Article Text

Abstract
Background—Expression of pro-inflammatory cytokines is increased in the intestinal lamina propria of patients with inflammatory bowel disease (IBD). Nuclear factor κB (NFκB) controls transcription of inflammation genes. On activation, NFκB is rapidly released from its cytoplasmic inhibitor (IκB), transmigrates into the nucleus, and binds to DNA response elements in gene promoter regions.
Aims—To investigate whether increased activation of NFκB is important in IBD and may be down-regulated by anti-inflammatory treatment.
Methods—Activation of NFκB was determined by western blot assessment and electrophoretic mobility shift assay in nuclear extracts of colonic biopsy samples as well as lamina propria mononuclear cells.
Results—Nuclear levels of NFκB p65 are increased in lamina propria biopsy specimens from patients with Crohn’s disease in comparison with patients with ulcerative colitis and controls. Increased activation of NFκB was detected in lamina propria mononuclear cells from patients with active IBD. Corticosteroids strongly inhibit intestinal NFκB activation in IBD in vivo and in vitro by stabilising the cytosolic inhibitor IκBα against activation induced degradation.
Conclusions—In both IBDs, but particularly Crohn’s disease, increased activation of NFκB may be involved in the regulation of the inflammatory response. Inhibition of NFκB activation may represent a mechanism by which steroids exert an anti-inflammatory effect in IBD.
- interleukin 1ß
- inflammatory bowel disease
- intestinal immunity
- signal transduction
- steroids
- tumour necrosis factor α
Statistics from Altmetric.com
- interleukin 1ß
- inflammatory bowel disease
- intestinal immunity
- signal transduction
- steroids
- tumour necrosis factor α
Recent studies have shown increased production of pro-inflammatory cytokines including interleukin (IL) 1ß and tumour necrosis factor α (TNF-α) in inflammatory bowel disease (IBD).1-6 Increased levels of TNF-α have also been reported in stools of children with active Crohn’s disease.7 The increased production of pro-inflammatory cytokines in the intestinal mucosa is thought to be an important factor in the pathophysiology of intestinal inflammation in IBD.4-6 However, the pivotal elements in the regulation of the increased inflammatory activity remain unclear. Important candidates are transcription factors that bind to gene promoter regions and are involved in the regulation of inflammation gene transcription. Nuclear factor κB (NFκB) was originally defined as a heterodimeric complex of two subunits, p65 and p50.8 ,9 The NFκB family includes the proteins p50 and its precursor p105 (NFκB1), p52 and its precursor p100 (NFκB2), p65 (RelA), c-Rel, and Rel B.8 ,9 Functionally active NFκB represents homo- or hetero-dimers of members of the Rel family of transcription factor proteins, which mostly include p65 as one of the molecules. The main function of NFκB appears to be the transcriptional regulation of certain genes as a transactivating factor. Binding of NFκB to specific DNA sequences located in gene promoter regions is a pivotal event in the regulation of transcriptional events by the factor.8 ,9 NFκB was originally defined by its ability to bind to a 10 bp oligonucleotide motif first identified in the enhancer region of the immunoglobulin κ light chain gene. Using labelled oligonucleotides resembling this binding sequence, NFκB can be assessed semiquantitatively by the electrophoretic mobility shift assay (EMSA).10 The detection of specific DNA binding describes a crucial event in the mechanisms of transcriptional regulation by NFκB, although further steps are necessary for the factor to exert control over inflammation gene transcription. Most proteins of the NFκB family are constitutively present in the cytoplasm of mononuclear phagocytes. In resting cells, they are bound to inactivating molecules such as inhibitor κB α (IκBα) or large NFκB precursors—for example, p105.8 ,9 A host of activating stimuli including lipopolysaccharide (LPS) and TNF-α induce site specific phosphorylation of IκBα and consecutive rapid dissociation of the complex accompanied by proteolytic degradation of IκBα.11-13 The released NFκB proteins subsequently transmigrate from the cytoplasm into the nucleus where they can augment transcription of specific genes in synergy with other pro-inflammatory transcription factors including NF-IL6.8-11 Nuclear translocation of both NFκB p50 and p65 can be blocked by the serine protease inhibitor N-α-tosyl-phenylalanine chloromethyl ketone (TPCK), which stabilises IκBα against degradation.14 NFκB may play a central role in the regulation of chronic inflammation by controlling the transcription of inflammation genes. Many of the pro-inflammatory cytokine genes include NFκB binding sites, and most have been shown to be transcriptionally regulated by the factor.8 ,9 ,15 ,16 Most interestingly, NFκB dimers containing p65 appear to have profound pro-inflammatory activity, whereas the p50 homodimer (NFκB1) is either inactive or may even be involved in blocking NFκB sites in some inflammation gene promoters against binding of p65 dimers.16-18Overexpression of p50 homodimers has been suggested to be the mechanism of LPS refractoriness following repetitive stimulation of mononuclear phagocytes.19 Recent studies suggest that activation of NFκB involving the p65 subunit may be a key event in trinitrobenzenesulphonic acid (TNBS)/ethanol colitis.20Although not shown in vivo, a role for NFκB p65 activation in the regulation of intestinal inflammation in IBD seems likely. In this study we show increased levels of NFκB p65 in nuclear extracts of intestinal lamina propria biopsy samples from patients with IBD. Nuclear extracts from lamina propria of patients with Crohn’s disease, a disease in which a particular role for TNF-α has been suggested,2-7 contained substantially higher levels of NFκB p65 than those from patients with ulcerative colitis. Total mucosal levels of IκBα are not different in patients with IBD and controls. Biopsy specimens as well as isolated lamina propria mononuclear cells (LPMNCs) from patients with active IBD show enhanced activation of NFκB in comparison with controls. Therefore activation of NFκB, which involves the p65 subunit, may contribute to the enhanced expression of inflammation genes observed in IBD. Steroids protect IκBα from degradation in vitro and thereby reduce the amount of available functionally active nuclear NFκB by inhibiting translocation of the p65 subunit. Inhibition of NFκB driven transcriptional control of pro-inflammatory cytokines may be an important factor in the anti-inflammatory action of steroids in IBD.
Materials and methods
Fetal calf serum was purchased from Gibco (Grand Island, NY, USA) or Sigma (St Louis, MO, USA). Cytokine enzyme linked immunosorbent assay (ELISA) kits were obtained from Genzyme/Virotech (Rüsselsheim, Germany) and from DPC Biermann (Bad Nauheim, Germany). Recombinant TNF-α was purchased from Genzyme, and antibodies against IκBα and NFκB p65 from Santa Cruz (Heidelberg, Germany). All other chemicals were obtained from Sigma if not specified differently. TPCK was obtained from Calbiochem/Nitric Oxide Research (La Jolla, CA, USA).
PATIENTS
A total of 35 patients with active Crohn’s disease and 39 patients with active ulcerative colitis participated in the study (the exact numbers of patients being investigated in the different experiments conducted are indicated). All patients with IBD were attending the outpatient clinics of the Charité University Hospital in Berlin because of increased clinical activity. At the time that blood was withdrawn or mucosal biopsy samples were obtained, 25/35 patients with Crohn’s disease and 35/39 patients with ulcerative colitis were receiving treatment with oral salicylates (mesalazine (Salofalk, Claversal or Pentasa), salazosulfapyridine (Azulfidine or Colopleon), olsalazine (Dipentum)). None of the patients were being treated with steroids or cytotoxic drugs, immunosuppressives or antibiotics. All patients underwent sigmoidoscopy or colonoscopy for routine clinical evaluation. Only patients with inflammatory activity indicated by endoscopic assessment were included. Infection or parasites were excluded by stool cultures, microscopic stool examination, and serology (yersinia). Blood was withdrawn after informed consent, and the study was granted prior approval by the institutional review board. Normal controls were age and sex matched healthy volunteers. Disease specificity controls included patients with diverticulitis, salmonellosis, and infectious enterocolitis.
HUMAN LPMNCs
Colonic biopsy specimens were obtained during routine colonoscopy, and LPMNCs were isolated from intestinal mucosa biopsy samples as described previously.6 In brief, after overnight collagenase digestion, the cell suspension was collected, layered on a Ficoll-Hypaque gradient (specific gravity 1.077), and centrifuged at 400 g for 40 minutes. Cells were harvested from the interphase between medium and Ficoll, washed and resuspended in 10 ml phosphate buffered saline supplemented with 10% neonatal calf serum, centrifuged at 500 g for 15 minutes, and the cell pellet resuspended in culture medium. The percentage of macrophages was determined by both non-specific esterase detection (Sigma) and staining for CD68 (Dako Diagnostika GmbH, Hamburg, Germany) using the alkaline phosphatase-anti-alkaline phosphatase (APAAP) technique (Vector Laboratories, from Dako). Additional controls were performed by fluorescence activated flow cytometry (FACS) using the FITC-Leu-M3 antibody (Becton-Dickinson, Mountainview, CA, USA). Contamination of epithelial cells was found to be less than 0.5% by both morphological examination and cytokeratin antibody (Dako) staining. The percentage of macrophages, CD4 T cells, CD8 T cells, and B cells found was similar to that described in earlier flow cytometric studies.21
CELL CULTURES AND CYTOKINE ASSAYS
Cells were cultured at a concentration of 1 × 106/ml in RPMI 1640 (supplemented with 10% fetal calf serum, 1% pyruvate, 100 U/ml penicillin, and 50 μg/ml gentamicin). After 24 hours, supernatants were separated from cells by centrifugation, snap frozen and stored at −70°C until determination of cytokine levels. Supernatant concentrations of TNF-α, IL-1ß and IL-1ra were assessed using a specific sandwich ELISA. All samples were analysed in triplicate or duplicate. The amount of cross reactivity was assessed by comparison with the concentration yielding a 50% inhibition of binding. Specificity of the ELISAs was demonstrated by the finding that up to 100 pg/ml IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL-8, IL-10, IL-12, granulocyte colony stimulating factor, granulocyte-macrophage colony stimulating factor, and TNF-α showed no significant cross reactivity or interference. Sensitivity levels were between 2.0 pg/ml (IL-1ß) and 4.0 pg/ml (TNF-α), and intra- and inter-assay precision variability was below 3%.22 Repeated thawing and freezing of the cultured cells, which would release cytokines stored in the cytoplasm, did not increase the cytokine levels significantly. Therefore all cytokines synthesised were secreted into the supernatant. For analysis of transcription factor activation, cells from patients with IBD and normal control cells were incubated for 15–90 minutes with LPS (1 μg/ml).
EXTRACTS
Biopsy samples were snap frozen in liquid nitrogen at the time of removal and later mechanically homogenised in liquid nitrogen (Dounce homogeniser). Cells were pelleted and immediately fractionated. Nuclear and cytosolic extracts respectively were prepared by adaptation of previously described techniques through freeze/thaw cycles between crushed ice and liquid nitrogen.23 ,24 Cytosolic extracts were collected in an aqueous buffer containing 10 mM Hepes (pH 7.9), 1.5 mM MgCl2, and 10 mM KCl. Nuclear extracts were prepared by solubilising the remaining nuclei in a second buffer containing 20 mM Hepes (pH 7.9), 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, and 25% glycerol. Both buffers were supplemented with 1 mM dithiothreitol, 0.5 mM phenylmethanesulphonyl fluoride, 1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 mM benzamidine, 1 mM sodium vanadate, and 1 mM NaF. The purity of the extract fractions was tested by marker proteins exclusively expressed in one of the compartments (IκBα and fructose aldolase as markers for the cytosol and OCT-1 as a nuclear marker).25-27 For the assessment of total expression levels of the proteins studied, western blot experiments were performed utilising snap frozen cell pellets or homogenised biopsy specimens which were lysed in boiling extraction buffer (1% sodium dodecyl sulphate, 10 mM Tris, pH 7.5, 1 mM sodium vanadate). Insoluble material was removed by centrifugation of samples for 20 minutes at 27 000 g. Total protein concentration in a small aliquot of sample was assessed using either the method of Lowry or a modified Bradford protein assay (Bio-Rad, Munich, Germany), and all samples were adjusted to an equal protein content before analysis.28 In addition, equal loading of proteins was controlled by total protein staining of all membranes with colloidal gold (Bio-Rad, Hercules, CA, USA).29 Only membranes with equal amounts of protein loaded were used for analysis. In parallel, total histone protein was assessed in nuclear extracts (antibody from Boehringer-Mannheim), and found to yield results identical with those from total protein staining.
EMSA FOR THE DETECTION OF SPECIFIC OLIGONUCLEOTIDE BINDING OF ACTIVATED NFκB
EMSA was used to detect specific binding of activated NFκB by the formation of DNA complexes.10 ,23 ,30 Nuclear protein-DNA binding studies were carried out for 30 minutes at 24°C in a 20 μl reaction volume containing 20 mM Hepes, pH 7.9, 10% glycerol, 5 mM MgCl2, 0.1 mM EDTA, 0.5 mM dithiothreitol, 1μg poly(dI-dC), 1 fmol digoxigenin-labelled DNA probe, and 5 μg nuclear protein. Gel electrophoresis was performed as described by Houet al 23 in an adaptation of methods described previously.10 ,30 A DNA probe, as described previously, was prepared by annealing the following two consensus oligonucleotides,31 which were labelled at the 5′ end with digoxigenin (Biometra, Mannheim, Germany and Boehringer-Mannheim):
- (i)
- 5′-AGT TGA GGG GAC TTT CCC AGG C-3′
- (ii)
- 3′-TCA ACT CCC CTG AAA GGG TCC G-5′.
A 6% non-denaturing polyacrylamide gel was used for electrophoretic separation. DNA probes bound to NFκB were retarded, whereas unbound (free) DNA probes migrated to the bottom of the electropherogram. Supershift assays with a polyclonal p65 antibody were carried out to confirm the presence of NFκB p65 as part of the complex. After blotting to a membrane, labelled oligonucleotides were detected by alkaline phosphatase anti-digoxigenin (Fab)2 fragments (Boehringer-Mannheim). In some experiments oligonucleotides were labelled with [32P]dATP using a random primed DNA labelling kit from Boehringer-Mannheim. Radioactive gels were fixed in an aqueous solution of 10% ethanol and 10% acetic acid before being dried by heat exposure. It was found that the non-radioactive detection system established was equal in sensitivity and specificity to conventional radiolabelled oligonucleotides.
WESTERN BLOT ASSESSMENT OF NFκB P65 AND IκB
A 5–15 μl volume of cell lysate (5–10 μg total protein) was separated on a 12% denaturing polyacrylamide gel. Separated proteins were transferred to a polyvinylidene difluoride membrane (20 V; 75 minutes; transfer buffer 25 mM Tris, 190 mM glycine, 20% methanol, 0.5% sodium dodecyl sulphate) by electroblotter (Bio-Rad, Hercules, CA, USA). The membrane was placed into blocking buffer (5% non-fat milk in 10 mM Tris/HCl, pH 7.5, 100 mM NaCl, 0.1% Tween 20) for one hour at room temperature. Blocking buffer was decanted and the membrane was incubated with primary antibody (diluted in blocking buffer) on a shaker at room temperature (one hour). After being washed (10 mM Tris/HCl, pH 7.5, 100 mM NaCl, 0.1% Tween 20), the membrane was incubated with a peroxidase conjugated secondary antibody, which was diluted in 5% non-fat milk in wash buffer (one hour; room temperature; gentle shaking). After being washed, the membrane was placed in a hybridisation bag containing Western View (Dianova/Transduction Laboratories) working solution. The bands were quantified by densitometry (Lumi-Imager; Boehringer-Mannheim). Equal loading of gels was controlled by assessing total protein staining of all membranes with colloidal gold.29
EXPRESSION OF DATA
Results are expressed as mean (SD) if not indicated otherwise. All experiments were carried out three or more times. Normal distribution of data was evaluated by calculating Lilliefors probabilities based on the Kolmogorov-Smirnov test.32 ,33 Statistical significance of the differences for non-normally distributed data was tested with the Mann Whitney U test or the Wilcoxon matched pairs test.33 ,34
Results
INCREASED EXPRESSION OF NFκB P65 IN NUCLEAR EXTRACTS FROM COLONIC BIOPSY SAMPLES OF PATIENTS WITH IBD
Nuclear protein from colonic biopsy samples was extracted. Nuclear levels of NFκB p65 protein were evaluated by western blot in colonic biopsy samples of ten patients with active Crohn’s disease, 14 patients with active ulcerative colitis, six disease specificity controls, and seven normal controls (table 1). Samples were diluted to equal concentrations of total protein before analysis. Patients with Crohn’s disease exhibited higher levels of nuclear NFκB p65 than those with ulcerative colitis (p = 0.035), disease specificity controls (p = 0.0312), and normal volunteers (p = 0.0012). In patients with ulcerative colitis, more nuclear NFκB p65 was found than in normal controls (p = 0.038), but no statistical difference was seen in comparison with disease specificity controls. The increased expression of nuclear NFκB p65 in disease specificity controls did not reach significance in comparison with normal controls (fig 1). Alternatively, biopsy specimens from the same sites were homogenised for the assessment of total tissue concentrations of NFκB p65 protein. Total levels of NFκB p65 were not different in patients with Crohn’s disease, those with ulcerative colitis, normal controls, and disease specificity controls (fig 2), and no differences in IκBα levels were seen between the groups (fig 2).

NFκB p65 in nuclear extracts from colonic biospsy samples. Colonic biopsy specimens from 10 patients with active Crohn’s disease (CD), 14 patients with active ulcerative colitis (UC), six disease specificity controls (DSC), and seven normal controls (NC) were extracted using a procedure to isolate the nuclear compartment. Densitometric readings from western blot assessments of NFκB p65 are demonstrated by the bar graph (mean (SD)). Before electrophoresis, samples were adjusted to have equal contents of total protein. The inset shows a representative sample of the original blots. Individuals are marked on the blot with U (ulcerative colitis), C (Crohn’s disease), D (disease specificity controls), or N (normal volunteer). The characteristics of the patients and localisation of the biopsy samples are shown in table 1. The disease specificity control patients shown in the blot are patients 28, 25, 27, and 29 (from left to right).

Total NFκB p65 and IκBα in colonic biospy tissue. Colonic biopsy specimens were homogenised and analysed by western blot without prior compartmentalised extraction. Densitometric readings are shown by the bar graph (mean (SD)). The filled bars show levels of total NFκB p65 (left y axis), and the shaded bars levels of IκBα (right y axis). Before electrophoresis, samples were adjusted to have equal contents of total protein. No statistical differences were seen between patients with active Crohn’s disease (CD, n = 6), patients with active ulcerative colitis (UC, n = 6), disease specificity controls (DSC, n = 6), and normal controls (NC, n = 5). The inset shows representative blots (C = Crohn’s disease, U = ulcerative colitis, D = disease specificity control, and N = normal control). Biopsy specimens were taken from the same location as those used for nuclear extracts (fig 1).
DNA BINDING OF NUCLEAR NFκB IS INCREASED IN EXTRACTS FROM LAMINA PROPRIA BIOPSY SAMPLES AS WELL AS IN LPMNCs ISOLATED FROM COLONIC BIOPSY SPECIMENS FROM PATIENTS WITH IBD
The increased levels of NFκB p65 in nuclear extracts in Crohn’s disease indicate that there may also be increased amounts of NFκB dimers which are capable of specific binding to consensus oligonucleotides. Figure 3 shows a representative gel shift experiment demonstrating increased amounts of DNA binding of NFκB in nuclear extracts from biopsy specimens (fig 3A) as well as isolated LPMNCs (fig3B). In all Crohn’s disease biopsy samples as well as unstimulated LPMNCs, greatly increased levels of nuclear NFκB could be detected by its specific oligonucleotide binding activity. In contrast with normal controls, most patients with ulcerative colitis also showed increased amounts of NFκB. The presence of p65 as part of the complex was confirmed by supershift assays (not shown). The parallel assessment of nuclear p65 levels by western blot again showed higher levels of NFκB p65 in nuclear extracts from Crohn’s disease LPMNCs than in ulcerative colitis or controls (fig 3B).
