


Abstract
CX516 (BDP-12) and CX546, two first-generation benzamide-type AMPA receptor modulators, were compared with regard to their influence on AMPA receptor-mediated currents, autaptic responses in cultured hippocampal neurons, hippocampal excitatory postsynaptic currents, synaptic field potentials, and agonist binding. The two drugs exhibited comparable potencies in most tests but differed in their efficacy and in their relative impact on various response parameters. CX546 greatly prolonged the duration of synaptic responses, and it slowed 10-fold the deactivation of excised-patch currents following 1-ms pulses of glutamate. The effects of CX516 on those measures were, by comparison, small; however, the drug was equally or more efficacious than CX546 in increasing the amplitude of synaptic responses. This double dissociation suggests that amplitude and duration of synaptic responses are governed by different aspects of receptor kinetics, which are differentially modified by the two drugs. These effects can be reproduced in receptor simulations if one assumes that CX516 preferentially accelerates channel opening while CX546 slows channel closing. In binding tests, CX546 caused an approximately 2-fold increase in the affinity for radiolabeled agonists, whereas CX516 was ineffective. More importantly, even millimolar concentrations of CX516 did not influence the dose-response relation for CX546, suggesting the possibility that they bind to different sites. Taken together, the evidence suggests that benzamide modulators from the Ampakine family form two subgroups with different modes and sites of action. Of these, CX516-type drugs may have the greater therapeutic utility because of their limited efficacy in prolonging synaptic responses and in attenuating receptor desensitization.
AMPA receptor pharmacology has grown enormously during the last 10 years with the discovery of compounds that allosterically modulate these receptors. Two structurally distinct types of such compounds were initially found in short succession, namely, aniracetam (Ito et al., 1990) and the benzothiadiazides diazoxide and cyclothiazide (Yamada and Rothman, 1992; Yamada and Tang, 1993). Although both subtypes generally potentiate AMPA receptor responses, the nature of their interaction with the AMPA receptor was found to differ in many respects. Thus, aniracetam and cyclothiazide were differentially sensitive to amino acid modifications in the receptor, and they had distinct preference patterns for receptor subunits (Johansen et al., 1995; Partin et al., 1996). Such differences are likely to extend to structurally related compounds, in particular to a family of benzamide compounds called Ampakines, which were developed using aniracetam as the lead compound (Arai et al., 1994, 1996a,b, 2000; Staubli et al., 1994a,b). Indeed, Ampakine modulators and cyclothiazide were shown to differ substantially in their effect on AMPA receptor-mediated responses (Arai et al., 1996a; Arai and Lynch, 1998a,b). In excised-patch studies, Ampakines generally were more effective in prolonging response decay upon glutamate removal (“deactivation”) and less effective in blocking receptor desensitization, and in hippocampal slices they produced more prominent augmentation and prolongation of synaptic transmission (Arai and Lynch, 1998a,b).
Ampakine ligands represent perhaps the most systematically developed subgroup of AMPA receptor modulators. Whereas early compounds such as 1-BCP (also called BDP and CX465; Arai et al., 1994) were of modest potency, newer members of this drug family act at micromolar concentrations (Arai et al., 2000). Their effects showed a remarkable consistency across multiple system levels from receptor biophysics to synaptic transmission (Arai et al., 1994, 1996a,b, 2000), gene expression (Holst et al., 1998; Lauterborn et al., 2000), neural activity (Staubli et al., 1994a,b; Hampson et al., 1998), and animal as well as human behavior (e.g., Granger et al., 1993; Larson et al., 1995, 1996; Lynch et al., 1996; Goff et al., 2001). However, cumulative evidence also indicated that there is some degree of functional diversity even within this drug family. In patch experiments, for instance, CX554 (BDP-20) reduced the rate of AMPA receptor desensitization more than 10-fold (Arai et al., 1996a), whereas CX516 (BDP-12) had only modest effects (Arai et al., 1996b). Also, EPSPs recorded in hippocampal slices in the presence of CX554 showed a prominent increase in the duration of the response (Arai and Lynch, 1998a), whereas CX516 mainly seemed to enhance amplitude (Arai et al., 1996b). Variation with regard to these measures was seen also in comparisons between other Ampakines, but most produced effects resembling those of CX554. For example, CX614 (Arai et al., 2000), with about 10 times higher potency than CX554, was effective in blocking desensitization in patch experiments and in prolonging synaptic responses. Another compound of this type was CX546 [1-(1,4-benzodioxan-6-ylcarbonyl)piperidine], which structurally resembles CX614 except for being sterically less constrained (see Fig.1, below). When examined for its effect on hippocampal synaptic responses, CX546 exhibited an unprecedented ability to prolong response duration while changes in amplitude often were minor. CX546 thus appeared to display features almost complementary to those of CX516.

Chemical structure of Ampakine modulators.
The present study was designed to examine these differences in greater detail by comparing the effects of CX516 and CX546 in parallel across a set of preparations. CX546 has been shown to increase neurotrophin expression in forebrain neurons (Lauterborn et al., 2000). Its ability to potentiate AMPA receptor currents has been demonstrated in some preparations (Baumbarger et al., 2001; Nagarajan et al., 2001), but its effects on synaptic responses and agonist binding have not yet been adequately characterized. Some properties of CX516 (BDP-12) have been described in an earlier study (Arai et al., 1996b). The first question to be addressed was whether the differences between the effects of these two compounds are sufficiently robust to manifest themselves across different preparations and recording techniques. This was examined specifically by comparing their effects on field responses in CA1 of hippocampal slices with those on excitatory postsynaptic currents (EPSCs) and autaptic responses in cultured hippocampal neurons, and with those on AMPA receptor currents in patches excised from CA1 pyramidal neurons. The second group of experiments then addressed the question whether CX516 and CX546 act through the same site on the receptor. This was examined by measuring the allosteric influence of the drugs on the binding of a radiolabeled agonist and by testing whether such effects reveal evidence for competitive interaction.
Materials and Methods
Patch-Clamp Recordings from Pyramidal Neurons in the Field CA1.
Patch-clamp studies were carried out with outside-out patches excised from pyramidal neurons in field CA1 of organotypic hippocampal slices (Arai et al., 1996a, 2000). The slice cultures were prepared from 13- to 14-day-old Sprague-Dawley rats (Harlan, Indianapolis, IN) and grown for 2 weeks on cellulose membrane inserts (Millicell-CM; Millipore Corporation, Bedford, MA). Patches were excised in a medium containing 125 mM NaCl, 2.5 mM KCl, 1.25 mM KH2PO4, 2 mM CaCl2, 1 mM MgCl2, 5 mM NaHCO3, 25 mM d-glucose, and 20 mM HEPES (pH 7.3) and relocated to a chamber perfused with recording medium containing 130 mM NaCl, 3.5 mM KCl, 20 mM HEPES, 0.01 mM MK-801, and 0.05 mM d-AP5. Patch pipettes had a resistance of 3 to 8 MΩ and were filled with a solution of 65 mM CsF, 65 mM CsCl, 10 mM EGTA, 2 mM MgCl2, 2 mM ATP disodium salt, and 10 mM HEPES (pH 7.3).
A piezo-device was employed to switch solutions applied to the patch within less than a millisecond. In brief, background medium and agonist-containing medium were flowing continuously through two lines of a bifurcated pipette that was moved by a piezo-device across a distance of 50 μm in 0.4 ms (Arai et al., 1996a,b). Data were acquired with a patch amplifier (AxoPatch-1D; Axon Instruments, Inc., Foster City, CA) at a filter frequency of 5 kHz and digitized at 10 kHz with PClamp/Digidata 1200 (Axon Instruments, Inc.). The holding potential was −50 mV. The drugs were applied at the same concentration in both background and glutamate lines; background flow lines were switched at least 15 s before applying the first glutamate pulse. Typically, five responses were collected and averaged for each condition. Measurement with each patch was alternated repeatedly between control (A, glutamate alone) and test conditions (B, glutamate + drug). For data analysis, response B was compared with the average of the responses A taken before and after response B, and peak and steady-state currents recorded in the presence of drug were normalized to those without drug. CX546 and CX516 solutions were prepared from 1 M stock solutions in dimethyl sulfoxide (DMSO); the same final concentrations of DMSO were included in all drug and control solutions.
Whole-Cell Recording from CA1 Pyramidal Neurons in Hippocampal Slices.
Sprague-Dawley rats of postnatal days 15 to 21 (Harlan) were decapitated under anesthesia, following National Institutes of Health guidelines and an institutionally approved protocol. Transverse hippocampal slices (400 μm) were prepared using a Vibratome (Leica Microsystems, Inc., Deerfield, IL). The slices were submerged in oxygenated artificial cerebrospinal fluid (ACSF) infused at 0.5 ml/min; the experiments were carried out at ambient temperature. The ACSF contained 124 mM NaCl, 3 mM KCl, 1.25 mM NaH2PO4, 2 mM CaCl2, 1 mM MgSO4, 5 mM NaHCO3, 10 mM glucose, and 10 mM HEPES (pH 7.4). The pipette solution contained 130 mM CsF, 10 mM EGTA/K, 20 mM HEPES, and 2 mM ATP-Mg (pH 7.35, adjusted with KOH). Whole-cell recording was made from pyramidal cells in field CA1 with voltage-clamp configuration; the cells were visualized with an infrared microscope (BX50 WI; Olympus, Tokyo, Japan) with differential interference contrast configuration. Synaptic responses were recorded using borosilicate glass electrodes (2–5 MΩ) in response to activation of Schaffer-commissural fibers stimulated by a bipolar nichrome electrode in stratum radiatum. After establishing a stable baseline, the perfusion line was switched to one containing the drug; solution exchange in the recording chamber was complete within 3 min. EPSCs were recorded with AxoPatch 200B. The signals were filtered at 5 kHz and digitized at 10 kHz with Digidata1200/pClamp 7. The holding potential was −70 mV.
Whole-Cell Recording from Primary Cultures Prepared from the Hippocampus.
Neuronal cultures were prepared with a slight modification of the method of Baughman et al. (1991), as described inArai et al. (2000). In brief, hippocampi were isolated from E16- to E18-day-old Sprague-Dawley rats, cut into small pieces, and incubated with 0.05% trypsin/0.53 mM EDTA at 37°C for 30 min. After centrifugation (900 rpm), the tissue pellet was suspended in plating minimal essential medium with 5% fetal calf serum, penicillin/streptomycin, 10 μM MK-801, and 100 μM AP5, and gently triturated until the cells were completely dispersed. Cells were plated onto a recording chamber (Nalge Nunc, Naperville, IL) with microislands coated with poly-d-lysine (0.02 mg/ml) and grown for 10 to 25 days at 37°C. Whole-cell recordings were made from solitary neurons on microislands exhibiting mature morphology. The extracellular recording solution contained 140 mM NaCl, 4 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 5 mM NaHCO3, 10 mM glucose, and 20 mM HEPES (pH 7.37), and was supplemented with 50 μM picrotoxin, 10 μM MK-801, and 100 μM AP5. The intrapipette solution contained 130 mM CsF, 10 mM EGTA, 2 mM ATP disodium salt, and 10 mM HEPES (pH 7.4). The holding potential was −60 mV. Autaptic responses were evoked by clamping the membrane potential at +20 mV for 1 ms. Neurons from which recordings were made were identified immunohistochemically.
Extracellular Recording in Hippocampal Slices.
Transverse hippocampal slices (400 μm) were prepared from male Sprague-Dawley rats (150–200 g; Charles River Laboratories, Wilmington, MA) as described elsewhere (Arai et al., 1996b). The slices were placed in an interface chamber, which was perfused at 0.5 ml/min with oxygenated ACSF containing 124 mM NaCl, 3 mM KCl, 1.25 mM KH2PO4, 3.4 mM CaCl2, 2.5 mM MgSO4, 26 mM NaHCO3, and 10 mM d-glucose, and exposed to humidified 95% O2/5% CO2. Field EPSPs were recorded from the stratum radiatum in response to activation of Schaffer-commissural fibers in the same stratum. The input-output relation of the synaptic response was first established to determine the maximum EPSP amplitude without spike component, and the stimulation intensity was adjusted to 50% of the maximum EPSP amplitude. After establishing a stable baseline, the perfusion line was switched to one containing CX516 or CX546.
Binding Assays.
Rat brain membranes were prepared from the telencephalon according to conventional procedures that involved 1) homogenization in an isotonic sucrose solution and differential centrifugation to obtain a P2 pellet fraction, 2) osmotic lysis, and 3) repeated washing by centrifugation and resuspending in the buffer in which most binding experiments were carried out (100 mM HEPES/Tris, 50 μM EGTA, pH 7.4) (see Arai et al., 2000 for details). Aliquots were frozen at −80°C; after thawing, the membranes were tip-sonicated and washed at least twice by centrifugation. For some assays, the membranes were suspended in an ACSF-type buffer containing 124 mM NaCl, 3 mM KCl, 1 mM KH2PO4, 2 mM MgSO4, 1 mM CaCl2, 5 mM NaHCO3, 10 mM HEPES, and 20 mM glucose (pH 7.4). Synaptoneurosomes were prepared according to the method ofHollingsworth et al. (1985) with minor modifications. In brief, rat cortex was minced in the ice-cold ACSF-type buffer described above, gently homogenized by hand in a Dounce tissue grinder, and filtered through cloth and Millipore filters of decreasing pore size (5 μm final pore size). The synaptoneurosomes were then washed three times by centrifugation (3000g) and resuspended gently in the ACSF buffer. Binding assays were generally conducted at 25°C with the centrifugation method. Aliquots of the membrane suspension (100 μg of protein in 200 μl) were incubated for 45 min with typically 20 to 50 nM radiolabeled compound and appropriate additions. Sets of 24 samples were then centrifuged for 20 min at 25,000g in a Beckman JA-18.1 rotor (Beckman Coulter, Inc., Fullerton, CA) with temperature settings such that the rotor temperature was maintained around 25°C. Ten minutes after the centrifugation, the supernatant was aspirated and the pellet was quickly rinsed with ice-cold buffered saline containing 50 mM KSCN (wash buffer). The pellets were dissolved in 20 μl of tissue solubilizer BTS-450 from Beckman Coulter before adding acidified scintillation fluid. Drugs were added from 100-fold concentrated solutions in DMSO; separate mixing tests were used to verify that these procedures did not result in drug precipitation. Control samples received the equivalent amount of DMSO (usually 1%); binding was changed by less than 10% at this solvent concentration. Background values (“nonspecific binding”) were measured by inclusion of 5 mM l-glutamate and subtracted from total binding; separate background values were determined for incubations with and without drug. Protein content was determined according to the method of Bradford (1976) with the reagent available from Bio-Rad (Hercules, CA) and with bovine serum albumin as standard. Binding curves were fitted to the data points through nonlinear regression using the Prism program (GraphPad Software, Inc., San Diego, CA). [3H]CNQX was purchased from PerkinElmer Life Sciences (Boston, MA) and [3H]fluorowillardiine from Tocris Cookson, Inc. (Ballwin, MO). Other reagents were from the usual commercial sources.
Results
The Ampakine ligands compared in the present study, CX516 and CX546, are shown in Fig. 1, together with congeners described in earlier reports (Arai et al., 1994, 1996a,2000). Both compounds contain a benzamide core but differ in the nature of the fused heterocycle. The nitrogen-containing ring in CX516 is aromatic in nature and, thus, the bicyclic quinoxaline group is completely planar. In CX546, the ethylene carbons joining the oxygens lie outside the plane defined by the benzene ring. It should be noted that whereas the benzene ring of CX546 is electron-rich because of the two attached oxygen atoms, the corresponding ring of CX516 is electron-deficient due to the nitrogen heterocycle. This fundamental difference could have significant impact on π-π interactions with aromatic amino acid side chains that could be present at the receptor binding site. Many other Ampakine modulators resemble CX546 in that they contain oxygen heterocycles fused to the benzene ring of the pharmacophoric benzamide structure.
Effects of CX546 on AMPA Receptor Currents in Excised Patches.
Outside-out patches were excised from CA1 pyramidal cells of cultured hippocampal slices and exposed to long-duration (800-ms) pulses of 1 mM glutamate (Fig. 2, top left). Patches were equilibrated at the selected drug concentration 30 s before applying the glutamate test pulses. In the absence of drug, responses rapidly desensitized until they reached a steady-state value of about 5% of the peak current. Increasing the CX546 concentration progressively raised the steady-state current and the peak current (Fig. 2, group data) without significant effect on the time constant for the decay from the peak to the steady-state. This indicates that desensitization is effectively blocked in those receptors that have bound the drug and that increasing the drug concentration mainly changes the proportion between drug-free and drug-occupied receptors. The threshold concentration for increasing the steady-state current was in the order of 30 μM. Since the drug effect did not reach saturation at the highest concentration tested (2 mM), the EC50 estimate of 1 to 2 mM is only approximate. A consistent feature at higher drug concentration was a delayed increase in the peak current during the first 100 ms of glutamate application, as shown on an expanded scale on the right. For the purpose of comparison, a dose-response curve for CX516 similar to that shown previously (Arai et al., 1996b) was included in Fig. 2. It is evident that the two drugs differ greatly in their efficacy of enhancing the steady-state current, which in the case of CX516 did not exceed 30% of the peak current, even at the near-saturating concentration of 5 mM. CX516 had considerably higher potency, however, for enhancing the steady-state current level with an EC50 in the order of 150 μM.

Effects of CX546 on AMPA receptor-mediated currents in excised patches. A, representative set of responses from a single patch at 0, 30, 100, and 1000 μM CX546. Membrane patches were excised from pyramidal neurons in field CA1 of hippocampal slices. The patches were equilibrated with recording solution without or with CX546 prior to application of glutamate; the drug was included at the same concentrations in the background flow solutions and in the glutamate-containing solutions. The horizontal bar indicates the application of 1 mM glutamate (800 ms). The region indicated with arrowheads is expanded on the right. B, concentration-response relations for the steady-state current of responses to 800-ms applications of 1 mM l-glutamate. Steady-state currents were normalized to the peak current measured without drug. The data represent the mean and S.E.M. of four to eight experiments with CX546 and four to nine experiments with CX516 (n = 9 for 1 and 5 mM CX516). The average steady-state current in the absence of drug was 5.1 ± 0.6%. The EC50 value for CX546 is estimated from sigmoidal curve fitting to be on the order of 1.6 mM. The EC50 value for CX516 is 150 μM (with a 95% confidence interval of 44–500 μM). The data for CX516 are taken from the experiments shown in Arai et al. (1996b) but have been reanalyzed to be expressed as percentage of the peak current; they have been included in the figure for the purpose of comparison. The lower graph shows the CX516 data on an expanded Y-scale.
Effects of CX516 and CX546 on AMPA Receptor-Mediated Responses in Different Physiological Preparations.
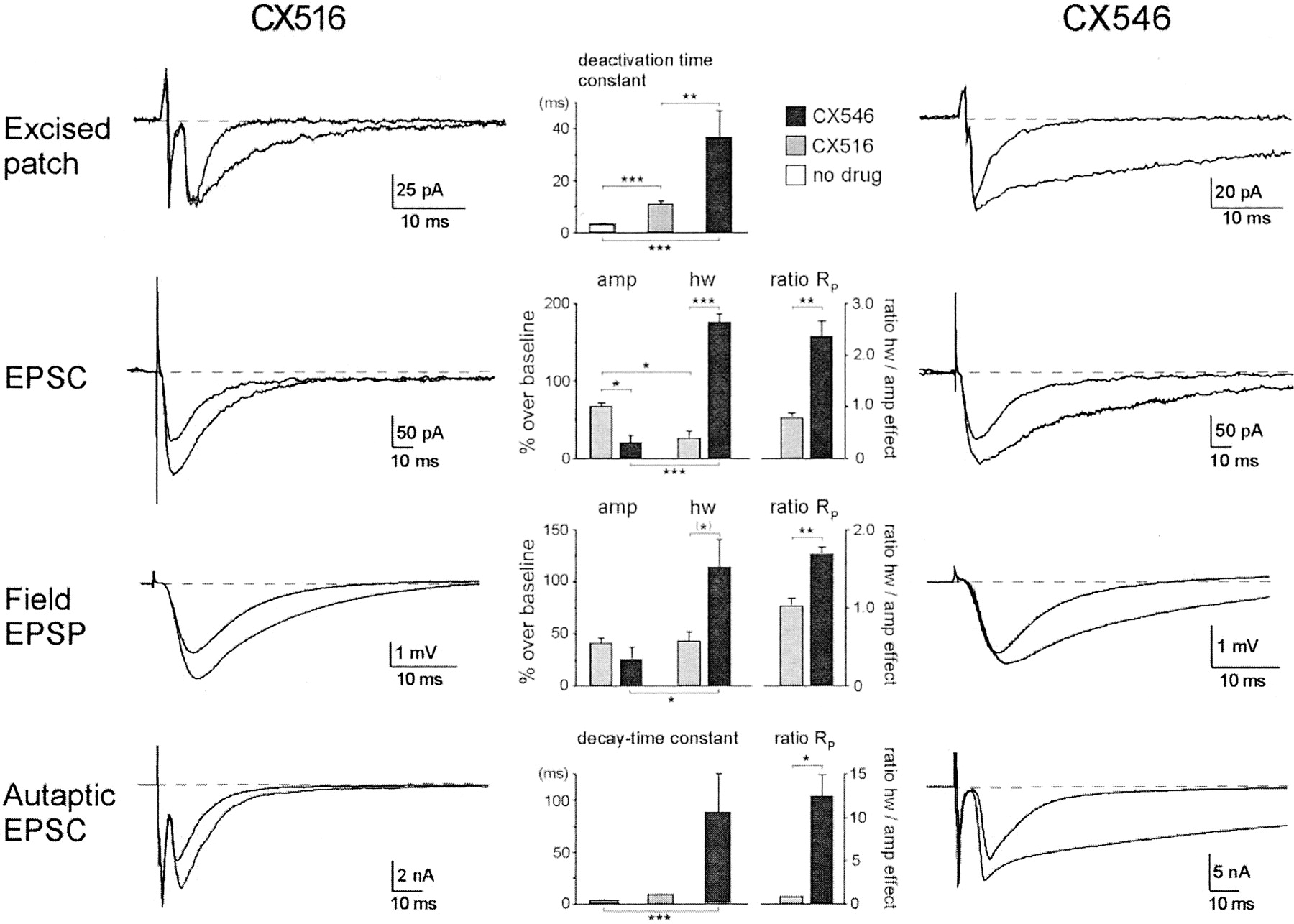
Figure 2 showed that CX516 and CX546 differ in their efficacy to modulate AMPA receptor kinetics. The following tests examined whether this is associated with manifest differences in their effect on synaptic responses. The top row in Fig.3 compares the effects on excised-patch responses to glutamate pulses (10 mM) of 1 ms duration. In the absence of drug, responses typically decayed to baseline with a time constant of 2 to 4 ms (Arai et al., 1996b). In the presence of CX546, this decay phase of the response was prolonged and fitted best with a two-exponential function. The fast component had a decay time constant of 3.4 ms, which was similar to that of the control response (3.5 ± 0.5 ms, n = 8), and its amplitude contribution became smaller with increasing drug concentration, indicating that it originated from a subpopulation of receptors that had not bound the drug. The slow component, which thus presumably represents receptors with drug bound to them, exhibited a decay time constant of 37 ± 10 ms (n = 4) at a CX546 concentration of 1 mM. This result indicates that the drug slows deactivation at least 10-fold. The effect of CX516, even at the high concentration of 5 mM, was much more modest (Fig. 3, top left), in agreement with our previous report (Arai et al., 1996b). The decay time constant was in this case increased from 3.5 ± 0.5 ms to 11.2 ± 1.3 ms (n = 8), or by a factor of 3.2. Amplitudes were minimally changed in these tests because a near-saturating concentration of glutamate was employed.

Comparison of the effects of CX516 and CX546 on AMPA receptor-mediated responses in various preparations. Traces recorded in the presence and absence of the drugs are shown superimposed on the left for CX516 and on the right for CX546. The graphs in the middle show the corresponding group data. Top row, effects of 5 mM CX516 and 1 mM CX546 on responses to a 1-ms pulse of 10 mM glutamate in a patch excised from a pyramidal neuron in field CA1 of a hippocampal slice. Holding potential −50 mV. The graph in the middle shows the group data for the deactivation time constant (in milliseconds) in the presence and absence of the drugs. The decay phase of the control responses and of the responses in the presence of CX516 was adequately fitted with single-exponential functions, whereas responses in the presence of CX546 were best fitted with two-exponential functions. The latter yielded a dominant component with a slow decay time constant and a small component (∼20% of amplitude) with a fast decay time constant comparable with that obtained under control conditions. The average time constant for the slow component (mean and S.E.M.;n = 4) is shown in the bar graph, together with the values for control responses (n = 8) and for CX516 (n = 8). Second row, effect of 2 mM CX516 and 400 μM CX546 on EPSCs recorded from CA1 pyramidal neurons in hippocampal slices. Synaptic responses before and after 10 min of drug infusion are superimposed. Holding potential −70 mV. The bar graph in the middle illustrates the percentage increase in the amplitude (amp) and half-width (HW) of the EPSCs over baseline (mean and S.E.M.,n = 4 for each drug). The columns on the right (ratio RP) indicate the average preference for amplitude or half-width effect across all trials with either CX516 or CX546. For this purpose, the ratios RHW (half-width with drug)/(half-width before drug) and RA (amplitude with drug)/(amplitude before drug) were calculated for each trial and then divided by each other to obtain a preference ratio RP= RHW/RA, where RP = 1 indicates equal increases in both amplitude and half-width. The RP values from all trials were then averaged and plotted as mean and S.E.M. Statistical significance is indicated throughout as follows: ★, p < 0.05, ★★,p < 0.01, ★★★, p < 0.001 (t test, two-tailed); (★), p< 0.05 (t test, one-tailed). Third row, effect of 1 mM CX516 and 200 μM CX546 on field EPSPs recorded in field CA1 of hippocampal slices (mean and S.E.M. of four and six experiments, respectively). Fourth row, effects of 5 mM CX516 and 1 mM CX546 on autaptic responses in hippocampal neurons cultured on poly(lysine) microislands. Holding potential −70 mV. Responses obtained in the absence of the drugs and in the presence of CX516 were best fit with a single-exponential function and those in the presence of CX546 with a two-exponential function. In the latter case, the fast component (4.3 ± 0.6 ms) had essentially the same time constant as the control responses (4.1 ± 1.1 ms). Summary data for the time constant of the slow component and for responses in the presence of CX516 are shown in the bar graph (mean and S.E.M. of 3–11 experiments).
The second row of Fig. 3 shows the effects of the two drugs on synaptically evoked responses using whole-cell recordings from CA1 pyramidal neurons of hippocampal slices. To avoid generalized discharges in the presence of CX546, drug concentrations were reduced, but the ratio between them was kept the same. CX546 proved again to be much more effective in prolonging response duration. The EPSC half-width was increased by 178 ± 11% (n = 4) in the presence of 400 μM CX546 versus 28 ± 10% (n = 4) in the presence of 2 mM CX516 (p < 0.001, t test). Interestingly, however, the latter compound had a much larger effect on response amplitude (68 ± 4%) when compared with CX546 (24 ± 9%;p < 0.05). This difference was characterized further by calculating for each drug trial a parameter, RP, which represents the preference for enhancing half-width more than amplitude (RP > 1) or vice versa (RP < 1; see Fig. 3 legend). The difference in this ratio (2.33 ± 0.30 for CX546 versus 0.77 ± 0.09 for CX516) was highly significant (p < 0.01). Similar trends were seen in field recordings from the same region (third row). CX516 at 1 mM prominently enhanced the EPSP amplitude by 43 ± 5% and it increased the halfwidth of the response by 44 ± 10% (n = 4). These effects are comparable with those reported in a previous study for this drug concentration (Arai et al., 1996b). CX546 at 200 μM increased the amplitude to a lesser degree (28 ± 11%), but it prolonged responses by a factor of 2.2 (115 ± 27% over baseline half-width, n = 6). The magnitude of this effect was comparable in primed and nonprimed responses (not shown), indicating that inhibitory postsynaptic potentials did not contribute in any significant way to the differences in these apparent drug effects. The difference between the preference ratios RP is again highly significant (1.66 ± 0.09 for CX546 versus 1.01 ± 0.10 for CX516; p< 0.01), although less pronounced than with whole-cell recording.
The last row of Fig. 3 compares the effects of the two drugs on synaptically evoked responses in hippocampal primary cultures. Hippocampal neurons cultured on microislands of poly(lysine) coating tend to make synapses onto themselves (Baughman et al., 1991; see alsoArai et al., 2000). In these cells, a brief voltage jump to a depolarizing holding potential produces a rapid inward sodium current that is followed by a synaptically mediated current known as autaptic response. These responses, which are completely abolished by CNQX (Arai et al., 2000), typically reached their peak around 4 to 10 ms after the rapid sodium current. The decay phase of the control response was in general fitted best with a single-exponential function with a time constant of 4.1 ± 1.1 ms (n = 11, range 1.5- 12.5 ms). Drugs like CX546 could be examined in this system at higher concentrations than in intact tissue because lower synapse density reduced the risk of uncontrolled discharges. Application of 1 mM CX546 greatly prolonged the autaptic response. As in the excised-patch experiments, the decay phase of the responses clearly exhibited two exponential components, one of which had a time constant similar to that of the control response (4.3 ± 0.6 ms) and, thus, presumably reflects a subpopulation of receptors which have not bound the drug. The decay time constant of the dominant slow component was 89 ± 37 ms (range 38–162 ms), which indicates that the drug again had slowed deactivation at least by a factor of 10. The effects of CX516 were markedly different. Even at a concentration as high as 5 mM, it increased the decay time constant to only 10.0 ± 0.4 ms or by a factor of 2.4. Given that both drugs were tested at concentrations near or above their half-saturating concentrations, these results indicate that CX546 has a much greater ability to prolong response duration when compared with CX516. Taken together, these data show that the two benzamides have highly distinct effects on the wave form of synaptic transmission and that the differences are consistent across preparations.
Allosteric Effects of Ampakine Modulators on Agonist Binding.
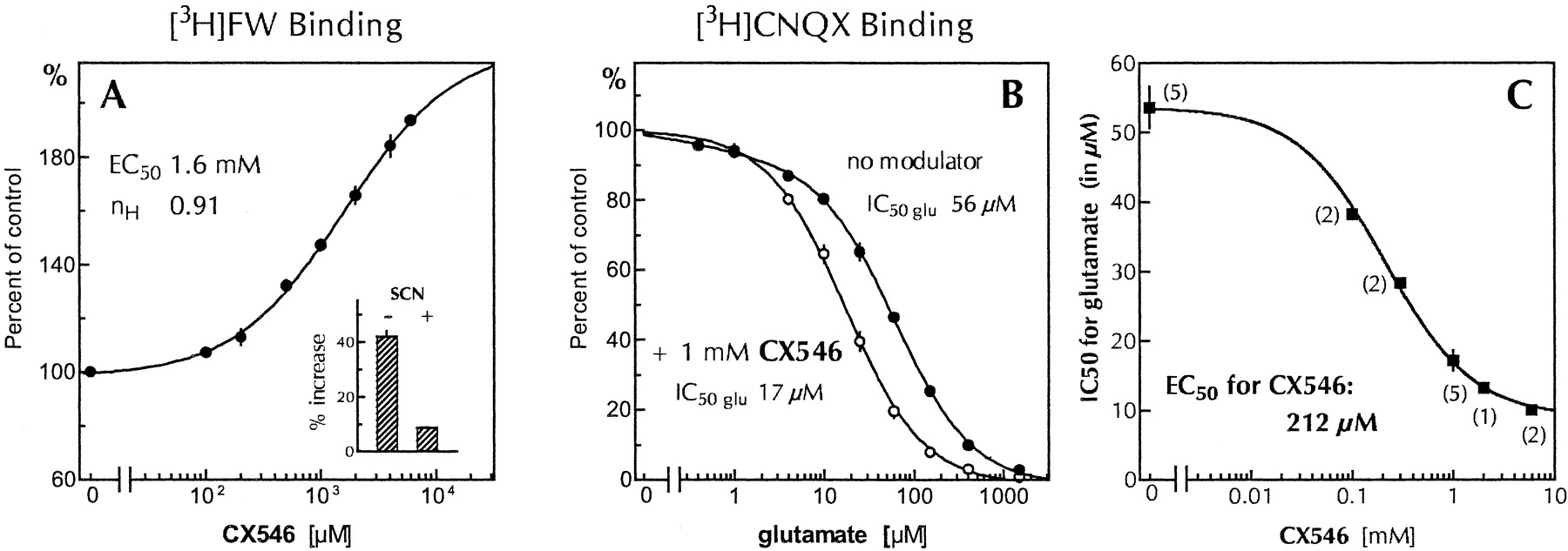
Most binding tests were carried out at 25°C with crude synaptic membranes from rat telencephalon. Figure4A shows that binding of the agonist [3H]fluorowillardiine to the AMPA receptor was substantially increased in the presence of CX546. The maximal increase was near 100% and thus larger than that seen with other Ampakine modulators (Arai et al., 1996a, 2000). EC50estimates for this effect varied across experiments between 0.8 and 2 mM, mainly due to solubility limitations, which prevented reliable determination of the upper asymptote. Hill coefficients were consistently near 1, as with most other AMPA receptor modulators.

