prostaglandin E2.41 There has been little information on prostaglandins generated in mouse stomach. We previously reported that intragastric administration of a mild irritant generated prostaglandin I2 and prostaglandin E2 in mouse stomach.41 It is widely known that prostaglandin E2 and prostaglandin I2 have a sensitising effect on sensory nerves.17 In terms of sensitising activity, prostaglandin I2 was found to be more potent than prostaglandin E2 in our previous study, which used a dog model whose blood pressure elevation occurred as a nociceptive response after administration of a potent pain inducer, bradykinin.42 We recently reported that in IP receptor knockout mice of the same type as those used in the present experiment, prostaglandin I2 was predominant in potentiating the pain sensation.25 In the present study, capsaicin significantly increased prostaglandin I2 levels but not prostaglandin E2 levels. Furthermore, we showed that IP receptor signalling is important in the protective action of capsaicin as this protective action was eliminated in IP−/− mice. The site of generation of prostaglandin I2 was not examined in the present study. As JTE522, a COX-2 selective inhibitor, did not affect the preventive action (data not shown), it is plausible that the generated prostaglandin I2 was COX-1 derived. It was reported previously that strong immunoreactivity to COX-1 was localised in the mucous neck cells of the gastric mucosa, and that only weak reactivity to this enzyme is found in the mucous cell types in the cardiac and pyloric glands of the stomach and in Brunner’s glands of the duodenum in rats.43 Another study also reported COX-1 immunoreactivity in the apical cytoplasm of mucous neck cells.44 The stomach expressed an abundance of prostaglandin I2 synthase although its localisation was not fully elucidated.45 Furthermore, our in situ hybridisation study revealed that IP receptor mRNA was expressed in the neurones of the dorsal root ganglion, suggesting that these neural IP receptors have significant roles.46
Immunohistochemical study of gastric CGRP has revealed that the immunoreactivity of CGRP is located mainly in spinal sensory nerve endings47 which originate from the dorsal root ganglia, and has shown positive reactions to PGP 9.5 antibody specific to nerve fibres.4 Thus CGRP containing afferent nerves may have IP receptors. Considering the localisations of these participants in this protective action, it is reasonable to surmise that prostaglandins, of which prostaglandin I2 has a labile nature, can act in an autocrine or paracrine fashion (or both) on CGRP containing nerve fibres and so facilitate release of CGRP. The cloned vanilloid receptor 1 (VR1) is recognised as a common molecular target for protons, noxious heat, and capsaicin. VR1 immunopositive nerve endings were predominantly found in the mucous neck cells of the proliferation zone, and around blood vessels in the submucosa in rat stomach.48 There has not been precise information on the localisation of VR1 in the mouse stomach although mice lacking VR1 were developed.49 In spite of the lack of information on the localisation of VR1 in mice, VR1 and CGRP may be colocalised in the mouse stomach as release of CGRP is increased by capsaicin.
In the present study, we used EP3 deficient mice for comparison as EP3 receptor subtype stimulation was reported to be involved in prostaglandin E2 induced sensitisation of polymodal receptors in response to bradykinin and heat.50 As shown in the present study, prostaglandin E2 was not increased in mouse stomach after stimulation with capsaicin. Judging from the increase after capsaicin stimulation, PGI2 may be the predominant prostaglandin that exhibits a protective action. In fact, IP knockout mice lack the protective action of capsaicin (fig 6) whereas perfusion with capsaicin reduced ethanol induced lesions significantly in EP3−/−. IP receptor signalling is known to increase intracellular cAMP levels in various cells. This increase in cAMP levels may explain sensitisation of the release of CGRP in response to capsaicin. Small amounts of CGRP mRNA and CGRP are also found in non-neural cells but the precise role of these CGRP positive cells in cytoprotective activity and their responsiveness to capsaicin have not been fully investigated.5
In summary, the present experiments showed that endogenous prostaglandin I2 has a modulating action on capsaicin induced CGRP release and facilitates the protective action of capsaicin against ethanol induced injury. The sensitising effect of prostaglandin I2 on CGRP containing nerves may be a crucial mechanism in the prevention of gastric mucosal injury.
Acknowledgments
We thank Michiko Ogino and Osamu Katsumata for technical assistance. We express our thanks to Mr C W P Reynolds for correcting the English of this manuscript. This work was supported by grants from an Integrative Research Program of the Graduate School of Medical Science, Kitasato University, and from Parents’ Association Grant of Kitasato University School of Medicine. This work was also supported by a research grant (#15390084), by a “High-tech Research Center” grant, and by a grant from the 21st Century COE Program, from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT).