Article Text

Abstract
BACKGROUND Substantial evidence implicates the neuropeptide substance P (SP) in mucosal immunoinflammatory responses. Autoradiographic studies have suggested a disturbance in SP receptor expression in inflammatory bowel disease (IBD).
AIMS Because of technical limitations such as poor cellular resolution with autoradiography, we used molecular methods to specifically localise the cellular expression of the neurokinin-1 receptor (NK-1R) in IBD colon, and to quantitate NK-1R mRNA expression levels therein.
METHODS In situ hybridisation and immunohistochemistry were used to localise NK-1R mRNA and protein, respectively, in normal, ulcerative colitis (UC), and Crohn's disease (CD) colonic resections. NK-1R mRNA expression levels of normal, UC, and CD mucosal biopsies were quantitated by competitive reverse transcription-polymerase chain reaction.
RESULTS NK-1R expression was localised to lamina propria mononuclear cells, epithelium, submucosal vasculature, smooth muscle, and myenteric plexus of normal and IBD colon. No ectopic NK-1R expression was observed in IBD. However, we found increased numbers of NK-1R expressing lymphoid cells in IBD tissue, aberrant negative epithelial expression of NK-1R in UC, and increased expression of NK-1R in CD myenteric plexus. Quantitation of NK-1R mRNA expression in IBD colonic mucosal biopsies revealed marked upregulation of NK-1R mRNA levels compared with non-inflamed mucosal expression levels (p<0.01).
CONCLUSIONS This report demonstrates the strategic localisation and upregulation of NK-1R expression in IBD colon, and thereby suggests the involvement of substance P in the pathophysiological symptoms of IBD.
- substance P
- neurokinin-1 receptor
- inflammatory bowel disease
Abbreviations used in this paper
- CD
- Crohn's disease
- IBD
- inflammatory bowel disease
- LPMC
- lamina propria mononuclear cells
- NK-1R
- neurokinin 1 receptor
- RT-PCR
- reverse transcription-polymerase chain reaction
- qcRT-PCR
- quantitative competitive RT-PCR
- SP
- substance P
- UC
- ulcerative colitis
- IL
- interleukin
- TNF-α
- tumour necrosis factor α
- PBS
- phosphate buffered saline
Statistics from Altmetric.com
Substantial evidence indicates that the neuropeptide substance P (SP) plays a role in inflammatory and immune responses.1 A spectrum of immunoregulatory effects has been described for this sensory neuropeptide.2 SP has been shown to influence lymphocyte traffic,3 lymphocyte proliferation,4 ,5 immunoglobulin production,4 ,6 chemotaxis of neutrophils,7phagocytosis of macrophages,8 and mast cell degranulation.9 Monocytes produce the inflammatory cytokines interleukin (IL)-1, IL-6, IL-10, and tumour necrosis factor α (TNF-α) in response to stimulation with SP.10 ,11The gastrointestinal tract is one of the most abundant sources of SP in the body.12 SP has been identified in extrinsic sensory neurones13 and in intrinsic enteric neurones of the gut,14 where it is known to modulate smooth muscle contractility, epithelial ion transport, vascular permeability, and immune function.12 ,15
Several lines of evidence implicate SP in the pathophysiology of certain experimental models of inflammatory bowel disease (IBD).16 Elevated levels of SP have been associated withTrichinella spiralis induced enteritis of rats17 and mice.18 Blockade of SP with either SP antibodies18 or with the SP antagonist CP 96,34519 markedly reduced jejunal inflammation of this parasitic infection. SP has also been shown to play an important role in Clostridium difficile toxin A enterocolitis.20 Administration of the specific SP receptor antagonists CP 96,34520 or CP 99,994,21 or desensitisation of sensory nerves with capsaicin21 ,22 inhibited toxin A mediated fluid secretion and intestinal inflammation. SP receptor gene knockout mice were protected from epithelial cell damage, and the inflammatory and secretory changes induced by toxin A.23 In the cotton-top tamarin model of spontaneous colitis, blockade of neurokinin-1 receptors by the SP antagonist LY-303870 blunted inflammation.24
Elevated levels of SP have been described in ulcerative colitis (UC),25-28 and have been shown to correlate with disease activity.25 ,26 Several findings report an increased concentration of SP nerve fibres in the colon of UC patients.29-31 Recently, Watanabe et al found that the linear density of SP fibres increased significantly in UC patients with active disease.31 Other findings demonstrated a decrease in SP nerves in severe inflammatory lesions of UC.32 Conflicting results have also been reported for Crohn's disease (CD). Mucosal SP levels were found to be significantly decreased in CD patients.25 Others have reported no significant difference between mucosal SP levels from CD patients and those of controls.27 An increased density of SP immunoreactive fibres has been demonstrated in CD colon.29 Compelling evidence for a disturbance in SP immunomodulation in IBD was described by Mantyh et al who demonstrated autoradiographically 1000-fold upregulation of SP binding sites in the lymphoid follicles and vasculature of patients with IBD.33 They also reported increased expression of SP binding sites on enteric neurones of patients with active and quiescent CD, but not on enteric neurones of patients with UC.34
Interpretation of autoradiographic studies is hampered by such technical limitations as poor resolution, internalisation of receptors, ligand degradation, and promiscuity of ligand binding. Rapid agonist induced endocytosis of the SP (neurokinin-1) receptor has been observed both in vivo and in vitro.35 ,36 SP can also bind with lower affinity to many non-neurokinin receptors.37-39 The issue of SP receptor expression in IBD therefore needed to be readdressed. In the present study we used in situ hybridisation and immunohistochemistry to localise the cellular expression of neurokinin-1 receptor (NK-1R) mRNA and protein, respectively, in inflamed colonic tissue from IBD patients. We also used quantitative competitive reverse transcription-polymerase chain reaction (qcRT-PCR) to quantitate NK-1R mRNA expression levels in biopsies from non-inflamed and inflamed (IBD) colonic mucosa.
Materials and methods
TISSUE SPECIMENS
To obtain full thickness colonic sections for NK-1R immunohistology and in situ mRNA localisation, diseased colonic tissue was obtained from five patients undergoing surgical resection for UC (two women and three men; mean age 40 years) and from five patients undergoing surgical resection for CD (two women and three men; mean age 38 years). At the time of resection, three of the UC surgical patients and two of the CD surgical patients were receiving corticosteroid treatment. Control colonic tissue was obtained from five patients undergoing surgical resection for colorectal adenocarcinoma (four women and one man; mean age 72 years). For control samples, only histologically normal tissue from an uninvolved area of the colon was used. None of the control surgical patients was receiving corticosteroid treatment.
For NK-1R mRNA quantitation, colonic biopsies were taken from a group of patients undergoing investigative colonoscopy. These were different patients to those from whom resection specimens were obtained for NK-1R localisation. Biopsies were obtained from active lesions of 10 patients with UC (three women and seven men; mean age 37 years) and from active lesions of 10 patients with CD (three women and seven men; mean age 28 years). At the time of endoscopy, four of the UC patients and four of the CD patients were receiving corticosteroid treatment. Control colonic biopsies were obtained from nine patients with no inflammatory disease activity (four women and five men; mean age 49 years). None of the control patients was receiving corticosteroid treatment. All patients were diagnosed using clinical, radiographic, endoscopic, and histological criteria.40 Biopsies were snap frozen in liquid nitrogen and stored at −70°C until further use. All protocols were approved by the University Teaching Hospitals Ethics Committee (Cork, Ireland).
LOCALISATION OF NK-1R mRNA EXPRESSION BY IN SITU HYBRIDISATION
In situ hybridisation was performed on paraffin embedded surgically resected human colon sections (4 μm thick), mounted on aminopropylethoxysilane treated slides. Following deparaffinisation and rehydration, prehybridisation treatments involved washing 2×5 minutes each in (i) phosphate buffered saline (PBS), (ii) PBS, 0.1 M glycine, (iii) PBS, 0.3% Triton X100, and (iv) PBS again. Sections were digested for 30 minutes at 37°C with proteinase K (10 μg/ml in 100 mM Tris HCl, 50 mM EDTA, pH 8.0), fixed for five minutes at 4°C in 4% paraformaldehyde; PBS and then acetylated for 2×5 minutes in fresh 0.25% acetic anhydride; and 0.1 M triethanolamine (pH 8.0). Sections were incubated at 37°C for 10 minutes in a prehybridisation buffer consisting of 50% formamide in 4×saline trisodium citrate (SSC) buffer. A digoxigenin labelled antisense RNA hybridisation probe (324 bp) corresponding to codons 230–338 of the human NK-1R cDNA sequence was synthesised from a recombinant plasmid clone (also used to construct the competitive standard for qcRT-PCR) by in vitro transcription with digoxigenin-11-UTP (Boehringer Mannheim) and T7 RNA polymerase. The nucleotide sequence of the NK-1R probe showed insignificant homology to any other sequences in the EMBL DNA sequence database. Hybridisation was performed at 42°C overnight in hybridisation buffer (50% formamide, 10% dextran sulphate, 1×Denhardt's reagent, 4×SSC, 10 mM DTT, 500 μg/ml yeast tRNA, and 100 μg/ml of heat denatured herring sperm DNA) containing 1 ng/ml digoxigenin labelled riboprobe. After hybridisation, tissues were washed with increasing stringency to 0.1×SSC at 37°C. Hybridised probe was detected immunologically using alkaline phosphatase conjugated sheep antidigoxigenin antibody (Boehringer Mannheim, Mannheim, Germany) and visualised with NBT-BCIP (purple/black precipitating product). Control slides involved competitive inhibition of hybridisation with a 10-fold excess of unlabelled antisense riboprobe. This resulted in a marked reduction in signal intensity thus confirming the specificity of the hybridisation.
IMMUNOHISTOCHEMICAL LOCALISATION OF NK-1R
The NK-1R antibody was raised in rabbits against a synthetic peptide (MDNVLPVDSDLSP) corresponding to the extracellular N terminal amino acids 1–13 of human NK-1R. The IgG fraction was affinity purified on an AH-Sepharose column to which the peptide had been coupled. Antibody specificity was confirmed by extensive radioimmunoassays and western blotting.
Immunohistochemistry was performed on paraffin embedded surgically resected human colon sections (4 μm thick), mounted on aminopropylethoxysilane treated slides. After deparaffinisation and rehydration, sections were microwaved in 10 mM citrate buffer, pH 6.0, at 350 W for five minutes to assist antigen retrieval. Sections were immediately cooled in TBS (50 mM Tris HCl, pH 7.6, 150 mM NaCl, 5 mM KCl) and then washed twice for five minutes in wash buffer (TBS, 0.001% saponin). Endogenous peroxidase activity was quenched with 0.9% hydrogen peroxide in distilled water for 30 minutes. Sections were then washed in wash buffer containing 1% normal goat serum (used for all subsequent wash steps). Non-specific binding sites were blocked by incubation in wash buffer containing 5% normal goat serum for one hour. Sections were washed twice and then incubated overnight at 4°C with the rabbit polyclonal antihuman NK-1R specific IgG at a dilution of 1:50 in 50 mM Tris HCl, pH 7.6, and 1% normal goat serum. Antibody binding was localised using a biotinylated goat antirabbit IgG followed by avidin-biotin conjugated horseradish peroxidase (Vectastain Elite ABC detection kit, Vector Laboratories, Burlingame, California, USA). Antibody binding was visualised using 3′,3′ diaminobenzidine (Vector Laboratories) yielding a brown reaction product. Staining with isotype matched rabbit IgG was performed as a negative control. A specificity control was performed involving preadsorption of NK-1R antibody with the immunising peptide (100 μM) for 16 hours at 4°C prior to staining of sections.
QUALITATIVE DETECTION OF NK-1R mRNA EXPRESSION BY RT-PCR
Total RNA was isolated by phenol chloroform extraction of guanidinium isothiocyanate lysates.41 cDNA was synthesised using approximately 100 ng of total RNA, 9 U of AMV reverse transcriptase (Promega Corp., Madison, Wisconsin, USA), 40 U of RNAsin (Promega Corp.), 500 μM of dNTPs, and 500 nM of SPR-specific antisense primer GGATTTCATTTCCAGCCCCT per 30 μl reaction for 90 minutes at 42oC.
NK-1R PCR was performed on the specific primed cDNA using the following sense and antisense primers, respectively: TGACCGCTACCACGAGCAAGTCTC and ATAGTCGCCGGCGCTGATGAAG corresponding to nucleotides 699–722 and 993–972 of human NK-1R cDNA, respectively. PCR primers were designed using the DNASTAR Lasergene Primerselect program (DNASTAR Inc., Madison, Wisconsin, USA). Primers were selected that showed insignificant homology to any other genes in the EMBL DNA sequence database. Primer pairs were chosen to span introns in their genomic sequences, thus ensuring mRNA specific amplification.
PCR was performed on 1% of the cDNA using a final concentration of 1.5 μM MgCl2, 50 μM dNTPs, 0.1 μM each primer, and 1 unit of Taq DNA polymerase (Promega Corp.) per 50 μl reaction. The thermal cycling programme involved denaturation at 96°C for 15 seconds, annealing at 60°C for 30 seconds, and extension at 72°C for one minute 30 seconds, for 40 cycles, followed by a final extension at 72°C for 10 minutes. Negative controls were performed by either omitting reverse transcriptase from cDNA synthesis or by omitting cDNA from the PCR amplifications. As a positive control, RNA from cells known to abundantly express NK-1R mRNA were used: IM-9 B lymphoblastoid cell line. Hot start PCR was used to increase the specificity of the amplification. PCR products were analysed by electrophoresis through 2% agarose gels and viewed under UV light after ethidium bromide staining. Product specificity was confirmed by DNA sequence analysis using an ABI Prism 310 Genetic Analyser (Perkin-Elmer, Norwalk, Connecticut, USA). Hae III digested φX174 DNA size markers were used.
QUANTITATION OF NK-1R mRNA EXPRESSION BY QUANTITATIVE COMPETITIVE RT-PCR (qcRT-PCR)
To facilitate quantitation of NK-1R mRNA, a competitive internal RNA standard was constructed.42 This standard was identical to the target NK-1R sequence except for an internal deletion of 71 bp. Construction of the competitive standard involved cloning a NK-1R PCR product (324 bp) into pBluescript (Stratagene, La Jolla, California, USA) at the EcoRV site. This NK-1R PCR product was obtained using the following sense and antisense primers respectively: GACTCCTCTGACCGCTACCA and GGATTTCATTTCCAGCCCCT corresponding to nucleotides 691–710 and 1014–995 of the human NK-1R cDNA, respectively. Orientation of the PCR product insert within the plasmid was determined by restriction mapping with HinfI. Digestion at the BsrGI and Bgl II unique restriction sites within the cloned insert resulted in deletion of a 71 bp fragment. Sticky ends of the plasmid were then filled in by Klenow DNA polymerase (Promega Corp.) and blunt ended recircularisation of the plasmid was performed using T4 DNA ligase (Promega Corp.). Following propagation in E coli, the deleted recombinant plasmid was subjected to in vitro transcription with T3 RNA polymerase (Promega Corp.) to synthesise deleted sense RNA transcripts (257 bp) for use as a competitive standard in qcRT-PCR.
For qcRT-PCR, varying amounts of RNA standard transcripts of known concentration were spiked into aliquoted target RNA sample, and the mixtures were then subjected to RT-PCR, as described above. As the internal standard is spiked in at the cDNA synthesis step, competition for both reverse transcription and PCR amplification occurs. Equivalence of PCR products occurs when target and standard templates are present in equal initial concentrations, permitting quantitation of the target template. Equivalence was determined as the point where target and competitive standard PCR products were of equal band intensity. Results for NK-1R mRNA quantitation were expressed as number of NK-1R mRNA molecules per μg of total RNA isolated. Total RNA was quantified using a nucleic acid quantitation kit (Invitrogen BV, the Netherlands). The accuracy, sensitivity, and reproducibility of the qcRT-PCR assay have been validated in control experiments.42 The assay is sufficiently sensitive to quantitate 100 NK-1R mRNA copies (corresponding to 103NK-1R mRNA transcripts/μg RNA isolated).
A third band of slower electrophoretic mobility than the target and competitor PCR products was observed (at 360 bp) when target and competitor PCR products were both present at relatively high concentrations. To confirm that this was due to heteroduplex formation between one strand of target and one strand of competitor PCR products, digestion with the single stranded DNA specific nuclease, S1, was performed. Digestion was performed at 37°C for 30 minutes with 1 unit of S1 nuclease (Promega Corp.) per μg of RT-PCR products.
STATISTICAL ANALYSIS
Quantitative RT-PCR results were expressed as NK-1R mRNA molecules per μg of total RNA isolated. The non-parametric Mann-Whitney test was used for statistical comparison. Differences with p<0.05 were considered to be statistically significant.
Results
EXPRESSION OF NK-1 RECEPTOR IN NORMAL COLON
To evaluate the pathological implications of NK-1R in IBD it was first necessary to determine the cellular expression of NK-1R in normal colon. We have previously mapped expression of NK-1R in non-inflamed colonic mucosa.43 In the present report, we used the techniques of in situ hybridisation and immunohistochemistry to examine cellular expression of NK-1R mRNA and protein, respectively, in full thickness normal colon sections.
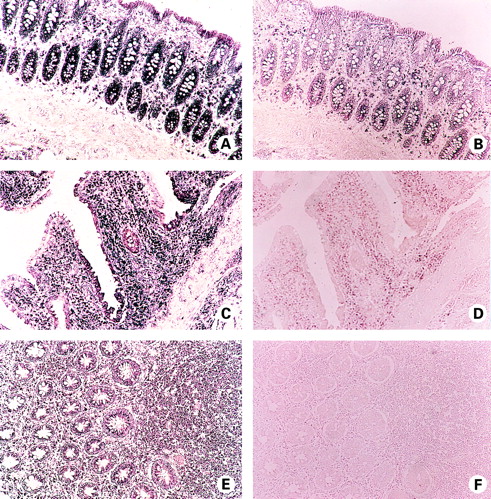
NK-1R mRNA expression localised to lamina propria mononuclear cells (LPMC), lymphoid follicles, and to surface and crypt epithelium of non-inflamed colon (fig 1A), thus confirming our previous results.43 mRNA expression also localised to submucosal arterioles and venules, circular and longitudinal smooth muscle layers, and to enteric neurones of the myenteric plexus (see fig 4A, C). Hybridisation specificity was confirmed as a 10-fold excess of unlabelled riboprobe resulted in a dramatic reduction in hybridisation signal intensity (fig 1B). Using immunohistochemistry, NK-1R protein expression gave a similar cellular distribution to that observed for NK-1R mRNA: NK-1R protein was detected in LPMC, lymphoid follicles, surface and crypt epithelium, submucosal arterioles and venules, longitudinal and circular smooth muscle, and in myenteric plexus neurones (figs 2A, 4E). The specificity of antibody binding was confirmed, as staining was competitively inhibited by preincubation of the primary antibody with the NK-1R-immunising peptide (fig 2B).

Localisation of neurokinin-1 receptor (NK-1R) mRNA expression in non-inflamed colon (A, B), in active ulcerative colitis (UC) colon (C, D), and in active Crohn's disease (CD) colon (E, F) by in situ hybridisation. (A) NK-1R mRNA (purple) is detected in lamina propria mononuclear cells (LPMC), and in surface and crypt epithelium of normal colon. (C) NK-1R mRNA expression (purple) by LPMC and epithelial cells of UC colon. Note the “patchy” positivity of surface epithelium. (E) Expression of NK-1R mRNA (purple) by LPMC and crypt epithelium of CD colon. Note the dense inflammatory infiltrate of NK-1R mRNA expressing cells. (B, D, F) Control hybridisation of consecutive sections of normal (B), UC (D), and CD (F) colon, with a 10-fold excess of unlabelled riboprobe. Signal intensity is dramatically reduced, confirming the specificity of hybridisation.

Localisation of neurokinin-1 receptor (NK-1R) expression in non-inflamed (A, C, and E), and inflamed Crohn's disease (CD) (B, D, F) myenteric plexus by in situ hybridisation (A–D), and immunohistochemistry (E, F). (A) NK-1R mRNA (purple) is detected in enteric neurones of the myenteric plexus of normal colon. (C) Higher magnification view of (A). (E) NK-1R protein (brown) staining is evident in the myenteric plexus of normal colon. Section was counterstained with haematoxylin (blue). (B) Expression of NK-1R mRNA by enteric neurones of the myenteric plexus of CD colon. Note the NK-1R positive cells of lymphoid morphology. (D) Higher magnification view of (B). (F) Immunohistochemical detection of NK-1R protein expression (brown) in the myenteric plexus of CD colon. Section was counterstained with haematoxylin (blue).

Immunohistochemical localisation of neurokinin-1 receptor (NK-1R) protein expression in non-inflamed colon (A, B), in active ulcerative colitis (UC) colon (C, D), and in active Crohn's disease (CD) colon (E, F). Sections are consecutive to those processed for in situ hybridisation in fig 1 and are shown at higher magnification. (A) NK-1R immunoperoxidase staining (brown) is detected in lamina propria mononuclear cells (LPMC), and in surface (top right of figure) and crypt epithelium of normal colon. (C) NK-1R expression (brown) is present in LPMC and in surface epithelium of UC colon. Note the crypt epithelium is negative for NK-1R expression (blue). (E) Expression of NK-1R protein (brown) by LPMC and crypt epithelial cells of CD colon. (B, D, F) Control staining of consecutive sections of normal (B), UC (D), and CD (F) colon. Specificity of antibody binding was confirmed, as preincubation of the primary antibody with the NK-1R immunising peptide inhibited staining. All sections were counterstained with haematoxylin (blue).
EXPRESSION OF NK-1 RECEPTOR IN IBD COLON
Using the techniques of in situ hybridisation and immunohistochemistry, NK-1R expression was localised to LPMC, lymphoid follicles, and epithelium of inflamed UC and CD colon (figs 1C, 1E, 2C,2E). Expression was also evident in submucosal arterioles and venules, in circular and longitudinal smooth muscle layers, and in enteric neurones of the myenteric plexus of both UC and CD resected colon (figs3C, 4B, 4D, 4F). Vascular expression of NK-1R was further localised to the endothelial lining and muscularis of submucosal arterioles and venules (fig 3C). Cellular expression of NK-1R protein exactly matched that observed for NK-1R mRNA. In other words, no ectopic expression of NK-1R was observed in IBD colon. Appropriate controls confirmed the specificity of riboprobe hybridisation (fig 1D, 1F) and antibody binding (fig 2D, 2F), respectively.