Effects of CX546 on the binding of AMPA receptor agonists. A, effect of CX546 on [3H]FW binding. Tests were carried out by incubating rat brain membranes (lysed and extensively washed P2 fraction) for 45 min at 25°C in the presence of 20 nM [3H]fluorowillardiine and the CX546 concentrations indicated on the x-axis. Incubations were terminated by centrifugation. CX546 was added from 100× stock solutions in DMSO; controls received 1% DMSO. The data points (mean and S.D.; n = 5) were fitted with a four-point logistic equation; the 95% confidence interval for the EC50 is 1.2 to 2.2 mM. Determination of the maximal drug effect and hence the EC50 value is constrained by solubility limitations. Inset, binding increase produced by 1 mM CX546 in the absence of thiocyanate (as in the parent graph) and in the presence of 50 mM thiocyanate (added as potassium salt), a chaotropic anion that increases binding affinity for AMPA receptor agonists. B, effect of CX546 on the binding affinity of the AMPA receptor for glutamate, as determined from the displacement of [3H]CNQX binding by increasing concentrations of glutamate. Brain membranes were incubated for 45 min at 25°C in the presence of 40 nM [3H]CNQX, the glutamate concentrations indicated on the x-axis, and 0 or 1 mM CX546. Binding in the presence of 10 mM glutamate was considered as nonspecific and was subtracted from all measurements. [3H]CNQX binding was minimally influenced by CX546; binding without glutamate was on average 1.6 pmol/mg protein in the absence of modulator and 1.7 pmol/mg protein in the presence of 1 mM CX546. For further analysis, binding in the absence of glutamate was set at 100% and percentage binding at each glutamate concentration was averaged (mean and S.D.) from five experiments at each modulator concentration. Curve fitting to the data without drug was statistically superior (p = 0.006) with the assumption of two sites. The IC50 of the dominant site (93%) was 56 μM; the second component (7%) with an IC50 below 1 μM probably represents the high-affinity variant of AMPA receptors with a possible contribution from kainate receptors. Data obtained in the presence of CX546 were adequately fitted by a single-site curve, probably indicating that the high-affinity component was not altered by the modulator and thus could no longer be separated computationally from the low affinity site. Cheng-Prusoff corrections yield K D values for glutamate of 51 μM (no modulator) and 15 μM (CX546). C, IC50 values for glutamate displacement of [3H]CNQX binding were obtained from experiments as shown in panel B, fitted with one-site sigmoidal curves (n H = 1), and re-plotted against the respective CX546 concentrations. Fitting a logistic equation to the data points provides a lower-limit IC50 for glutamate of 9 μM and an EC50 for CX546 of 212 μM (95% confidence interval 120–380 μM). Data are presented as means and S.D. from the number of experiments shown in parentheses.
Effects on the binding of the endogenous agonist glutamate cannot be determined in this manner because specific binding is too small. However, the affinity for glutamate can be determined by measuring the ability of the latter to displace the antagonist [3H]CNQX (Fig. 4B). In the absence of drug, glutamate displaced [3H]CNQX binding with an IC50 of 56 μM, which yields an affinity for glutamate of 51 μM after Cheng-Prusoff correction. CX546 shifted the displacement curve to the left; at a concentration of 1 mM, the IC50 value for glutamate was 17 μM (K D 15 μM). The IC50 values for glutamate determined over 0.3 to 6 mM CX546 were re-plotted in Fig. 4C as a function of the drug concentration. Fitting of a sigmoidal dose-response curve to the data points indicates that CX546 increases the affinity for glutamate 5.8 times and that it does so with an EC50 of about 200 μM. This affinity estimate is 5 to 10 times lower than the EC50 obtained from [3H]FW binding. Several factors may account for this difference. One is that [3H]CNQX binding mainly probes low-affinity AMPA receptors, which are believed to be synaptic (Hall et al., 1992), whereas FW binding accords higher weight to the high-affinity variants, which probably are less susceptible to the drug effect. In addition, the expected reciprocal enhancement between drug and agonist affinity would be more apparent in the displacement experiment, in which the agonist concentration was varied up to near-saturation, than in the FW assay in which the agonist occupied only a small fraction of the available receptors.
CX516, in obvious contrast, produced no reliable change in [3H]FW binding up to concentrations of 20 mM (Fig. 5A). These findings replicate previous observations with [3H]AMPA binding (Arai et al., 1996b). Binding experiments are usually conducted in nonphysiological buffers and with lysed membranes, and thus the possibility must be considered that receptor properties are altered in substantial ways. The binding tests with CX516 were therefore repeated with freshly prepared synaptoneurosomes, which remain metabolically active and maintain the receptor in a more authentic ionic and proteomic environment. Even under those test conditions, however, no significant effects on [3H]FW binding were detected (Fig. 5A).

Effects of CX516 on binding and interactions with other modulators. A, effect of CX516 on [3H]FW binding. Tests were carried out at the CX516 concentrations indicated on thex-axis with either the conventional membrane preparation at an incubation temperature of 25°C or with freshly prepared synaptoneurosomes at 0°C. Data are means and S.E.M. from two to six determinations at each drug concentration. B, tests for competitive interaction between CX516 and CX546. Saturation curves for the effect of CX546 were established in rat brain membranes in the absence and presence of 10 mM CX516. Assays were carried out as described in Fig.4A with 20 nM [3H]FW (no SCN−) at 25°C, except for the use of two different incubation media. The upper curves were obtained in assays in which the membranes were suspended in 100 mM HEPES/Tris, 100 μM EGTA, pH 7.4, a buffer routinely used for binding tests. The lower curves were obtained in a physiological medium (ACSF) containing 124 mM NaCl, 3 mM KCl, 1 mM KH2PO4, 2 mM MgSO4, 1 mM CaCl2, 5 mM NaHCO3, 10 mM HEPES, and 10 mM glucose. In each case, data points (means and S.E.M.) are averages from three experiments and were fitted with a logistic equation. C, lack of interaction with cyclothiazide (CTZ). Synaptoneurosomes were incubated for 45 min at 0°C with 50 nM [3H]FW and increasing concentrations of cyclothiazide in the absence and presence of 5 mM CX516. CX516 had no detectable effect on the dose-response relation for cyclothiazide; EC50 values for cyclothiazide were 31 μM and 30 μM in the absence and presence of CX516, respectively. D, lack of interaction of CX516 with GYKI 52466. Brain membranes were incubated for 45 min at 25°C with 50 nM [3H]FW and the agents indicated in the figure. All incubations contained 3.3 mM CX546. CX546 and CX516 were added from aqueous stock solutions, and GYKI was added from a 20 mM stock solution in DMSO. The data show means and their S.E.M. from triplicate determinations; comparable results were obtained in two additional experiments.