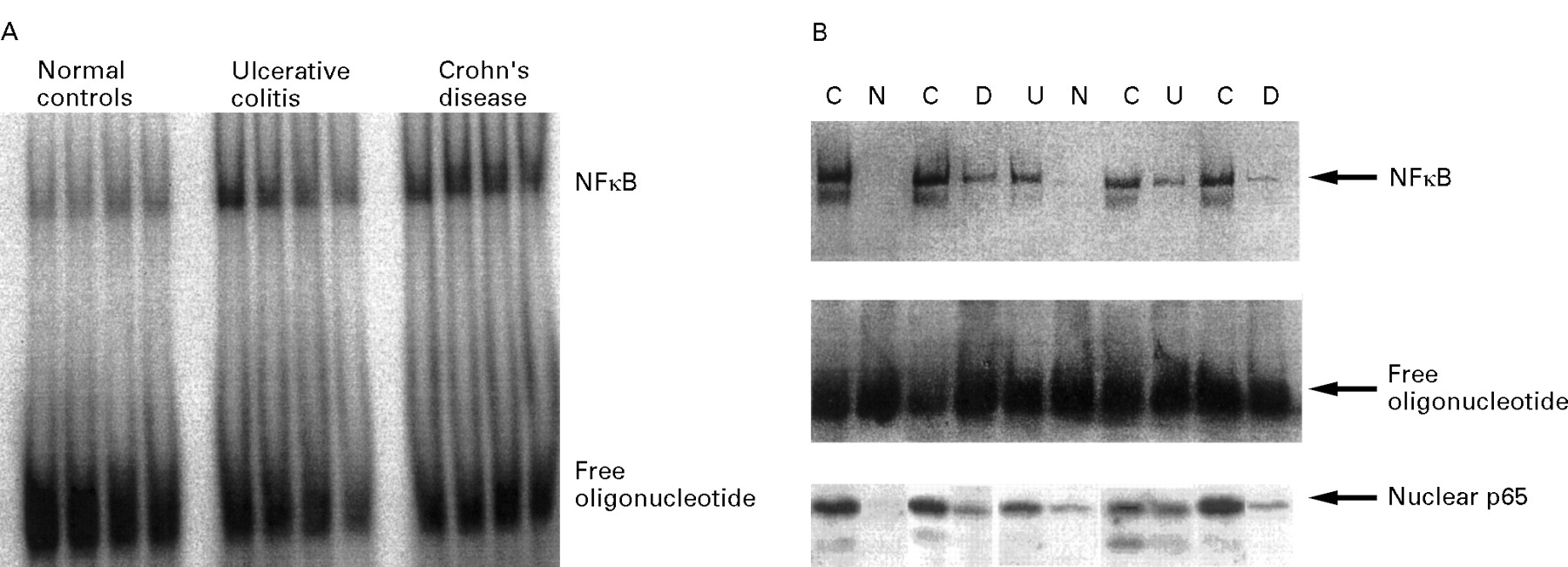
Detection of NFκB in the intestinal lamina propria. (A) Nuclear extracts were prepared from colonoscopic biopsy specimens. A representative radioactive electrophoretic mobility shift assay using consensus oligonucleotides to detect NFκB in nuclear extracts from intestinal tissue is shown. In both Crohn’s disease (n = 9) and ulcerative colitis (n = 8) increased levels of NFκB were found in comparison with disease specificity controls (n = 5) and healthy volunteers (n = 8). (B) Nuclear extracts were prepared from LPMNCs, which were freshly isolated from colonic biopsy specimens. A blot from a representative non-radioactive gel shift experiment with ten samples (C = Crohn’s disease; U = ulcerative colitis; D = disease specificity controls; N = normal controls) is shown. The highest levels of activated NFκB were seen in patients with active Crohn’s disease, with similar levels in all four samples. In only one of four normal control samples could low levels of oligonucleotide binding proteins be detected. Levels in patients with ulcerative colitis (four) as well as disease specificity controls (three) appeared to be increased as well, although not as much as in Crohn’s disease. The presence of NFκB p65 as part of the complex was controlled by supershift experiments (not shown). The levels of nuclear p65 determined in the same extracts by western blot are shown in the lower part of the figure.
INHIBITION OF NFκB TRANSLOCATION DOWN-REGULATES PRO-INFLAMMATORY CYTOKINE SECRETION BY IBD LPMNCs
The serine protease inhibitor TPCK inhibits activation of NFκB by preventing nuclear translocation of both p65 and p50 through stabilisation of IκBα.14 As table 2 shows, TPCK down-regulates pro-inflammatory cytokine secretion by IBD LPMNCs in a similar fashion to dexamethasone. As expected, dexamethasone as well as TPCK greatly reduced LPS stimulated nuclear levels of oligonucleotide binding of activated NFκB (fig 4A). Dexamethasone as well as TPCK stabilised IκBα against LPS induced proteolysis analogously to the effect previously described in peripheral blood mononuclear cells (fig4B).
Down-regulation of pro-inflammatory cytokine secretion by specific inhibition of NFκB activation

Inhibition of NFκB activation (A) and IκBα degradation (B) by steroids (dexamethasone) and N-α-tosyl-phenylalanine chloromethyl ketone (TPCK). Nuclear extracts were prepared from lamnia propria mononuclear cells obtained from colonoscopic biopsy samples (two patients with Crohn’s disease and two normal volunteer controls). Cells were cultured for 30 minutes in the presence of lipopolysaccharide (LPS). A representative blot from a patient with Crohn’s disease is shown. (A) Both dexamethasone (10 μM) and TPCK (30 μM) strongly reduced the amounts of NFκB available for binding to consensus oligonucleotides. (B) IκBα was assessed in total extracts by western blot. TPCK as well as dexamethasone stabilised IκBα against activation induced degradation.