Localisation of neurokinin-1 receptor (NK-1R) mRNA expression in active ulcerative colitis (UC) colon by in situ hybridisation. (A) NK-1R mRNA expression (purple) is detected in lamina propria mononuclear cells (LPMC) and in a lymphoid follicle of UC colon. Crypt epithelial cells negative for NK-1R mRNA are present. (C) Expression of NK-1R mRNA by submucosal arterioles and venules of UC colon. mRNA expression (purple) is evident in the endothelium and muscularis of blood vessels. (E) High magnification view of crypt epithelial cells negative for NK-1R mRNA expression, surrounded by NK-1R positive (purple) LPMC in UC colon. (B, D, F) Control hybridisation of consecutive sections of UC colon, with a 10-fold excess of unlabelled riboprobe. Signal intensity is dramatically reduced, confirming specificity of hybridisation.
In the non-inflamed colon, we have previously localised NK-1R expression to the majority of LPMC in situ, and to a broad range of LPMC subsets ex vivo, including CD4, CD45R0, CD45RA, CD8, CD19, and CD14 positive cells.43 The majority of LPMC of inflamed UC and CD colon were also positive for NK-1R expression (figs 1C, 1E, 2C,2E), as were lymphoid follicles (fig 3A), suggesting a subset unrestricted expression of NK-1R in LPMC of inflamed colon.
Aberrant negative epithelial expression of NK-1R was noted in active UC. As illustrated in fig 1C, NK-1R expressing surface epithelial cells are interspersed by NK-1R negative epithelial cells in UC colon. This observation of aberrant negative epithelial expression was also frequently detected in crypt epithelial cells of UC colon (figs 2C, 3A,3E). The loss of epithelial NK-1R expression occurred in all UC specimens (n=5), irrespective of corticosteroid treatment (n=3/5), and was very rarely observed in CD epithelia (figs 1E, 2E). Intense staining of the myenteric plexus was evident in CD colon compared with normal (fig 4) or UC colon (data not shown). This increased NK-1R detection could perhaps be attributed to hyperplastic neurones known to be characteristic of CD,44 or to lymphoid cells (NK-1R expressing) which have been shown to infiltrate the plexus in CD (fig4B, 4D).45
Localisation of NK-1R mRNA and protein were consistent for all specimens within each group (control, UC, and CD; n=5 each). Localisation of NK-1R expression in IBD patients was not affected by corticosteroid treatment (UC n=3/5; CD n=2/5).
NK-1R mRNA EXPRESSION IS UPREGULATED IN IBD COLONIC MUCOSA
We have previously developed a qcRT-PCR assay to accurately quantify NK-1R mRNA expression.42 Figure 5 illustrates quantitation of NK-1R mRNA expressed in a particular UC biopsy sample. Equivalence of PCR amplification products is seen at 4.1×103 NK-1R mRNA molecules, in which target (295 bp) and competitive standard (228 bp) PCR products are of equal band intensity. When adjusted for the amount of total RNA in the assay, this represents a level of expression of 4.1×104 NK-1R mRNA transcripts/μg RNA. The higher molecular weight band (∼360 bp) seen on the gel is due to heteroduplex formation between one strand each of target (295 bp) and competitor (228 bp) PCR products. The unannealed portion of the target strand, corresponding to the sequence deleted from the competitive standard, loops out to form a bulky secondary structure that results in slower electrophoretic mobility than either of the two linear products of the competitive PCR. Digestion with the single stranded DNA specific nuclease, S1, eliminated the 360 bp band (fig 5B). This confirmed that the 360 bp band was the predicted heteroduplex containing a single stranded “loop-out” amenable to S1 digestion. This type of heteroduplex is commonly observed in competitive PCR, but as the heteroduplex consists of a hybrid of one strand from target and competitor products, it does not bias the normal occurrence of equivalence and hence quantitation.42 ,46

Quantitation of neurokinin-1 receptor (NK-1R) mRNA expression in a mucosal biopsy from active ulcerative colitis (UC) colon. (A) Gel shows a representative NK-1R mRNA quantitation using qcRT-PCR. Competitive standards were spiked into the aliquoted biopsy RNA sample at concentrations ranging from 2.6×104molecules to 2.6×102 molecules (lanes 1–6; 2.5-fold dilution series). Equivalence (*) is seen at 4.1×103NK-1R mRNA molecules, in which target (295 bp) and competitive standard (228 bp) PCR products are of equal band intensity (lane 3). When adjusted for the amount of total RNA in the assay, this represents a level of expression of 4.1×104 NK-1R mRNA transcripts/μg RNA in this particular UC biopsy sample. HaeIII digested φX174 DNA size markers (M) were used. (B) Confirmation of heteroduplex formation. A third band of slower electrophoretic mobility (∼360 bp) than the target (295 bp) or competitor (228 bp) PCR products was observed when the concentration of both was relatively high (lane 3). This was absent when competitor or target were amplified separately (lanes 1 and 2, respectively). To confirm that the 360 bp band consisted of a heteroduplex between one strand each of target and competitor PCR products, the PCR products from lane 3 were subjected to digestion with the single stranded DNA specific nuclease, S1 (lane 4). This treatment eliminated the 360 bp band, confirming the presence of a single stranded “loop out” amenable to S1 digestion, which is characteristic of the predicted heteroduplex.