Given that the physiological effects of CX516 are generally less extensive than those of most other modulators, the possibility remains that the drug does bind to the receptor at a shared site, but that the change in receptor kinetics is very limited and too small to be detected in a binding assay. If so, then CX516 should at the very least impede the binding of the other drugs and hence shift their dose-response curves to the right. None of those tests, including some which were conducted in a physiological ACSF buffer, provided clear evidence for competitive interaction at CX516 concentrations that were active in physiological experiments (Fig. 5B). Even at the high CX516 concentration of 5 mM, the dose-response curve of CX546 was shifted by only a small factor (1.29 ± 0.06; n = 5); at 10 mM CX516, the EC50 value was shifted by a factor of 1.32 ± 0.13 (n = 3) in the standard HEPES/Tris buffer and by 1.0 ± 0.1 (n = 3) in the ACSF buffer. Likewise, no clear evidence was found that 5 mM CX516 competes with cyclothiazide (Fig. 5C; shift factors of 0.98 and 1.14 in synaptoneurosomes and brain membranes, respectively). In case of true competition, this would imply that the binding constant for CX516 is well above 20 mM. No further attempts were made to substantiate such competition because drugs begin to confer solvent-like nonspecific effects at these high concentrations.
The possibility was also considered that CX516 acts like an “inverse agonist” at the receptor site for the GYKI-type 2,3-benzodiazepines, which generally block AMPA receptors through an allosteric mechanism (Zorumski et al., 1993) but in some circumstances also produce more complex, facilitatory effects (Arai, 2001). GYKI 52466 does not affect binding of [3H]AMPA or [3H]FW under normal assay conditions (Kessler et al., 1996), but it reduces agonist binding substantially if measured in the presence of positive modulators like CX614 (Arai et al., 2000) and CX546 (Fig. 5D). If CX516 occupies the same site as GYKI 52466, then it should reverse this inhibitory action of the latter. No consistent evidence for such an interaction was found with CX516 concentrations up to 5 mM (Fig. 5D).
Lastly, no evidence was found that CX516 at 2 to 5 mM acts on other components of glutamatergic synapses including kainate receptors,N-methyl d-aspartate receptors, and sodium-coupled glutamate transport (Table1).
Lack of effect of CX516 on other glutamate receptors and on glutamate transport
Discussion
The present study has shown in direct comparisons that the two AMPA receptor modulators CX516 (BDP-12) and CX546 differ in the way they modulate AMPA receptor-mediated responses and that this may be related to fundamental differences in the way they interact with the receptor. Both compounds are first-generation Ampakines with low but overall similar potencies. EC50 values for the effect of CX546 on agonist binding were between 0.2 and 2 mM, depending on the experimental context, and estimates for physiological measures were within the same range in this study and in a published report (283 μM, in Baumbarger et al., 2001). CX516 increased the amplitude of synaptic responses with an EC50 of 180 μM (Arai et al., 1996b), although other response parameters often required higher drug concentrations. One obvious conclusion is that the differences between them are not in any way related to their potency. In fact, newer and more potent Ampakine modulators such as CX614 (Arai et al., 2000) often exhibit efficacies lower than that of CX546 for prolonging fast responses. Thus, although more potent variants of both subtypes are known, CX516 and CX546 remain valuable prototypes to examine the functional differences between what appear to be two basically different Ampakine subfamilies.
These differences were apparent across a wide spectrum of measures. Synaptically evoked responses revealed a remarkable ability of CX546 to prolong the duration of the response, the effects being several times larger than those produced by CX516. The latter, however, was equally or more effective in increasing the amplitude of the response. This dissociation was evident in extracellular recordings but was more salient when measuring EPSCs because higher CX546 concentrations could be employed (see also Lin et al., 2002). Similarly striking differences between the drugs were observed in excised-patch experiments with both fast and long applications of glutamate (Figs. 2and 3). Responses to 1-ms applications of glutamate, which were intended to mimic synaptic responses, were slowed in their decay by CX516, but the effect of CX546 was larger in that it prolonged response deactivation at least 10 times. Effects on responses to long applications of glutamate differed to similar degrees in that CX546 effectively prevented the desensitization normally seen under those conditions, whereas CX516 caused only modest changes in desensitization rate and steady-state current (see also Arai et al., 1996b). Finally, CX516 and CX546 differed in their impact on agonist binding. The apparent affinity for agonists was increased by CX546 to a larger degree than with any other Ampakine ligand, whereas CX516 was without detectable effect under a wide range of test conditions.
There have been other notable differences between these two drug types. When tested in hippocampal slices, drugs like CX546, such as CX614, generally appeared to have comparable EC50 values for enhancing EPSP amplitude and half-width (see, for example, Arai et al., 1994, 2000). In marked contrast, CX516 had no significant effect on EPSP half-width at a concentration of 180 μM, which half-maximally increased the amplitude of the response (Arai et al., 1996b), and concentrations of at least 1 mM were needed to produce even the modest half-width effects shown in Fig. 3. In a similar vein, steady-state currents in excised-patch responses were increased to about 30% of the peak current with an EC50 value of 150 μM, but other response parameters, such as the slowing of response deactivation, again required millimolar concentrations (Arai et al., 1996b). This indicates that the nature of the drug-receptor interaction may be more complex for CX516 than for other Ampakine modulators. Because only a subset of its effects are apparent in the hundred micromolar range, they may have been missed or underestimated in their importance in earlier comparisons of CX516 with other modulators that have more dramatic effects on receptor kinetics (Baumbarger et al., 2001; Nagarajan et al., 2001).
The double dissociation in the effect of the two compounds on amplitude versus duration of synaptic responses clearly indicates that their actions on AMPA receptors are qualitatively different and that CX516 is not just a “weaker” version of other benzamides, as suggested in some of these earlier studies. Although its efficacy with regard to many of the parameters analyzed in this study was much lower, this was clearly not the case for the amplitudes of synaptic responses, which were increased by CX516 to an equal or higher degree compared with CX546. That the two compounds act through separate mechanisms is further indicated by the binding data. The failure of CX516 to influence agonist binding is not readily attributed to its more modest action because even aniracetam and 1-BCP (Arai et al., 1994), two modulators with relatively weak effects, produced statistically reliable changes in binding (see Xiao et al., 1991, for aniracetam; M. Kessler, unpublished data, for 1-BCP). It therefore seems most likely that CX516 targets distinct aspects of receptor kinetics, which inherently have negligible impact on apparent agonist affinity and which produce circumscribed changes of modest size in most physiological response parameters.
