


Abstract
We have synthesized iodinated resiniferatoxin bearing a 4-hydroxy-5-iodo-3-methoxyphenylacetate ester (I-RTX) and have characterized its activity on rat and human TRPV1 (VR1) receptors, as well as in behavioral assays of nociception. In whole cell patch-clamp recordings from transfected cells the functional activity of I-RTX was determined. Currents activated by capsaicin exhibited characteristic outward rectification and were antagonized by capsazepine and I-RTX. On rat TRPV1 the affinity of I-RTX was 800-fold higher than that of capsazepine (IC50 = 0.7 and 562 nM, respectively) and 10-fold higher on rat versus human receptors (IC50 = 0.7 and 5.4 nM, respectively). The same difference was observed when comparing the inhibition of [3H]RTX binding to rat and human TRPV1 membranes for both RTX and I-RTX. Additional pharmacological differences were revealed using protons as the stimulus. Under these conditions capsazepine only partly blocked currents through rat TRPV1 receptors (by 70 to 80% block), yet was a full antagonist on human receptors. In contrast, I-RTX completely blocked proton-induced currents in both species and that activated by noxious heat. I-RTX also blocked capsaicin-induced firing of C-fibers in a rat in vitro skin-nerve assay. Despite this activity and the high affinity of I-RTX for rat TRPV1, only capsazepine proved to be an effective antagonist of capsaicin-induced paw flinching in rats. Thus, although I-RTX has limited utility for in vivo behavioral studies it is a high-affinity TRPV1 receptor antagonist that will be useful to characterize the functional properties of cloned and native vanilloid receptor subtypes in vitro.
The transient receptor potential TRPV1 vanilloid receptor (also known as VR1; see revised TRP channel nomenclature Montell et al., 2002) gates a nonselective cation channel that is expressed by sensory neurons and that can be activated by protons, heat, and capsaicin, the pungent ingredient of chili peppers (Caterina et al., 1997; Tominaga et al., 1998). Ligands acting at the TRPV1 vanilloid receptor subtype have the potential therapeutic utility to treat thermal hyperalgesia-related pain and some inflammatory conditions (for review, see Szallasi and Blumberg, 1999; Caterina and Julius, 2001). One of the first antagonists described for the capsaicin receptor was capsazepine (Bevan et al., 1992). This ligand has been used widely to explore the functional significance of TRPV1 receptors in pain. However it has relatively low micromolar affinity for TRPV1 receptors and because it also blocks voltage-gated calcium channels, this has made interpretation of functional data with this compound less straightforward (Docherty et al., 1997).
To date, one of the highest affinity ligands reported for the TRPV1 receptor is the natural plant product resiniferatoxin (RTX), which was first isolated from Euphorbia resinifera (Szallasi and Blumberg, 1989). Interestingly, it has been shown recently that iodination of this agonist RTX confers antagonist-like properties to the ligand without substantially affecting its affinity for rat TRPV1 receptors (Wahl et al., 2001). This discovery presents a new opportunity to explore the pharmacology of TRPV1 receptors. The purpose of the present study was severalfold. The first aim was to confirm the observation that iodination of RTX eliminates agonist activity without substantially affecting its binding affinity, to extend these observations to human TRPV1 receptors, and to determine its functional inhibitory effects against responses to both acidic pH and heat, two physiologically relevant activators of the channel. To achieve this we synthesized iodinated RTX bearing a 4-hydroxy-5-iodo-3-methoxyphenylacetate ester (I-RTX) and used radioligand binding and electrophysiological studies to characterize its activity on cloned human and rat TRPV1 receptors (Caterina et al., 1997). These pharmacological properties of I-RTX were then contrasted with that of capsazepine and morphine in rodent behavioral assays of nociception. I-RTX was also tested for direct agonist-like activity in wild-type and TRPV1 knockout (KO) mice to more fully characterize its utility and specificity of action. Some of these data have been presented previously in abstract form (Seabrook et al., 2001).
Materials and Methods
Preparation of I-RTX.
Iodo-resiniferatoxin was prepared in one synthetic step from resiniferatoxin by iodination using the sodium iodide/Chloramine-T reagent system (Kometani et al., 1985). Iodination occurs selectively ortho to the hydroxyl substituent and cleanly if the reaction is not forced to completion by adding excess reagents or using prolonged reaction times.