The mean level of expression of NK-1R mRNA in biopsies from non-inflamed mucosa was found to be 0.9×104 NK-1R mRNA transcripts/μg total RNA (n=9) (fig 6). Biopsies from active lesions of UC patients exhibited increased NK-1R mRNA levels and yielded a mean of 1.8×104 NK-1R transcripts/μg total RNA (n=10). However, this increase did not reach statistical significance (p<0.1). In active CD, the mean level of NK-1R mRNA expression in mucosal biopsies was 6.3×104 NK-1R transcripts/μg total RNA (n=10). This sevenfold upregulation in NK-1R mRNA relative to normal mucosal mRNA levels was statistically significant (p<0.01). NK-1R mRNA levels from CD mucosa were also significantly increased compared with expression levels from UC mucosa (p<0.01). IBD mucosal NK-1R mRNA levels displayed significant upregulation compared with normal mucosal expression levels (p<0.01). The biopsy levels of NK-1R mRNA were consistent within each group (control, UC, and CD) (fig 6), and in the IBD biopsies NK-1R mRNA level was independent of corticosteroid treatment (UC n=4/10; CD n=4/10). This agrees with previous findings that the level of colonic SP binding sites in IBD is not modified by corticosteroid or other anti-inflammatory therapies.34

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Neurokinin-1 receptor (NK-1R) mRNA levels in mucosal biopsies from normal colon (n=9), active ulcerative colitis (UC) colon (n=10), and active Crohn's disease (CD) colon (n=10). The bar indicates mean NK-1R mRNA expression in each group. *p<0.01.
Discussion
In this study, we used the definitive resolution of in situ hybridisation and immunohistochemistry to examine the cellular targets for SP binding via the neurokinin-1 receptor in normal compared with inflamed IBD colon. We also used the exquisite sensitivity of competitive RT-PCR to quantitate NK-1R expression at the mRNA level in biopsies of normal and IBD colonic mucosa. Our results demonstrated that although the phenotype of NK-1R expressing cells was not altered in IBD, increased numbers of NK-1R positive lymphoid cells were observed in IBD tissue. In addition, the pattern of epithelial NK-1R expression was found to be altered in UC while increased NK-1R expression was observed in the myenteric plexus of CD. Furthermore, we found that inflamed IBD mucosa exhibited significantly increased expression of NK-1R mRNA compared with normal mucosal levels.
Using the complementary techniques of in situ hybridisation and immunohistochemistry, NK-1R mRNA and protein expression were detected in LPMC, lymphoid follicles, surface and crypt epithelium, submucosal arterioles and venules, circular and longitudinal smooth muscle, and myenteric plexus of normal colonic resections. This cellular distribution of NK-1R expression complements recent work by Smithet al which demonstrated NK-1R immunoreactivity in circular muscle, myenteric and submucosal neurones, mucosal cells, and endothelial cells of human antrum and duodenum.47 We further localised the vascular expression of NK-1R to endothelium and smooth muscle of arterioles and venules. This finding agrees with reports of SP binding sites in human vascular endothelium.47 ,48
We did not observe any ectopic expression of NK-1R in IBD. However, we found increased numbers of NK-1R bearing lymphoid cells in IBD tissue, aberrant negative epithelial expression of NK-1R in UC, and increased NK-1R expression in CD myenteric plexus. We also found upregulated levels of NK-1R mRNA in IBD mucosal biopsies. Our observed increase in IBD NK-1R mRNA levels could be attributed to increased infiltration of NK-1R mRNA positive lymphoid cells in inflamed mucosa. Alternatively, cellular expression of NK-1R mRNA may be upregulated in LPMC from actively inflamed mucosa. Activation of macrophages with LPS has been shown to upregulate NK-1R mRNA expression.49 We have previously demonstrated upregulation of NK-1R mRNA expression on activation of the human IM-9 B lymphoblastoid cell line with PMA and ionomycin.50 Quantitation of NK-1R mRNA levels in LPMC isolated from normal or actively inflamed colonic mucosa did not reveal any differences in cellular expression levels.50 While this favours the view that NK-1R expression is not upregulated in individual LPMC during inflammation, it must also be considered that isolated LPMC may not be reflective of LPMC in situ in the mucosal microenvironment.