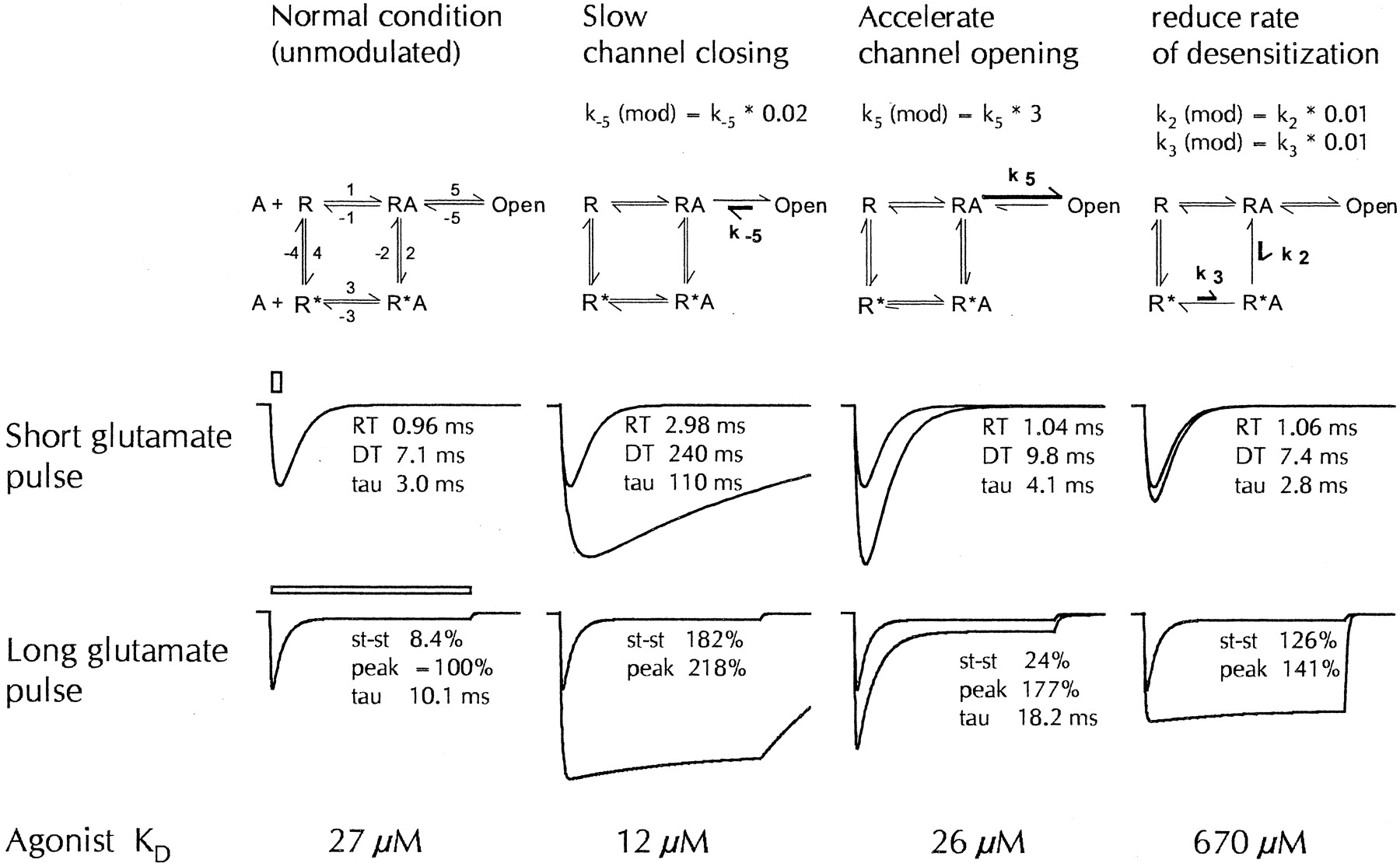
One such possibility would be that CX516 preferentially accelerates channel opening and that other benzamide modulators mainly slow channel closing. The consequences are illustrated in Fig.6 with simulations based on a standard kinetic receptor model (Ambros-Ingerson and Lynch, 1993). As shown in the second column, deceleration of channel closing drastically slows the rate at which fast responses deactivate. It should be noted that this manipulation also greatly reduces the apparent response desensitization observed during long glutamate applications, in essence because entrapment in the open state outweighs the competing “attraction” of the desensitized state, and that the binding affinity for agonists is substantially increased, as indicated by the 2-fold reduction of the K D value. All three effects are characteristic for CX546 and many other benzamide-type modulators. Changing, instead, the rate of channel opening produces effects that are quite distinct and closer in nature to those of CX516 (third column). Acceleration of channel opening as little as 3-fold prominently increases the response amplitude in this model (+93%) but exerts comparably weak effects on other response parameters such as the steady-state current (2.9-fold increase), time constant of desensitization (1.8-fold increase) or binding affinity (<5% change). The deactivation time constant is also minimally affected but nonetheless shows a clear increase over control (1.4-fold). Slowing the transition to the desensitized state (last column of Fig. 6) produces yet another set of effects that are reminiscent of those observed with cyclothiazide (Arai and Lynch, 1998a). These simulations evidently are for illustrative purposes only. Actual receptor kinetics is bound to be more complex because AMPA receptors have multiple ligand sites and open states (Rosenmund et al., 1998), and because drugs are likely to affect more than a single rate constant. Nonetheless, it seems at least plausible and in qualitative agreement with our observations that CX516 at submillimolar concentrations mainly accelerates channel opening with only small impact on channel closing, whereas CX546 primarily slows channel closing. Because CX546 is also very effective in blocking response desensitization, it is likely that this compound has additional effects on desensitization or resensitization rates that lead to a stabilization of the agonist-bound state, as suggested by Nagarajan et al. (2001).
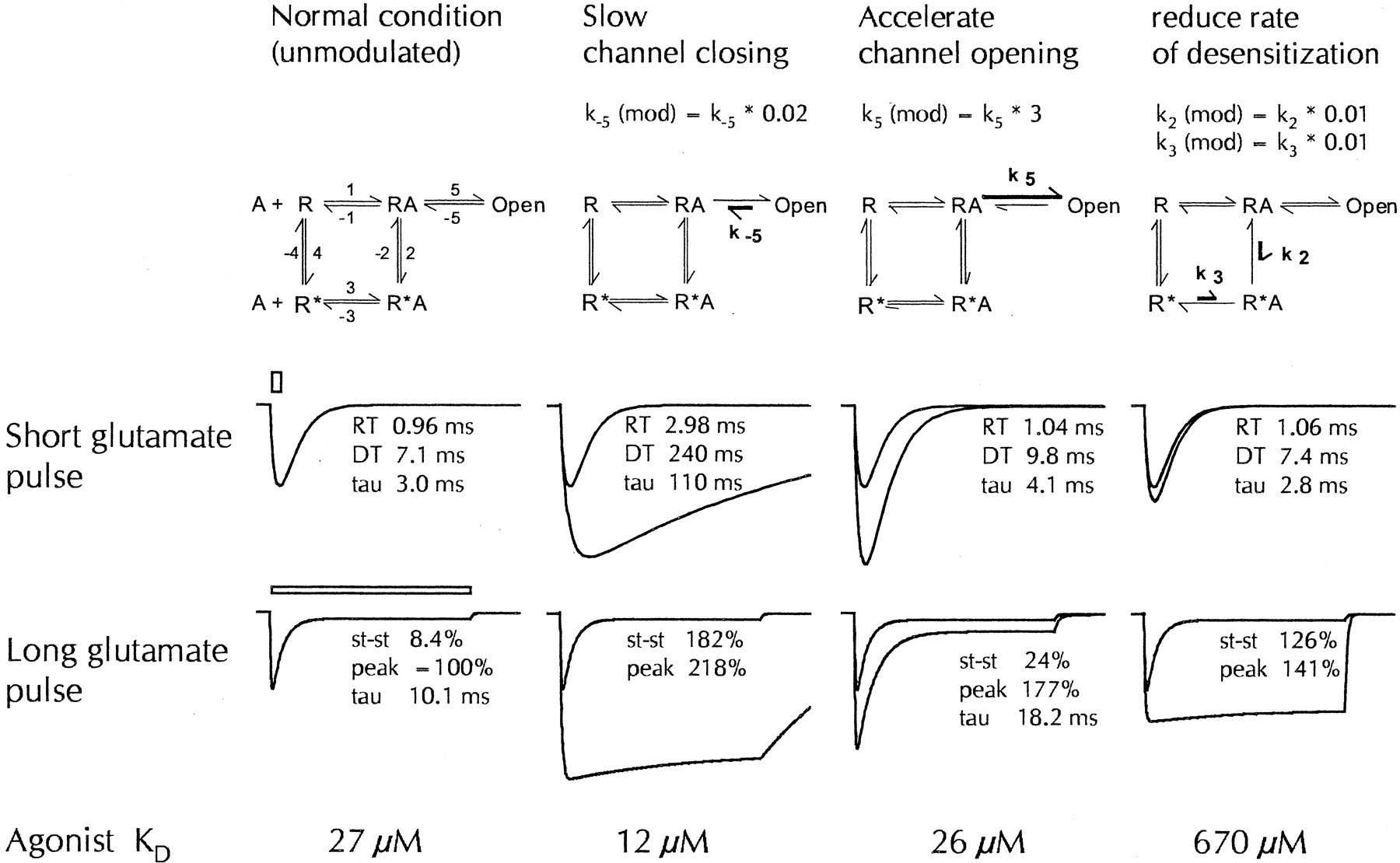
Simulation of drug effects using kinetic receptor models. Response curves were simulated using the five-state receptor model shown in the upper row (Ambros-Ingerson and Lynch, 1993) with sets of rate constants previously used (Arai et al., 1996a; Kessler et al., 1996). R represents the receptor, A an agonist; asterisks denote desensitized receptor states. The numbers next to the transition arrows indicate the rate constants, which were selected as follows:k 1 = 1.5 μM−1 · s−1,k 3 = 5 μM−1 · s−1; and k −1 = 10,000,k 2 = 1,500,k −2 = 6,k −3 = 10,k 4 = 3,k −4 = 40,k 5 = 5000, andk −5 = 700 s−1, respectively. The simplified schemes in columns 2 to 4 indicate which rate constants have been modified (bold arrows) in the simulations shown underneath. In the second column, the channel closing ratek −5 was assumed to be slowed by a factor of 50. In the third column, the channel opening rate (k 5) was increased 3-fold. In the fourth column, desensitization (k 2) was slowed 100 times and the rate constant k 3 was reduced by the same factor to maintain microreversibility. The second row shows simulated responses to very brief glutamate applications (1 mM peak concentration, decaying exponentially with a time constant of 2 ms); the sweep length is 40 ms. The leftmost column shows responses with the basic set of rate constants; in columns 2 to 4, responses without and with modification are superimposed. RT, 10–90% rise time; DT, 90–10% decay time; tau, decay time constant from monoexponential curve fit. The third row illustrates the corresponding responses obtained for a 200- ms application of 1 mM glutamate (horizontal bar) to assess response desensitization. st-st, steady-state current (all steady-state and peak currents are expressed as percentage of the peak current of the control response). The bottom row shows the equilibriumK D value for the agonist (glutamate), which has been shown to be a function of all rate constants and which was calculated using the equation given in Ambros-Ingerson and Lynch (1993).
