


Abstract
The metabotropic glutamate receptor subtype 5 (mGluR5) activates calcium mobilization and extracellular signal-regulated kinase 1/2 (ERK1/2) phosphorylation in cortical astrocytes. These are independent signaling systems, and they can be differentially regulated. We recently discovered two novel selective allosteric potentiators of mGluR5, 3,3′-difluorobenzaldazine (DFB) and N-{4-chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl) methyl]phenyl}-2-hydroxybenzamide (CPPHA). In studies of mGluR5 activation of calcium transients in recombinant systems, both DFB and CPPHA are without effect on baseline calcium levels, but they induce parallel leftward shifts in the concentration-response curve to agonists. However, it is conceivable that these compounds will have differential effects on different signaling pathways in native systems. Here, we examined the effects of CPPHA and DFB on mGluR5-induced calcium transients and ERK1/2 phosphorylation in cultured rat cortical astrocytes. Both potentiators induced parallel leftward shifts of the concentration-response curves of DHPG- and glutamate-induced calcium transients in astrocytes. These effects are identical to their effects on mGluR5 expressed in human embryonic kidney 293 or Chinese hamster ovary cells. DFB induced a similar shift of concentration-response curve of DHPG-induced ERK1/2 phosphorylation. Interestingly, CPPHA induced an increase in basal mGluR5-mediated ERK1/2 phosphorylation and potentiated the effect of low concentrations of agonists. In contrast, CPPHA significantly decreased ERK1/2 phosphorylation induced by high concentrations of agonists. Thus, CPPHA has qualitatively different effects on mGluR5-mediated calcium responses and ERK1/2 phosphorylation. Together, these data provide evidence that different allosteric potentiators can differentially modulate coupling of a single receptor to different signaling pathways.
Metabotropic glutamate receptors (mGluRs) are seven membrane-spanning G protein-coupled receptors that play a critical role in modulation of neuronal excitability and synaptic transmission in the central nervous system. To date, eight mGluR subtypes have been cloned (termed mGluR1–mGluR8), and these have been divided into three groups based on sequence homology, pharmacological profiles, and G protein coupling. Group I mGluRs (mGluR1 and mGluR5) couple primarily to phospholipase C through Gq/G11 to trigger phosphoinositide hydrolysis and intracellular calcium transients. Group II and group III mGluRs couple to inhibition of adenylyl cyclase and other effector systems through Gi/o (Conn and Pin, 1997). Group I mGluRs are often localized postsynaptically where they modulate ion channel activity, whereas group II and group III mGluRs are primarily localized presynaptically and regulate neurotransmitter release. mGluRs are members of family 3 G protein-coupled receptors (GPCRs), which possess a large extracellular agonist binding domain in the N terminus of the receptor. Even though a number of agonists and antagonists that bind to the N-terminal agonist binding domain (orthosteric site) have been developed, discovery of subtype-selective ligands at the orthosteric site has been challenging because of the high conservation of the agonist binding domain within the mGluRs. Recently, subtype selectivity has been achieved with compounds that bind to allosteric sites in the seven membrane-spanning region of the mGluRs (Johnson et al., 2004). These include both antagonists (Litschig et al., 1999; Varney et al., 1999; Pagano et al., 2000) and allosteric potentiators (Knoflach et al., 2001; Johnson et al., 2003; Maj et al., 2003; Marino et al., 2003; Mathiesen et al., 2003; O'Brien et al., 2003, 2004; Cube et al., 2005) that are highly selective for individual mGluR subtypes.
A large number of cellular and behavioral studies have led to the hypothesis that selective agonists of one mGluR subtype, mGluR5, may have exciting potential as novel antipsychotic and cognition-enhancing agents (Alagarsamy et al., 1999a,b; Mannaioni et al., 2001; Marino et al., 2001; Chavez-Noriega et al., 2002; Kinney et al., 2003; Campbell et al., 2004). Recently, we reported that two compounds, 3,3′-difluorobenzaldazine (DFB) (O'Brien et al., 2003) and N-[4-chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]phenyl]-2-hydroxybenzamide (CPPHA) (O'Brien et al., 2004), act as selective allosteric potentiators of mGluR5. These compounds have no agonist activity by themselves, but they potentiate activation of mGluR5-mediated calcium transients in transfected cell lines by glutamate. The discovery of allosteric potentiators of specific mGluR subtypes raises the exciting possibility that such compounds could be developed as novel tools or therapeutic agents to increase mGluR function. In the simplest view, these compounds could increase activity of the intended mGluR subtypes at any synapse or in any cell population in which these receptors are normally active. However, it is possible that these compounds will have a more complex profile of effects in native systems. mGluR5 can couple to multiple signaling pathways, and it is conceivable that allosteric potentiators of mGluRs could preferentially impact signaling through some pathways and not others. For instance, we reported that mGluR5 activates phosphoinositide hydrolysis and phosphorylation of extracellular signal-regulated kinase (ERK) 1/2 in cortical astrocytes by independent mechanisms (Peavy et al., 2001, 2002). Furthermore, these responses to mGluR5 activation can be differentially regulated by protein kinase C (Peavy et al., 2002). Interestingly, a growing body of evidence suggests that different traditional orthosteric agonists can differentially activate different signaling pathways of a single GPCR, a phenomenon referred to as agonist receptor trafficking (Berg et al., 1998; Brink et al., 2000; Gazi et al., 2003). This could have important implications for development of allosteric potentiators. If allosteric potentiators differentially regulate specific responses to mGluR5 activation, it will be critical to determine the effect of the compounds on specific responses that are thought to be relevant for an intended therapeutic action in native systems. Likewise, this could provide an opportunity to develop highly specific agents that selectively activate a desired response to receptor activation without potentiating other responses. Thus, it will be critical to develop an understanding of the effects of DFB, CPPHA, and other allosteric potentiators on different signaling pathways activated by mGluR5 activation in native systems. We now report that DFB has similar effects on mGluR5-mediated calcium transients and ERK1/2 phosphorylation in cortical astrocytes, whereas CPPHA has differential effects on these two signaling pathways.
Materials and Methods
Drugs. CPPHA was synthesized as described previously (O'Brien et al., 2004). All other ligands were obtained from Tocris Cookson Inc. (Ellisville, MO).
Cell Culture. Rat cortical astrocytes were prepared as described by Peavy et al. (2001). In brief, neocortices from 2- to 4-day-old Sprague-Dawley rat pups were dissected and dissociated in DMEM by trituration with 1-ml pipette tips. The cells were then centrifuged and resuspended in DMEM [containing 1 mM sodium pyruvate, 2 mM l-glutamine, and PenStrep (100 units/ml penicillin and 0.1 mg/ml streptomycin; Invitrogen, Carlsbad, CA)] supplemented with 10% FBS in T75 tissue culture flasks; the medium was changed the next day. Cell cultures were maintained at 37°C in an atmosphere of 95% air, 5% CO2 for 6 to 8 days. Cells were shaken overnight (280–310 rpm) to remove oligodendrocytes and microgliocytes. For ERK1/2 phosphorylation assay, the cells were then trypsinized and replated into poly-d-lysine-precoated 12-well plates at a density of about 6 × 106 cells/well in full DMEM with 10% FBS. The second day, the medium was switched to full DMEM with G-5 supplement (Invitrogen) containing epidermal growth factor (10 ng/ml), basic fibroblast growth factor (5 ng/ml), insulin (5 μg/ml), and other factors. The cells were nearly confluent within 2 days and resembled the polygonal astrocytic appearance in vivo. Three days after the addition of G-5 supplement and 20 h before experiments, the medium was aspirated, and the cells were washed three times with 1× Hanks' balanced salt solution, and 1 ml of glutamine-free DMEM was added to each well.
Calcium Fluorescence Assay. Secondary astrocytes were replated into poly-d-lysine-precoated 96-well plates in full DMEM [containing 1 mM sodium pyruvate, 2 mM l-glutamine, and PenStrep (100 units/ml penicillin and 0.1 mg/ml streptomycin)] supplemented with 10% FBS and was switched to G-5-containing medium the next day. The day before each experiment, medium was switched to glutamine-free DMEM with 10% dialyzed FBS. On the day of the assay, cells were washed with 2 × 100 μl of assay buffer [1× Hanks' balanced salt solution buffer containing 20 mM HEPES (Invitrogen), 2.5 mM probenecid (Sigma-Aldrich, St. Louis, MO), and 0.1% bovine serum albumin (Sigma-Aldrich)]. The cells were loaded with calcium-sensitive dye from FLIPR calcium 3 assay kit (Molecular Devices, Sunnyvale, CA) for 1 h in cell culture incubator, and then cells were washed with 2 × 100 μl of assay buffer and 180 μl of assay buffer was added to each well. Cells were excited at 485 nm, and fluorescence increases at 525 nm due to calcium transients were measured by 96-well fluorometric imaging plate reader (FlexStation; Molecular Devices). Assay buffer (20 μl) containing DFB or CPPHA or vehicle (dimethyl sulfoxide) was added 5 min before the addition of 20 μl of assay buffer containing different concentrations of agonist. When glutamate was used, AMPA and NMDA receptor antagonists CNQX (10 μM) and l-AP5 (20 μM) were added 20 min before the addition of potentiators.
ERK1/2 Phosphorylation Assay. On the day of each assay, cells were first treated with potentiators, in the presence or absence of CNQX (10 μM) and l-AP5 (20 μM) or mGluR5 antagonists and then stimulated with agonist. At the end of stimulation, medium containing the drug was aspirated, and 200 μl of ice-cold lysis buffer (containing 50 mM Tris-HCl, 50 mM NaCl, 5 mM EDTA, 10 mM EGTA, 1 mM Na3VO4, 2 mM Na4P2O7·10 H2O, 4 mM magnesium para-nitrophenyl phosphate, and 1 mM phenylmethylsulfonyl fluoride plus 10 μg/ml leupeptin and 2 μg/ml aprotinin) was added to each well. Cells were frozen at –80°C and underwent three thaw and refreeze cycles. Cells were scraped into clean tubes, the samples were centrifuged, and the supernatant was collected. Equal amounts of supernatant from each sample were mixed with 3× lithium dodecyl sulfate sample buffer, subjected to SDS-polyacrylamide gel electrophoresis, and transferred to nitrocellulose membranes. Membranes were first blocked and then stained with primary rabbit anti-p44/42 mitogen-activated protein kinase (ERK1/2) polyclonal antibody mixed with primary mouse anti-phospho-p44/42 mitogen-activated protein kinase (phospho-ERK1/2) monoclonal antibody (Cell Signaling Technology Inc. Beverly, MA). After washing three times, membranes were subsequently stained with fluorescent dye Alexa Fluor 680-conjugated secondary goat anti-mouse IgG (H+L) (Invitrogen) mixed with fluorescent dye IRDye800-conjugated secondary goat anti-rabbit IgG (H+L) (Rockland, Gilbertsville, PA). Membranes were scanned using Odyssey Imaging System (LI-COR, Lincoln, NE). ERK1/2 phosphorylation (phosphorylated ERK1/2) is first normalized to total ERK1/2 and then expressed as percentage of maximal response or -fold above control.
Data Analysis. Data were expressed as mean ± S.E.M., and curve fitting was performed with GraphPad Prism 3 (GraphPad Software Inc., San Diego, CA). Student's t test was used to evaluate significance of differences between mean values for each study, and differences were considered significant for p < 0.05.
Results
DFB Has Similar Allosteric Effects on mGluR5-Mediated Calcium Transients and ERK1/2 Phosphorylation in Cultured Rat Cortical Astrocytes. DFB was first identified as a potentiator of mGluR5 in a high-throughput calcium fluorescence assay in CHO cells (O'Brien et al., 2003). In CHO cells, DFB has no agonist effect, but it potentiates responses to activation of human and rat mGluR5 when measuring the calcium fluorescence response to the agonists DHPG, glutamate, and quisqualate (O'Brien et al., 2003). Overexpression of receptors in heterologous systems may alter the intracellular environment, including receptor-G protein stoichiometry, which may lead to results that do not always reflect responses in native systems. Thus, it is important to verify studies in CHO cells in a native system. Rat cortical astrocytes have been reported to predominantly express mGluR5 (Peavy et al., 2002) and to provide an ideal native system to characterize the effects of allosteric potentiators of this receptor. Thus, we determined the effect of DFB on mGluR5-mediated calcium transients induced by DHPG and glutamate in cortical astrocytes. Consistent with studies in CHO cells, DFB has no agonist effect on mGluR5 in astrocytes (Fig. 1A), but it potentiates the response to an EC20 concentration of DHPG (Fig. 1A) and induces a parallel shift to the left of the DHPG and glutamate concentration-response curves (Fig. 1, B and C). DFB has similar efficacy at potentiating DHPG and glutamate responses in astrocytes as in human and rat mGluR5 expressed in CHO cells (O'Brien et al., 2003). At 100 μM, DFB induces a 2.23- and 2.41-fold shift in the DHPG and glutamate concentration-response curves, respectively. This is similar in magnitude to the shift in human and rat mGluR5 CHO cells (O'Brien et al., 2003). We also measured the effects of DFB and CPPHA on calcium transients induced by glutamate with the presence of AMPA receptor and NMDA receptor antagonists CNQX and l-AP5. Data (not shown) indicates that there is no AMPA- and NMDA receptor-contributed component in calcium transients induced in astrocytes by glutamate under various conditions.
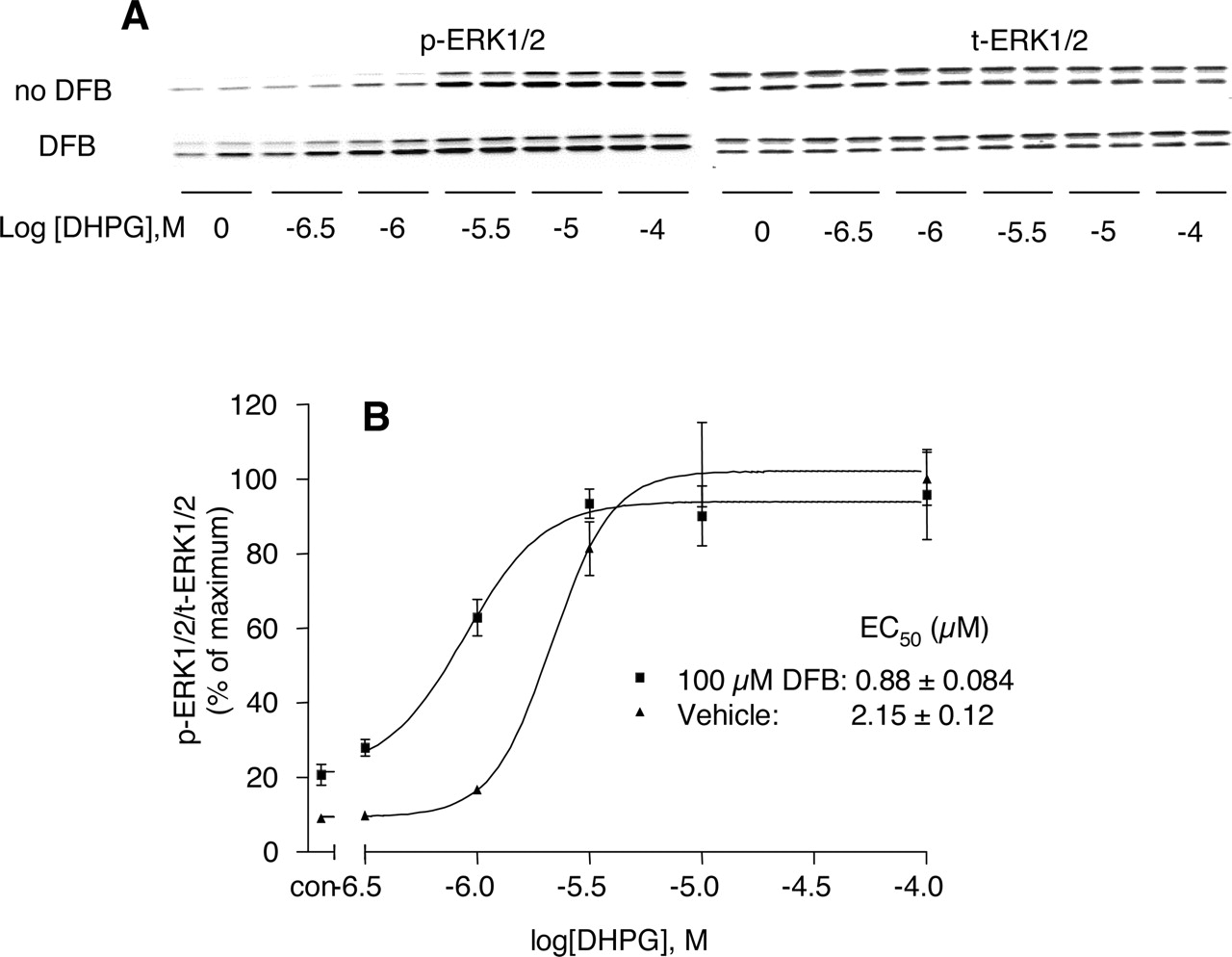
We previously reported that mGluR5-mediated calcium transients in rat cortical astrocytes can be regulated by protein kinase C, whereas protein kinase C does not regulate mGluR5-induced ERK1/2 phosphorylation in these cells (Peavy et al., 2002). Furthermore, we found that mGluR5-mediated ERK1/2 phosphorylation is independent of activation of phospholipase C and that it is mediated by transactivation of the epidermal growth factor receptor (Peavy et al., 2001). These studies suggest that these are independent signaling pathways and that these two responses to mGluR5 can be differentially regulated. Thus, it is conceivable that allosteric potentiators of mGluR5 could differentially regulate these two responses to mGluR5 activation. However, as shown in Fig. 2, A and B, DFB induced a similar potentiation of DHPG-induced ERK1/2 phosphorylation as was seen with calcium transients. Unfortunately, measurement of glutamate-induced ERK1/2 phosphorylation is complicated by the fact that glutamate transporters are expressed at high levels in rat cortical astrocytes and that they are capable of reducing extracellular glutamate concentrations over the 10-min period of glutamate addition used in the ERK1/2 phosphorylation assay. Nonetheless, we determined the effects of DFB on the ERK1/2 phosphorylation response to glutamate, and we found that DFB slightly potentiates the response at low concentrations of glutamate, but it has no significant effect on the maximal ERK1/2 phosphorylation response (data not shown). Thus, DFB potentiates agonist-induced calcium mobilization and ERK1/2 phosphorylation and shifts the concentration-response curves to the left, but it has no significant effect on the maximal ERK1/2 phosphorylation response.
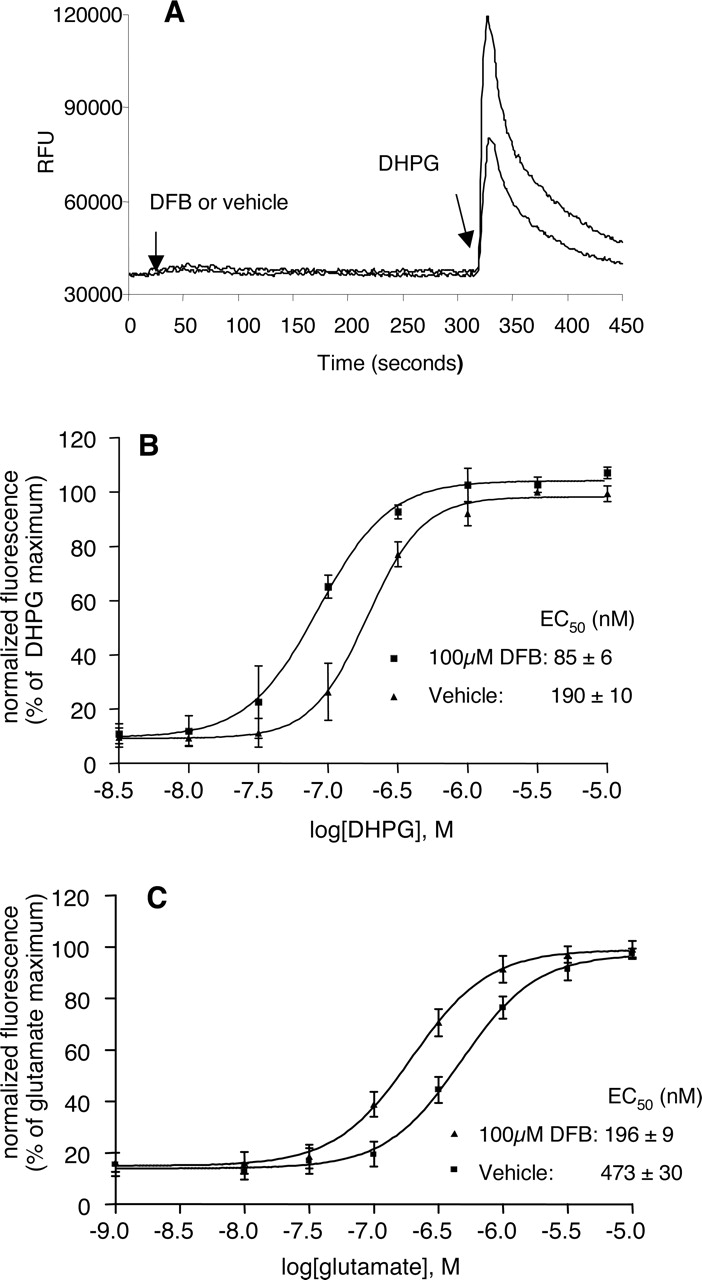
DFB potentiates mGluR5-mediated calcium transients. Secondary rat cortical astrocytes were cultured in 96-well plates in G-5 supplement-containing medium for 3 days, changed to glutamine-free DMEM the day before assay, and the next day they were loaded with the calcium-sensitive dye from FLIPR calcium 3 assay kit. DFB (100 μM) was added to cells after 16 s of baseline determination. Five minutes later, a range of concentrations of agonist were added, and then cells were excited at 485 nm and fluorescence at 525 nm was monitored with the FlexStation. When glutamate was used as agonist, CNQX (10 μM) and l-AP5 (20 μM) were added 20 min before the addition of DFB to eliminate any potential AMPA- and NMDA receptor-mediated component in calcium transients. A, example trace from fluorescence assay for calcium transients, demonstrating the potentiation of the DHPG-induced calcium response in cortical astrocytes by DFB. B, potentiation of DHPG-induced calcium transients by DFB. Curves were generated from three independent experiments. Points represent mean ± S.E.M. C, potentiation of glutamate-induced calcium transients by DFB. Curves were generated from three independent experiments. Points represent mean ± S.E.M.
CPPHA Has Qualitatively Different Effects on Calcium Transients and ERK1/2 Phosphorylation in Rat Cortical Astrocytes. We recently reported that a novel compound, CPPHA, is a second allosteric potentiator of mGluR5 that is structurally distinct from DFB (O'Brien et al., 2004). Interestingly, CPPHA has functional effects on calcium transients in mGluR5-expressing CHO cells that are similar to those of DFB. However, unlike DFB, this compound does not bind to the binding site that is occupied by allosteric antagonists such as MPEP. Thus, CPPHA and DFB are likely to potentiate mGluR5 responses by actions on different allosteric sites. Since CPPHA and DFB act at different sites on mGluR5, it is conceivable that these two compounds could have differential effects on this receptor in astrocytes. Consistent with previous studies in CHO cells, CPPHA has no agonist effect on mGluR5, but it potentiated DHPG- and glutamate-induced calcium transients in astrocytes (Fig. 3). As with DFB, the response to CPPHA on mGluR5-mediated calcium mobilization in astrocytes was similar to that seen in CHO cells (O'Brien et al., 2004). Thus, CPPHA induced a parallel shift in the DHPG and glutamate concentration-response curves with a response of a 5.79- and 7.38-fold shift, respectively, at 10 μM CPPHA.

DFB potentiates mGluR5-mediated increases in ERK1/2 phosphorylation in cortical astrocytes. Secondary rat cortical astrocytes were cultured in 12-well plates in G-5 supplement-containing medium for 3 days, and then the cells were starved in glutamine-free DMEM for 20 h. The day of assay, cells were incubated with 100 μM DFB for 10 min and then stimulated with agonist for 10 min. ERK1/2 phosphorylation was measured with Western blotting for dually phosphorylated ERK1/2 and normalized with Western blotting for total ERK1/2 and then expressed as percentage of maximal ERK1/2 phosphorylation. A, representative Western blot analysis in duplicate. B, densitometry analysis of the potentiation of DHPG-induced ERK1/2 phosphorylation by DFB from four independent experiments; points represent mean ± S.E.M.

CPPHA potentiates mGluR5-mediated calcium transients. Secondary rat cortical astrocytes were cultured in 96-well plates in G-5 supplement-containing medium for 3 days and changed to glutamine-free DMEM the day before assay. The next day, cells were loaded with the calcium-sensitive dye from FLIPR calcium 3 assay kit and placed in FlexStation. CPPHA (10 μM) was added to cells after 16 s of baseline determination. Five minutes later, a range of concentrations of agonist was added, and then cells were excited at 485 nm and the fluorescence was measured at 525 nm. When glutamate was used, CNQX (10 μM) and l-AP5 (20 μM) were added 20 min before the addition of CPPHA. A, example trace from fluorescence assay for calcium transients demonstrating the potentiation of the calcium response in cortical astrocytes by CPPHA. B, potentiation of DHPG-induced calcium transients by CPPHA. Curves were generated from four independent experiments. Points represent mean ± S.E.M. C, potentiation of glutamate-induced calcium transients by CPPHA. Curves were generated from three independent experiments. Points represent mean ± S.E.M.
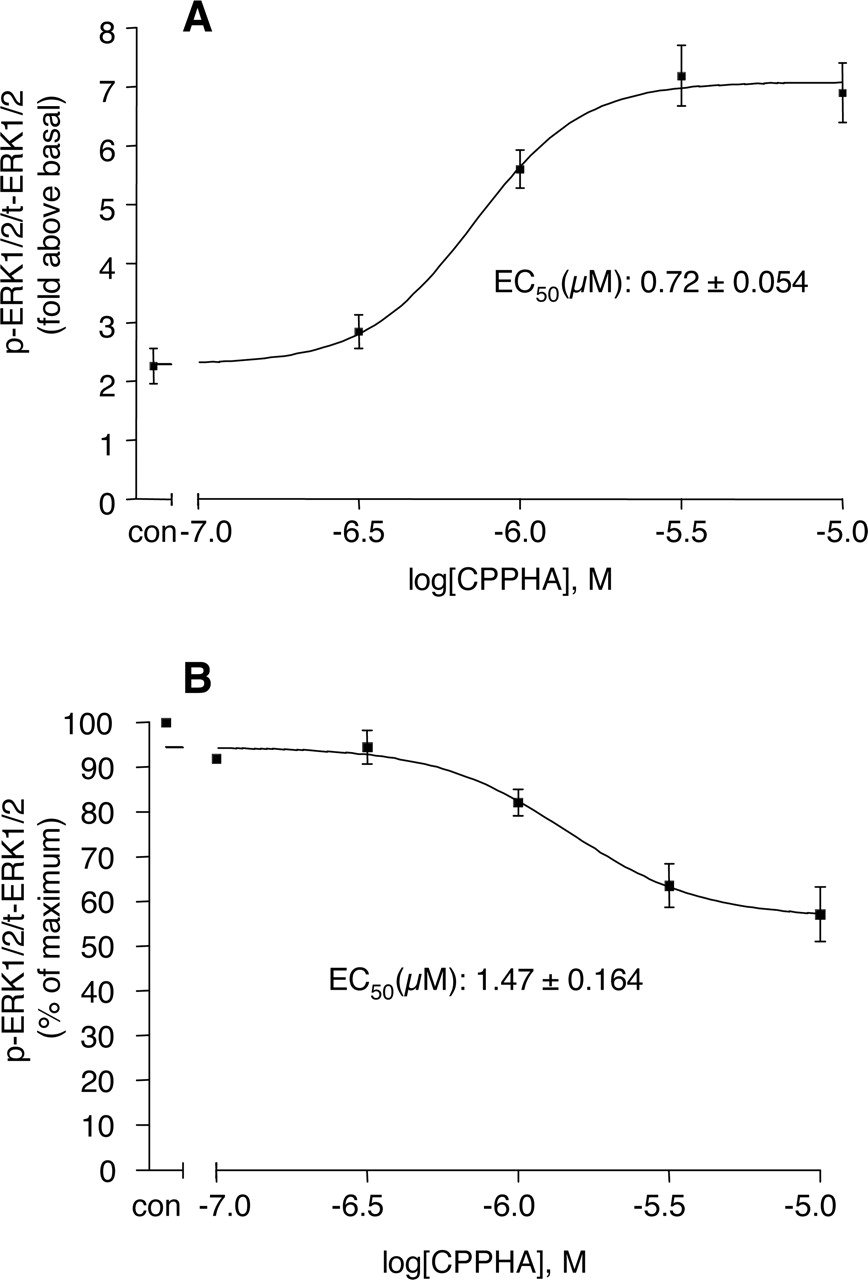
Interestingly, the effect of CPPHA on mGluR5-mediated ERK1/2 phosphorylation is qualitatively different from its effect on calcium mobilization. As shown in Fig. 4, A and B, 10 μM CPPHA potentiates the ERK1/2 phosphorylation response to low concentrations of DHPG in a manner similar to that seen with DFB. In contrast, 10 μM CPPHA inhibits ERK1/2 phosphorylation induced by high concentrations of DHPG (Fig. 4, A and B). Thus, CPPHA tends to flatten the concentration-response relationship for DHPG-induced increases in ERK1/2 phosphorylation rather than shifting the concentration-response curve to the left. Concentration-response studies of the effect of CPPHA revealed that CPPHA potentiates the ERK1/2 phosphorylation response to 1 μM DHPG with an EC50 value of 0.72 μM (Fig. 5A), and it inhibits the response to 10 μM DHPG with an EC50 value of 1.47 μM (Fig. 5B). We also determined the effect of CPPHA on glutamate-induced ERK1/2 phosphorylation. As discussed above, the presence of glutamate transporters makes measurement of glutamate-induced activation of ERK1/2 phosphorylation more difficult. However, consistent with studies with DHPG, we also found that CPPHA potentiated the ERK1/2 phosphorylation response to low concentrations of glutamate, but it inhibited the response to high concentrations of glutamate (data not shown).
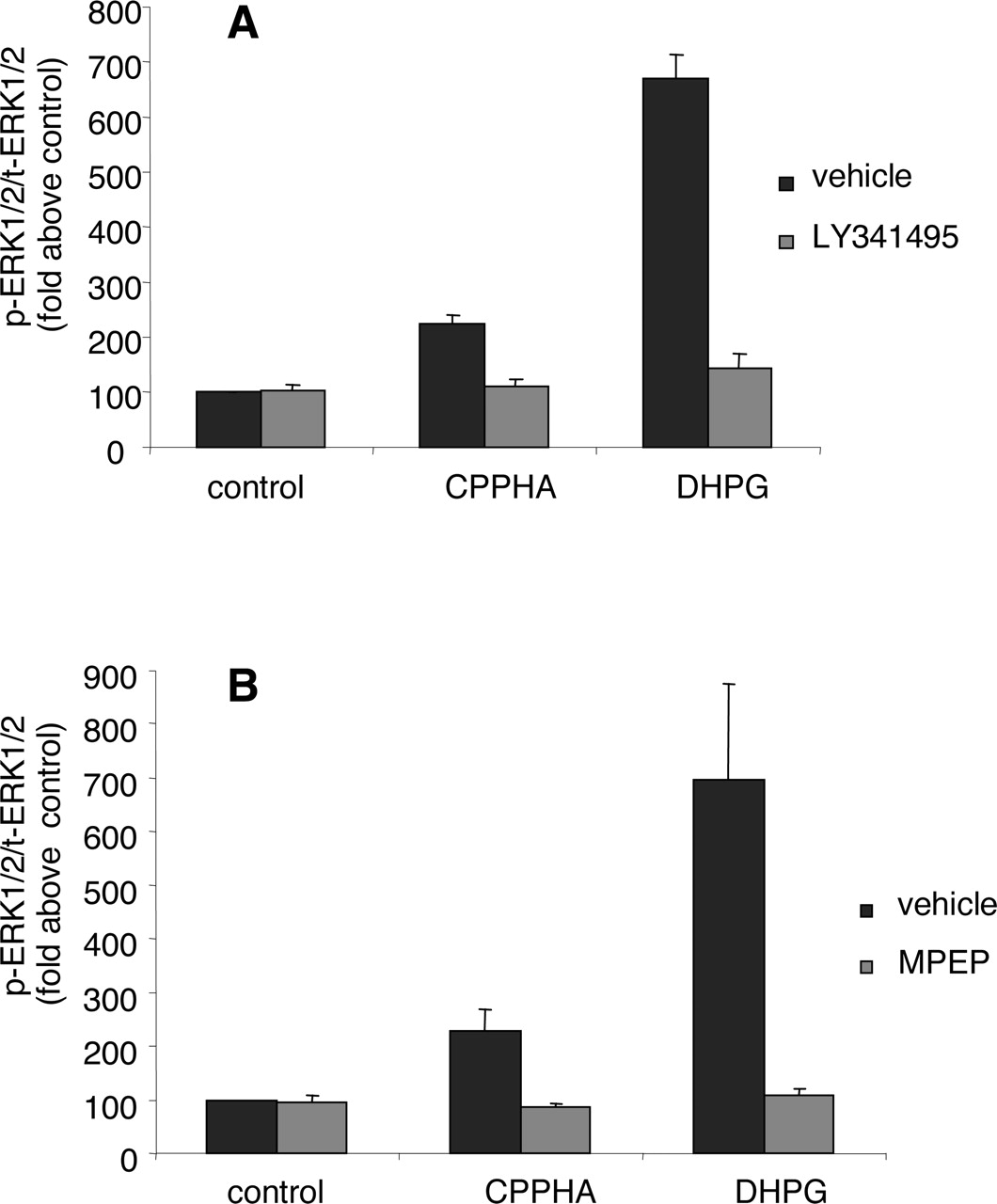
In addition to potentiating the ERK1/2 phosphorylation response to low concentrations of DHPG, CPPHA also induced a slight increase in ERK1/2 phosphorylation when added in the absence of DHPG (Fig. 4, A and B). This CPPHA-induced increase in basal ERK1/2 phosphorylation was completely blocked by both the noncompetitive mGluR5 antagonist MPEP and competitive antagonist LY341495 (Fig. 6), suggesting that this basal response to CPPHA is mediated by mGluR5.

CPPHA flattens the agonist concentration-response curve of mGluR5-mediated ERK1/2 phosphorylation in cortical astrocytes. Secondary rat cortical astrocytes were cultured in 12-well plates in G-5 supplement-containing medium for 3 days and then starved in glutamine-free DMEM for 20 h. The day of assay, cells were incubated with 10 μM CPPHA for 10 min and then stimulated with agonist for 10 min. ERK1/2 phosphorylation was measured with Western blotting for dually phosphorylated ERK1/2 and normalized with Western blotting for total ERK1/2 and then expressed as percentage of maximal ERK1/2 phosphorylation. A, representative Western blot analysis in duplicate. B, densitometry analysis of the modulation of DHPG-induced ERK1/2 phosphorylation by CPPHA from three independent experiments; points represent mean ± S.E.M.

Concentration-response relationship of CPPHA-induced potentiation and inhibition of ERK1/2 phosphorylation responses in cortical astrocytes induced by different concentrations of DHPG. A, CPPHA potentiates 1 μM DHPG-stimulated ERK1/2 phosphorylation. B, CPPHA inhibits 10 μM DHPG-stimulated ERK1/2 phosphorylation. Points represent mean ± S.E.M. of three independent experiments in duplicate.
The finding that CPPHA inhibits the ERK1/2 phosphorylation response to high concentrations of DHPG is intriguing and suggests that CPPHA could have fundamentally different effects on downstream physiological responses to activation of mGluR5 that are mediated by ERK1/2 phosphorylation versus calcium mobilization. However, the mechanism by which CPPHA inhibits this response to DHPG is not clear. It is possible that CPPHA shifts the kinetics of the ERK1/2 phosphorylation response. If so, CPPHA could potentiate the peak response to DHPG, but the response may already be desensitized at the 10-min time point used in these studies. Thus, we determined the time course of DHPG-induced ERK1/2 phosphorylation in the absence and presence of 10 μM CPPHA (Fig. 7) and 100 μM DFB, respectively. CPPHA significantly inhibited 10 μM DHPG-induced ERK1/2 phosphorylation in each time point after 2 min (Fig. 7), and it did not shift the time point of the peak response. Consistent with the studies of DFB effects with 10 min agonist incubation, DFB has no significant effect at any time point on ERK1/2 phosphorylation induced by DHPG at a dose that triggers maximal response (data not shown).

Blockade of CPPHA-induced increases in basal ERK1/2 phosphorylation by competitive and noncompetitive antagonists of mGluR5. ERK1/2 phosphorylation was measured as outlined in Fig. 4. Cells were preincubated with 100 μM LY341495 (A) or 10 μM MPEP (B) for 30 min and then treated with 10 μM CPPHA for 10 min. Points represent mean ± S.E.M. of three independent experiments determined in duplicate.
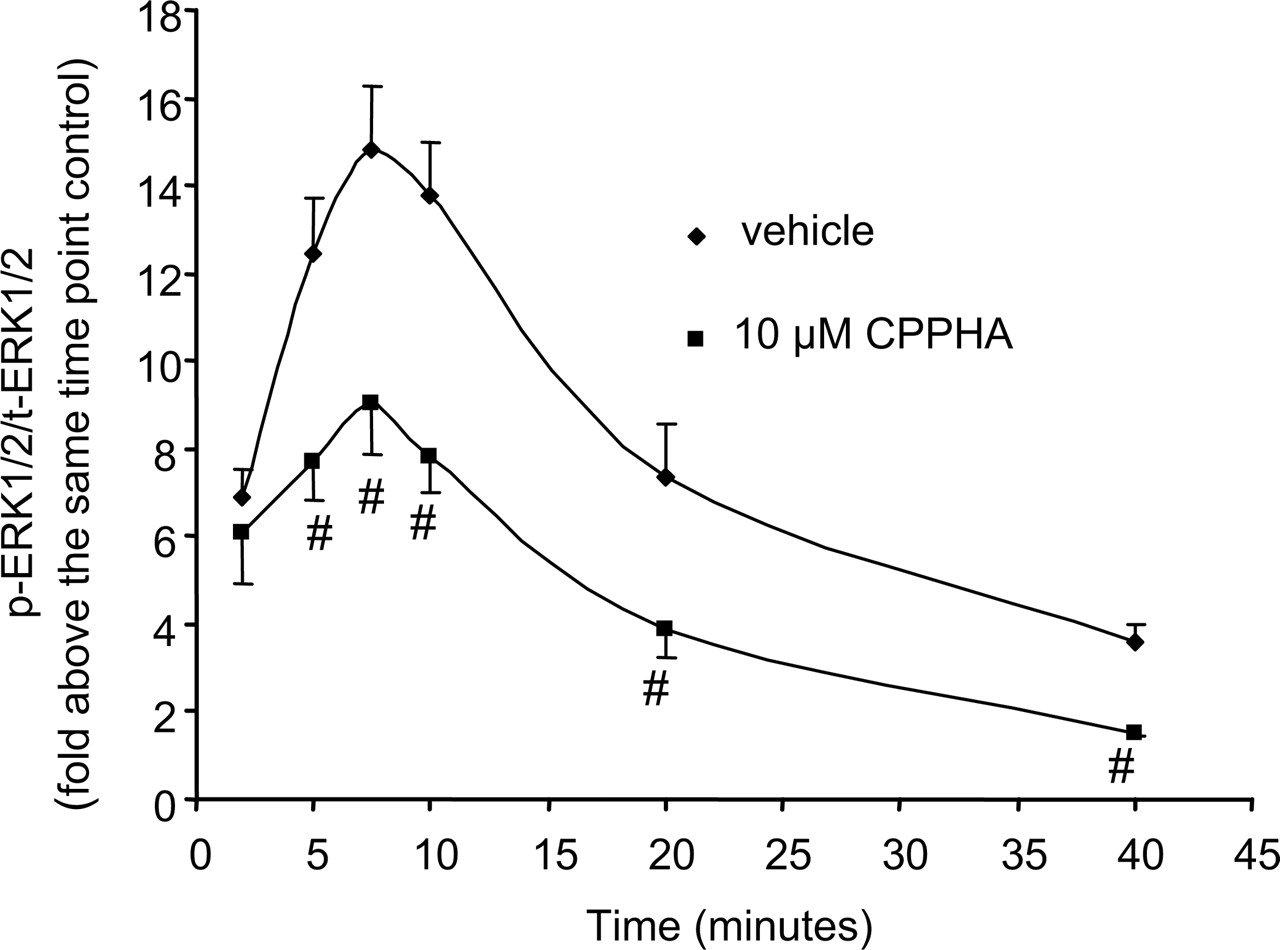
Time course of ERK1/2 phosphorylation. Cultured rat cortical astrocytes were stimulated with mGluR5 agonist for the time periods indicated in the presence and absence of CPPHA (10 μM). ERK1/2 phosphorylation was first normalized to total ERK1/2 and then expressed as fold above the same time point control (cells incubated with vehicle). Points represent mean ± S.E.M. from at least four independent experiments. #, p < 0.05.
Discussion
Recent years have seen tremendous advances in the discovery of allosteric modulators of GPCRs. Although allosteric regulators have been discovered for all major GPCR families, progress has been especially exciting for the family 3 GPCRs, such as the mGluRs, where a number of highly potent and selective allosteric potentiators with drug-like properties have been identified. Allosteric potentiators could have a number of potential advantages over direct-acting agonists as potential therapeutic agents. For example, orthosteric neurotransmitter binding sites are typically highly conserved across receptor subtypes for a single neurotransmitter, making it difficult to develop highly selective orthosteric site ligands. In addition, there are a number of problems associated with the use of direct-acting agonists as drugs. These include adverse effects associated with excessive activation of the receptor and loss of activity dependence of receptor activation. Allosteric potentiators have provided a proven approach for avoiding these problems in developing activators of ligand-gated ion channels. The classic example of this is the use of benzodiazepines as allosteric potentiators of GABA-A receptors that provide an effective and safe approach to treatment of anxiety disorders without inducing the potentially lethal effects of direct-acting GABA receptor agonists (Mohler et al., 2002). The emerging expansion of this approach to GPCRs has been an exciting and fundamental advance in our approach to discovery of novel classes of activators of this important receptor class. However, we are only beginning to understand the effects of allosteric potentiators of GPCRs, and the effects of these compounds on different signaling pathways in native systems have not been rigorously examined. Signaling by GPCRs is highly complex and involves coupling to multiple G proteins and other signaling partners and activation of multiple intracellular transduction pathways. In the simplest view, allosteric potentiators of GPCRs could induce a similar increase in activity of the intended receptor subtype in any cell population and on any response that is normally regulated by the receptor. However, it is possible that these compounds will have a more complex profile of effects and could differentially impact signaling through some pathways and not others.
We now report the effects of two novel allosteric potentiators of mGluR5, DFB and CPPHA, on mGluR5-induced ERK1/2 phosphorylation and calcium mobilization, two independent signaling mechanisms that are activated by mGluR5 in cortical astrocytes. Interestingly, these two potentiators behave similarly when measuring mGluR5 agonist-induced calcium transients. For this response, both DFB and CPPHA induce a parallel leftward shift of the agonist concentration-response curves. This response to DFB and CPPHA is similar to the response that we previously reported for mGluR5 expressed in CHO cells (O'Brien et al., 2003, 2004). Thus, the robust allosteric potentiator response is not unique to recombinant systems where mGluR5 is overexpressed, but it is also seen in cultured astrocytes that natively express mGluR5.
Interestingly, DFB induced a leftward shift of the agonist concentration-response curve when measuring DHPG-induced ERK1/2 phosphorylation that was similar to that observed when measuring calcium transients. In contrast, CPPHA induced qualitatively different effects on calcium mobilization and ERK1/2 phosphorylation. CPPHA induced a parallel leftward shift in the DHPG and glutamate concentration-response curves for calcium mobilization, but it flattened the concentration-response curves when measuring ERK1/2 phosphorylation. Thus, in the presence of low concentrations of agonist, CPPHA potentiates ERK1/2 phosphorylation, whereas CPPHA significantly attenuated the ERK1/2 phosphorylation response to a high concentration of agonist. Based on this, an in vivo response to an allosteric modulator in the CPPHA class could depend on the signaling pathway involved in mediating a given response. Indeed, it is possible that a compound having the effects described for CPPHA could actually have the opposite effects of those that would be predicted for a receptor activator, especially in conditions where there is high activation of a receptor by an endogenous agonist. In general, a compound with effects shown for CPPHA on ERK1/2 phosphorylation would be predicted to stabilize responses mediated by this pathway so that it enhances responses in conditions of low activity but inhibits responses in settings where there is a high level of endogenous receptor activation. This type of effect could be useful in some settings in that it may provide a protection against toxicity that could occur with excessive activation of some neurotransmitter receptors. However, this could also be a disadvantage in cases where there is a need to maximally potentiate a given pathway induced by receptor activation. Thus, when considering potential in vivo effects of allosteric potentiators, it will be critical to consider the possibility of differential effects on different responses to receptor activation.
The mechanism by which CPPHA inhibits ERK1/2 phosphorylation responses to mGluR5 activation are not entirely clear. However, CPPHA significantly inhibited ERK1/2 phosphorylation at each time point of the time course of DHPG-induced ERK1/2 phosphorylation after 2 min. This suggests that the inhibitory effect of CPPHA at 10 min was not due to a shift in the response kinetics so that there was an earlier peak in the maximal response to agonist. Importantly, this effect of CPPHA on ERK1/2 phosphorylation is not a general effect of allosteric potentiators, because DFB potentiates the ERK1/2 phosphorylation response to low concentrations of DHPG, but it does not inhibit the response to high concentrations of the agonist.
In recent years, concepts about GPCR function have dramatically changed from a view of these receptors as simple on/off switches to a view of complex signaling molecules in which equilibrium between active and inactive states for coupling to different signaling pathways can be differentially regulated by ligands (Liggett, 2002). Studies of β2 adrenergic receptors (Seifert et al., 1999), μ opioid receptors (Whistler et al., 1999), angiotensin II receptors (Hunyady et al., 1994), chemokine receptors (Blanpain et al., 2002), and complement factor 5a receptor (Whistler et al., 2002) show that GPCRs can exhibit multiple subsets of active conformations that link to different and independent signaling pathways. Furthermore, traditional orthosteric agonists can differentially activate different signaling pathways of a single GPCR, a phenomenon referred to as agonist receptor trafficking (Berg et al., 1998; Brink et al., 2000; Gazi et al., 2003). Based on this, it is not surprising that allosteric ligands bind to the same or distinct regions of the receptors to differentially regulate different responses to receptor activation.
The finding that DFB and CPPHA can have differential effects on different signaling pathways has important implications for the potential effects of these compounds on neuronal excitability and synaptic transmission in specific neuronal circuits. We and a number of other investigators have found that activation of mGluR5 can have a variety of effects on different neuronal populations, including cell depolarization, modulation of different potassium currents, potentiation of NMDA receptor currents, and many other responses. It is likely that these responses are mediated by different signaling mechanisms and could be differentially regulated (for review, see Valenti et al., 2002). These findings also have important implications for considering allosteric potentiators as potential therapeutic agents. Activators of mGluR5 have been proposed as potential therapeutic agents for treatment of schizophrenia and other disorders involving impaired cognitive function (Alagarsamy et al., 1999a,b; Mannaioni et al., 2001; Marino et al., 2001; Chavez-Noriega et al., 2002; Kinney et al., 2003; Campbell et al., 2004). However, previous studies raise the possibility that excessive activation of mGluR5 could also have adverse effects, including increased anxiety (Spooren et al., 2000; Tatarczynska et al., 2001), increased pain sensitivity (Salt and Binns, 2000; Bhave et al., 2001; Karim et al., 2001), and epileptiform seizure activity (Zhao et al., 2004). Different neuronal populations and signaling pathways are likely to be differentially involved in the multiple effects of mGluR activation. For example, ERK1/2 phosphorylation is thought to be required for mGluR5-mediated effects on inflammatory pain (Karim et al., 2001). It is conceivable that allosteric regulators could be developed to preferentially regulate a specific pathway to achieve a desired response. This possibility raises the critical importance of continued studies focused on developing a better understanding of the signaling pathways involved in different responses to mGluR5 activation, coupled with careful analysis of the effects of different classes of allosteric ligands on these pathways in native systems.
Footnotes
-
This study was supported by grants from National Institute of Neurological Disorders and Stroke, National Institute of Mental Health, National Alliance for Research on Schizophrenia and Depression and the Stanley Foundation (to P.J.C.), and a National Institutes of Health National Research Service Award postdoctoral fellowship (to A.L.R.).
-
doi:10.1124/jpet.105.090308.
-
ABBREVIATIONS: mGluR, metabotropic glutamate receptor; GPCR, G protein-coupled receptor; DFB, 3,3′-difluorobenzaldazine; CPPHA, N-[4-chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]phenyl]-2-hydroxybenzamide; ERK, extracellular signal-regulated kinase; DMEM, Dulbecco's modified Eagle's medium; FBS, fetal bovine serum; FLIPR, fluorometric imaging plate reader; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; NMDA, N-methyl-d-aspartate; CNQX, 6-cyano-2,3-dihydroxy-7-nitroquinoxaline; l-AP5, l-(+)-2-amino-5-phosphonopentanoic acid; CHO, Chinese hamster ovary; MPEP, 2-methyl-6-(phenylethynyl)-pyridine; LY341495, (2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl) propanoic acid.
- Received May 31, 2005.
- Accepted August 30, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}