Aberrant negative epithelial expression of NK-1R was observed in UC colonic resections and was rarely detected in CD. Negative epithelial expression of NK-1R in UC may perhaps account for the lower NK-1R mRNA levels detected in UC mucosal biopsies compared with CD mucosal biopsies (p<0.01). Intestinal epithelial expression of NK-1R mRNA has been reported to be upregulated in Clostridium difficile toxin A enteritis.51 C difficile infection of the gut has also been shown to stimulate local expression of proinflammatory cytokines.52 We have recently demonstrated induction of NK-1R expression in human colonic epithelial cell lines by the proinflammatory Th1 cytokines interferon γ, TNF-α, and IL-1β (manuscript submitted). A Th1 cytokine profile was associated with CD.53 ,54 Cytokine patterns in UC have not demonstrated a distinct Th1 or Th2 phenotype.54 ,55 Differences in cytokine profiles may possibly explain the observed differences in epithelial NK-1R expression between CD and UC.
There are marked structural abnormalities of the enteric nervous system in IBD.56 Changes are more pronounced in CD and involve hyperplasia of ganglion cells, extensive axonal degeneration, and infiltration of the plexus with plasma cells, mast cells, and lymphocytes.44 ,57 Examination of NK-1R expression by enteric neurones revealed intense staining of CD myenteric plexus compared with normal myenteric plexus. Upregulation of SP binding sites on enteric neurones has been described in CD.34 Increased NK-1R detection in CD myenteric plexus could be attributed to hyperplasia of NK-1R positive ganglion cells, infiltration of NK-1R positive lymphoid cells, or increased nociception. Nociception induced upregulation of NK-1R mRNA has been reported in dorsal root ganglion neurones of rats with hind paw inflammation.58 ,59
In conclusion, we have localised cellular expression of NK-1R to LPMC, lymphoid follicles, epithelia, submucosal vasculature, smooth muscle, and myenteric plexus of IBD colon. In all specimens, localisation of NK-1R mRNA by in situ hybridisation and NK-1R protein by immunohistochemistry were in perfect concordance, confirming cellular sites of NK-1R expression. We have also demonstrated increased numbers of NK-1R bearing cells in IBD tissue, aberrant negative epithelial expression of NK-1R in UC, increased expression of NK-1R in CD myenteric plexus, and upregulated levels of NK-1R mRNA in IBD mucosal biopsies. The greater upregulation of NK-1R mRNA in CD versus UC biopsies, as determined by quantitative RT-PCR, may reflect the more uniform epithelial expression and myenteric plexus upregulation of NK-1R observed in situ in CD. This is also supported by our observation of no marked differences in the level of NK-1R positive inflammatory cells between CD and UC. The increased expression of NK-1R in CD versus UC suggests that substance P may be relatively more important in the pathophysiology of CD than previously suggested by reports that SP itself is elevated more in UC.25-28 The increased expression of NK-1R in the CD myenteric plexus might indicate that primary neural mechanisms may be relatively more important in the pathophysiology of CD than UC. Our results suggest that there are differences between CD and UC in how the effects of SP are regulated. Increased neuronal expression and more uniform epithelial expression of NK-1R in CD may imply greater responsiveness to limited amounts of SP. On the other hand, lack of upregulation of NK-1R in UC myenteric plexus might favour lower responsiveness to the higher levels of SP present in UC. Epithelial cells in UC may also downregulate their responsiveness to SP by loss of NK-1R expression. Our findings demonstrate the likely cellular targets for pharmacological NK-1R antagonists, and thereby suggest the potential beneficial effects of such agents on the many pathophysiological symptoms of IBD.
Acknowledgments
Grant support was obtained from the following: Health Research Board of Ireland; Wellcome Trust; Bing Foundation; Peptide Biochemistry Core of CURE: Digestive Diseases Research Centre (DK 41301); VA Greater Los Angeles Health Care System (90073).
Abbreviations used in this paper
- CD
- Crohn's disease
- IBD
- inflammatory bowel disease
- LPMC
- lamina propria mononuclear cells
- NK-1R
- neurokinin 1 receptor
- RT-PCR
- reverse transcription-polymerase chain reaction
- qcRT-PCR
- quantitative competitive RT-PCR
- SP
- substance P
- UC
- ulcerative colitis
- IL
- interleukin
- TNF-α
- tumour necrosis factor α
- PBS
- phosphate buffered saline