The most surprising finding has been, however, that CX516 did not appear to compete with the binding of other modulators at physiologically meaningful concentrations. Even at CX516 concentrations as high as 10 mM, the EC50 for CX546 was shifted less than 2-fold, and competition tests with cyclothiazide and GYKI 52466 were similarly negative. The most straightforward explanation is that CX516 binds to a site on the receptor different from those accessed by the other modulators. Other explanations are possible, such as that AMPA receptors lost their affinity for CX516 during membrane isolation, but the absence of a drug effect in synaptoneurosomes makes this seem less likely. It is also conceivable that CX516 binds to only a subset of the available drug sites. AMPA receptors are multimeric proteins with each subunit possessing a benzamide modulator site (Hennegriff et al., 1997; Arai et al., 2000), and thus each functional receptor unit possesses several homologous drug sites. If CX516 for some reason were to bind to only one of them, it could affect physiological responses while remaining hard to detect in competition tests. Subunit selectivity would not be likely to account for this because patch and binding experiments with homomeric receptors did not find major differences between subunits (not shown). It seems possible, however, that unique subunit constellations in heteromeric receptors or secondary modifications such as phosphorylation bestow greatly increased affinity for CX516 to a selected subset of the receptor subunits. More data will be needed to select from these alternatives. However, even if CX516 were eventually found to act through a site that is common to all Ampakine compounds (as assumed in some of the above interpretations), the conclusion would nonetheless remain valid that its interaction with the receptor differs in basic ways from that of other compounds such as CX546 or CX614, which clearly produced parallel effects on physiology as well as binding, indicating that membrane isolation, particular subunit constellations, or post-translational modifications have little impact on their interaction with the receptor.
A question of considerable interest will be how the differences among AMPA receptor modulators manifest themselves at the system level. The only report in which both drug types were examined across behavioral tests did not find obvious disparities (Davis et al., 1997), but the study was not specifically designed to reveal such differences. It also seems likely that not all brain circuits and associated behaviors are equally sensitive regarding specifics of drug action. Neurons that summate asynchronous synaptic events over longer time frames, such as the slow hippocampal theta wave, may not be susceptible to particular changes in the wave form of individual transmission events, whereas circuits designed to respond rapidly to coincident inputs may experience very distinct shifts in their integrative properties. Thus, differences between drugs like CX516 and CX546 might be obvious only in some behaviors, and tasks that rely on the hippocampus are, perhaps, not among them.
Differences between the two drug types may have other important consequences, however. Drugs that prominently increase response duration may be more likely to cause progressive depolarization due to cumulative EPSP summation and thus to destabilize networks with recurrent excitable connections such as the hippocampal area CA3. CX516 was indeed well tolerated by hippocampal slices at very high concentrations, whereas CX546 often caused epileptic discharges even at concentrations below half-saturation. These observations probably extrapolate to the behavioral level because CX516 generally seemed to have a more desirable therapeutic index with behavioral thresholds around or below 10 mg/kg (Davis et al., 1997; Baumbarger et al., 2001) and no overt seizures at dosages up to 500 mg/kg (G. Rogers, unpublished observation). Thus, from a therapeutic perspective, drugs with action profiles like that of CX516, i.e., with an intrinsically limited efficacy in augmenting synaptic responses, may be more desirable than those that produce drastic changes in AMPA receptor kinetics.
In conclusion, Ampakine drugs do not act in a unitary fashion, and it seems advisable to distinguish at least two major subcategories with clearly distinct effects on AMPA receptor function. Whether the two groups recognize independent sites on the receptor or act through a common site but in highly distinctive ways remains to be established. Since we have previously shown that cyclothiazide and CX614 (Arai et al., 2000) do not appear to interact in a fully competitive way, the lack of interaction of CX516 with either of these drugs augments further the complexity of the pharmacology of these up-modulators. It also remains to be explored how newer sulfonamide-type modulators such as PEPA (Sekiguchi et al., 1997), S18986 (Desos et al., 1996), biarylsulfonamides (Ornstein et al., 2000; Linden et al., 2001), and D1 (Arai et al., 2002; Phillips et al., 2002) relate to these subcategories. It will also be of interest to study in the paradigms used here newer analogs of CX516 that are functionally similar yet possess micromolar affinities. Further insight into the relationships between these drugs will hopefully come from crystallographic analyses as employed by Sun et al. (2002), who have been able to determine the site of action of cyclothiazide by forming cocrystals between drug and receptor. Regardless of the particulars of the receptor-drug interactions, the present data indicate an enormous functional diversity even within a single drug class, and it is likely this diversity will be of importance for the physiological and therapeutic effects of these drugs.
Footnotes
- Received June 14, 2002.
- Accepted August 15, 2002.
This research was supported by grants awarded to A.C.A from the National Science Foundation (IBN-9806215), the National Institutes of Health (NS41020), and the Central Research Committee of Southern Illinois University (201-08).
DOI: 10.1124/jpet.102.040360
Abbreviations
- AMPA
- R,S-(±)-α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- CX546
- 1-(1,4-benzodioxan-6-ylcarbonyl)piperidine
- CX516 [BDP-12]
- 1-(quinoxalin-6-ylcarbonyl)piperidine
- 1-BCP
- 1-(1,3-benzodioxol-5-ylcarbonyl)piperidine
- CX614
- 2H,3H,6aH-pyrrolidino[2",1"-3′,2′]1,3-oxazino[6′,5′-5,4]benzo[e]1,4-dioxan-10-one
- CX554
- 2H,5aH-pyrrolidino[2",1"-3′,2′]1,3-oxazino[6′,5′-5,4]benzo[d]1,3-dioxolan-9-one
- EPSP
- excitatory postsynaptic potential
- DMSO
- dimethyl sulfoxide
- ACSF
- artificial cerebrospinal fluid
- EPSC
- excitatory postsynaptic current
- MK-801
- dizocilpine maleate
- AP5
- 2-amino-5-phosphonopentanoic acid
- CNQX
- 6-cyano-7-nitro-quinoxaline-2,3-dione
- RP
- preference ratio
- FW
- fluorowillardiine
- GYKI
- GYKI 52466, 1-(4-aminophenyl)-4-methyl-7,8-methylenedioxy-5H-2,3-benzodiazepine hydrochloride
- CTZ
- cyclothiazide
- SCN−
- thiocyanate
- PEPA
- 4-[2-(phenylsulfonylamino)ethylthio]-2,6-difluoro-phenoxyacetamide
- S18986
- (S)-2,3-dihydro-[3,4]cyclopentano-1,2,4-benzothiadiazine-1,1-dioxide
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}