STEROID TREATMENT REDUCES ACTIVATION OF NFκB
Nuclear extracts were prepared from sigmoid biopsy samples from seven patients with highly active Crohn’s ileocolitis and additional inflammation in the sigma (CDAI 250–450), before they received steroid treatment. Patients were examined again after seven days of treatment with high dose systemic (60 mg prednisolone/day) and topical (5 mg betamethasone in 100 ml, twice daily) steroids. Nuclear levels of NFκB were decreased in all patients by steroid treatment (fig 5; p = 0.0156), whereas IκBα concentrations remained unchanged or were also decreased (data not shown).
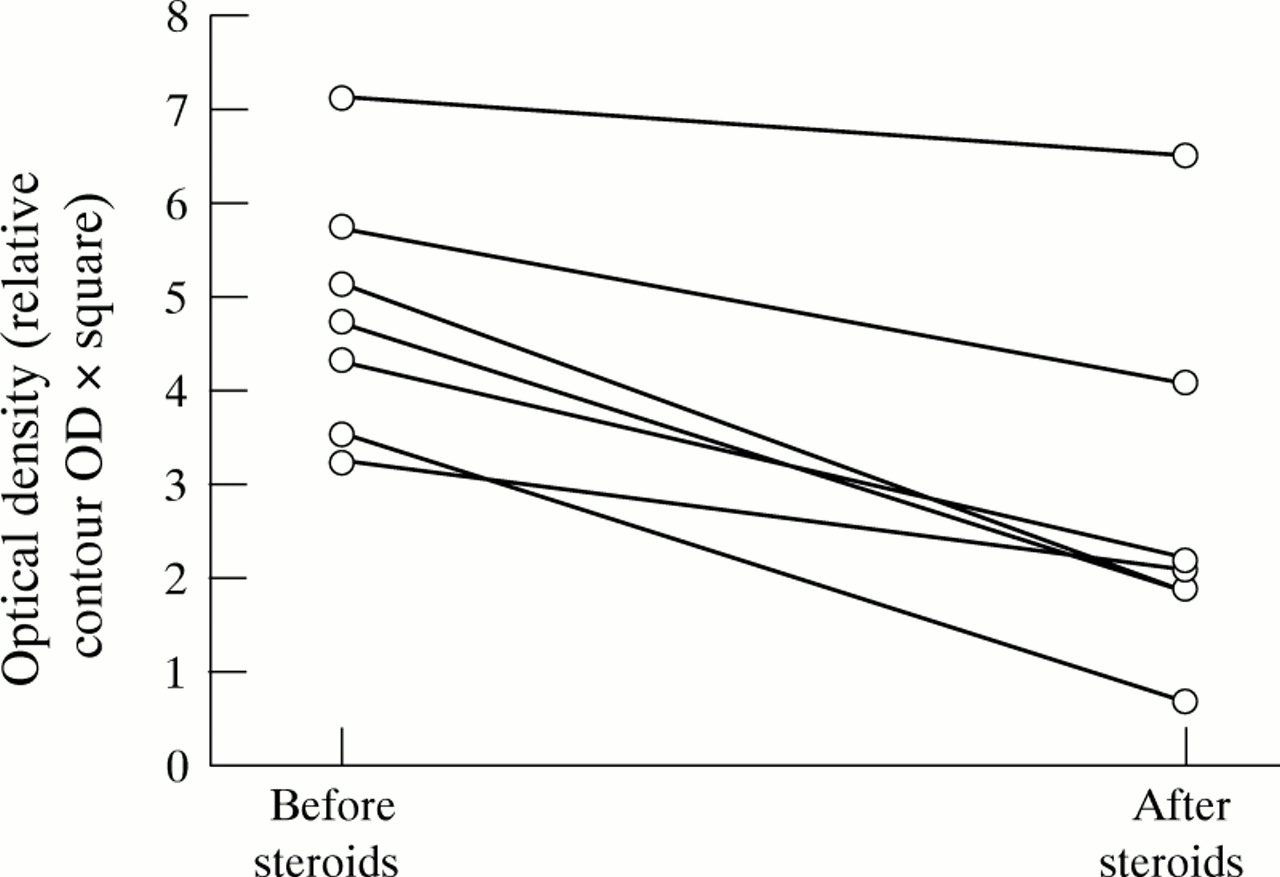
Nuclear NFκB p65 from colonoscopic biopsy specimens from patients with inflammatory bowel disease is decreased by steroid treatment. Sigmoid biopsy specimens were obtained from patients with moderately to highly active ileocolonic Crohn’s disease and involvement of the sigmoid (CDAI 250–450). Nuclear extracts were prepared and concentrations of NFκB p65 were assessed by western blot. Seven days of treatment with steroids (60 mg prednisolone/day per os in addition to 5 mg betamethasone twice daily as an enema) induced a significant reduction (p = 0.0156) in nuclear NFκB p65 concentration. IκBα levels remained unchanged (data not shown).
Discussion
Both IL-1ß and TNF-α have been suggested to be important mediators involved in the initiation and perpetuation of intestinal inflammation in IBD. Increased secretion by isolated IBD LPMNCs as well as whole biopsy samples has been shown in comparison with normal control cells. Activation of NFκB may be a pivotal event in pro-inflammatory signal transduction.8 ,9 Transcription factor members of the Rel-NFκB family, which in resting cells are bound to cytoplasmic inhibitory factors (for example IκB, NFκB p105), are released on activation induced degradation of their cytoplasmic inhibitors by the proteasome/ubiquitin complex.9 ,11-13 ,24 NFκB p65 hetero- or homo-dimers migrate to the nucleus and regulate transcriptional activity by binding to specific DNA sequences in promoter/enhancer regions of inflammation genes.8 ,9 We therefore investigated both the level of nuclear NFκB p65 protein and oligonucleotide binding of activated nuclear NFκB. Intestinal biopsy specimens were investigated because they reflect the in vivo state of NFκB activation without any potential alterations that may be caused by in vitro isolation procedures. We found increased amounts of NFκB p65 protein in nuclear extracts of mucosal biopsy samples of patients with Crohn’s disease and ulcerative colitis in comparison with both disease specificity controls and normal controls. The highest levels were seen consistently in lamina propria biopsy samples from patients with Crohn’s disease. Potential candidates responsible for the activation of the NFκB system in Crohn’s disease may include TNF-α. Recent immunological as well as therapeutic studies suggest that TNF-α plays a major role in the pathophysiology of intestinal inflammation in Crohn’s disease35 but it may be less important in chronic ulcerative colitis.36 The increased nuclear concentrations of NFκB p65 detected in IBD mucosal biopsy samples may partially be a product of TNF stimulation by non-immune cells—for example, epithelial cells. TNF induced involvement of NFκB in the regulation of inflammation genes in these cells may be an important regulatory mechanism in the inflammatory process. Several other cell types should also be discussed as potential contributors to the activation of the NFκB system seen in intestinal lamina propria in IBD. Protein concentrations (as assessed by western blot) of NFκB p65 and IκBα were not different in intestinal mucosa from patients with IBD, disease specificity controls and normal controls if total tissue extracts were not separated into cytoplasmic and nuclear extracts. This finding is expected because activation of NFκB results in a shift from the cytoplasm to the nucleus. Moreover, IκB concentrations, which are decreased by degradation in response to a stimulus, rapidly returned to their original levels in vitro within less than 90 minutes.9-13 Translocation into the nucleus and binding to specific DNA sequences are important events which are prerequisites for transcriptional control by NFκB. Enhanced detection of NFκB p65 protein should therefore be accompanied by increased availability of oligonucleotide binding of NFκB. Increased nuclear levels of consensus oligonucleotide binding of activated NFκB were detected in both Crohn’s disease and ulcerative colitis biopsy samples as well as LPMNCs in comparison with normal controls. Crohn’s disease LPMNCs again appeared to contain more NFκB p65 protein in nuclear extracts than LPMNCs from patients with ulcerative colitis. The heightened activation of NFκB could be a major regulator of pro-inflammatory cytokine secretion in IBD. However, a host of other activating stimuli, including TNF and LPS, may also induce the activation of NFκB as a secondary event. In order to dissect the regulatory role of NFκB in IBD, we tried to block the release of cytosolic NFκB proteins into the nuclear compartment using the serine protease inhibitor TPCK.14 TPCK strongly down-regulated the increased secretion of the pro-inflammatory cytokines TNF-α and IL-1ß in LPMNCs from patients with IBD. These experiments indicate a regulatory role for activated NFκB in the inflammatory cytokine response in IBD. Moreover, it appears that NFκB p65 (RelA), which has been suggested to have a greater pro-inflammatory role in the induction of inflammation gene expression than p50,16-19 is an important factor in the regulation of intestinal inflammation in IBD. Our results which demonstrate increased nuclear levels of NFκB p65 as well as activated oligonucleotide binding of NFκB in IBD support the concept of a perpetuating stimulation of the NFκB system, which is characterised by rapid activation and de-activation kinetics.8 ,9 In vitro exposure to steroids rapidly inhibits NFκB activation. Previous reports have shown that glucocorticoids use stabilisation of IκBα as part of their anti-inflammatory mechanism of action.37 ,38 Our study confirms that steroids cause in vitro reduction of activated NFκB in IBD LPMNCs. Moreover, in vivo treatment with high dose steroids leads to inhibition of NFκB p65 activation in Crohn’s disease, which paralleled clinical improvement of patients. However, this inhibition of NFκB activation was not accompanied by in vivo enhancement of IκBα, as would have been expected from in vitro systems.37 ,38 Studies currently being performed therefore examine the role of the IκB/NFκB system during clinical relapse of disease from remission and in experimental treatment situations in IBD (glucocorticoids, IL-10, anti-TNF antibodies). Activation of NFκB may play a central role in the initiation of the relapsing inflammatory process and moreover may be targeted by the therapeutic mechanism of various anti-inflammatory drugs including corticosteroids37 ,38 and aminosalicylates (fig6).39 Activation of NFκB may be a pivotal element in the pathophysiology of chronic intestinal inflammation, but is unlikely to be the primary cause of IBD.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
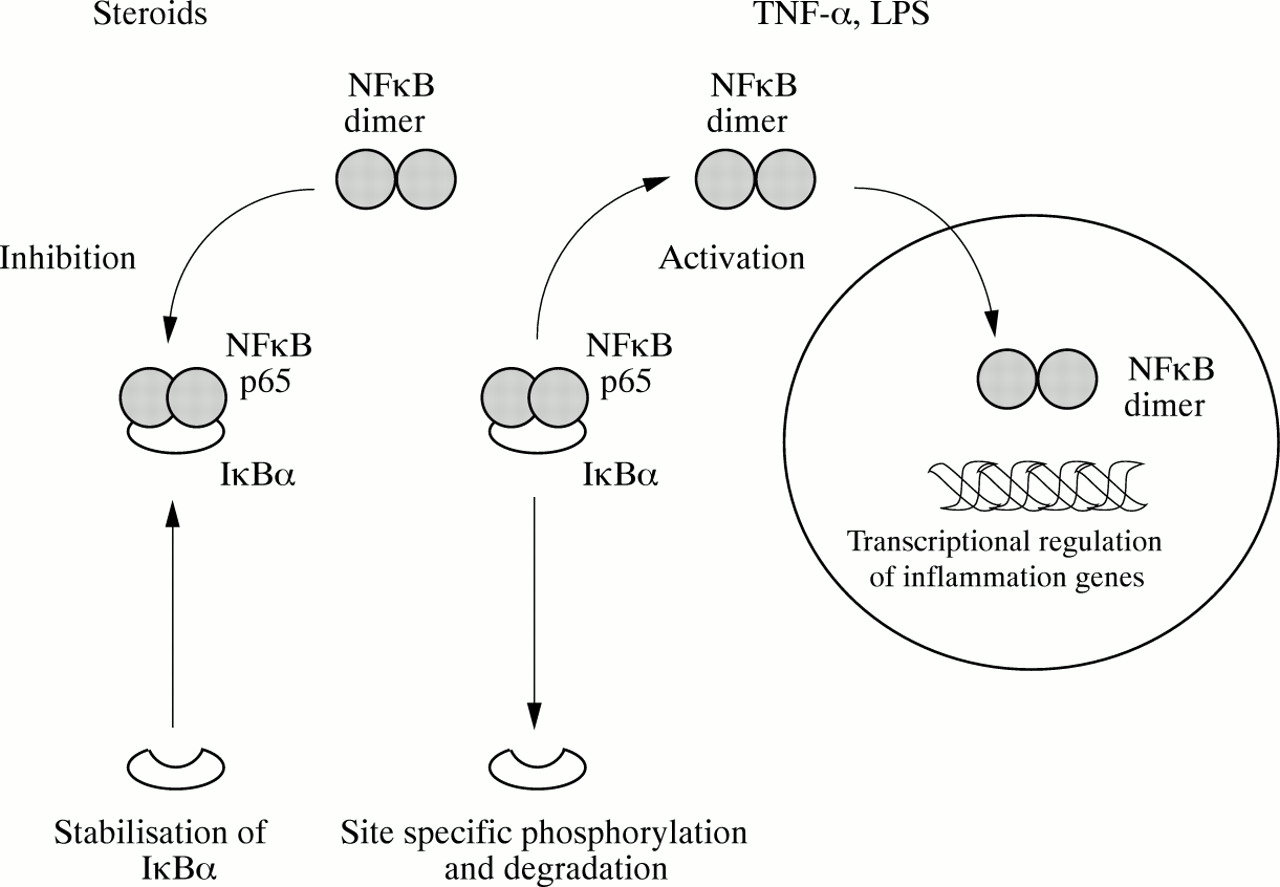
Schematic representation of NFκB activation and inhibition by steroids. Activating stimuli including tumour necrosis factor α (TNF-α) and lipopolysaccharide (LPS) promote degradation of IκBα and the subsequent release of NFκB p65 into the cytosolic compartment. NFκB p65 translocates into the nucleus and forms dimers as part of the activation process. Functionally active NFκB can then bind to specific sites in inflammation gene promoter regions and initiate transcription. Steroids appear to stabilise IκBα against activation induced degradation and thereby reduce the amount of functionally active NFκB available in the nucleus.
Acknowledgments
Parts of this work have been presented at the 5th European Gastroenterology Week in Paris, France (1996). This work was supported by a grant from the Deutsche Forschungsgemeinschaft (SCH 512/1–2 to S S), by an award from the German Crohn’s and Colitis Foundation (DCCV, to S N), by a student research prize from the Ministry of Science of Saxony, Germany (to J H) and by development grants from the Genzyme GmbH and Syngen Pharma GmbH. The authors gratefully appreciate critical advice from H Lochs (Berlin), A Raedler (Hamburg), and U Schindler (South San Francisco) as well as excellent technical help from A M Wenner and S Eidner. The contribution of the endoscopy staff as well as S Wedel in obtaining biopsy specimens and recruiting patients is gratefully acknowledged. The authors are indebted to the healthy volunteers who served as normal controls.