RTX (11.4 mg, 0.018 mmol) was dissolved in acetonitrile (3 ml). Phosphate buffer (4.6 ml, pH 7.2) was added, followed by sodium iodide (0.1 M in phosphate buffer, 200 μl, 0.020 mmol) and then chloramine T (0.1 M in phosphate buffer, 200 μl, 0.020 mmol). The reaction was stirred for 6 min and then sodium bisulfite solution (0.1 M in water, 2 ml, 0.2 mmol) was added and the mixture was extracted with ethyl acetate (3 × 5 ml). The organic extracts were evaporated then purified by preparative HPLC (ABZ+ column, 250 mm × 21 mm-i.d.; MeCN/0.1% aqueous trifluoroacetic acid) to give I-RTX, 3.0 mg, 22%. m/z 755 (M + 1)+, HPLC >98% (two systems), 1H NMR (500 MHz, δ CDCl3) 7.47 (1H, s), 7.39 (2H, d,J = 7), 7.33 to 7.24 (3H, m), 7.23 (1H, s), 6.80 (1H, s), 6.04 (1H, s), 5.91 (1H, s), 4.74 (1H, br. s), 4.63 (1H, d,J = 12), 4.55 (1H, d, J = 12), 4.24 (1H, s), 3.91 (3H, s), 3.54 (2H, s), 3.24 (2H, br. s), 3.12 (1H, m), 3.09 (1H, m), 2.58 (1H, pentet, J = 8), 2.46 (1H, d,J = 17), 2.16 (1H, dd, J = 14, 9), 2.07 (1H, d, J = 18), 2.03 (1H, s), 1.85 (3H, s), 1.57 (1H, m), 1.54 (3H, s), 0.98 (3H, d, J = 7). The product was shown to be of high purity (>98%) and completely free of unreacted resiniferatoxin. A second sample of I-RTX was obtained from Tocris Cookson (Ballwin, MO).
The physicochemical properties of I-RTX include a high calculated log P of between 6.6 (Advanced Chemistry Development Software Solaris version 4.67) and 8.7 (Klopman and Wang, 1991), with a molecular mass of 754 Da. The high lipophilicity of I-RTX is not surprising given the properties of the parent compound RTX, which bears two H donors, nine H acceptors, has a log P of 5.45 to 5.10 between pH 4 and 10, and a pK a of 9.92 with sparing solubility between pH 4 and 10 (calculated using Advanced Chemistry Development Software Solaris version 4.67). Also, RTX binds readily to plasma proteins, specifically α1-acid glycoprotein (Szallasi et al., 1992).
Electrophysiology.
Human embryonic kidney cells (tsA-201-AEQ17) were stably transfected with rat TRPV1 receptors as described previously (Grant et al., 2001). Cells were grown in Dulbecco's modified Eagle's medium with glutamine, 10% fetal bovine serum at 30°C, 5% CO2, and plated onto poly-d-lysine-coated glass coverslips. Human TRPV1-CHO KI cells were stably transfected using pCI-neo vector containing human TRPV1. After selection with geneticin, clones were assessed for functionality using a fluorometric imaging plate reader and single cell cloned by limiting dilution. Cells were grown in Iscoves modified Dulbecco's medium with glutamine, 10% fetal bovine serum, hypoxanthine (5 mM) + thymidine (0.8 mM) supplement at 30°C, 5% CO2, and plated onto glass coverslips. In some experiments rat TRPV1 was also transiently expressed in CHO-KI cells, and human TRPV1 in tsA-201 cells (where indicated underResults) for a direct comparison of the pharmacology of rat versus human receptors. Cells were incubated at 30°C because this is a heat-activated channel and cell viability was improved at this temperature. Coverslips with transfected cells were placed in a recording chamber and perfused at room temperature (22°C) at a rate of 1 ml/min. Recording of whole cell currents under voltage clamp were made with a Axopatch 200B amplifier (Axon Instruments, Union City, CA). Capacitance transients were canceled and series resistance compensation was >70%. Fire-polished patch pipettes (120TF-10; Harvard Apparatus, Kent, UK) had a tip diameter ∼1 μm, and resistances were approximately 2 to 3 MΩ. The intracellular pipette solution contained 110 mM CsF, 30 mM TEA-Cl, 20 mM Cs-BAPTA, 1 mM MgCl2, 2 mM Mg-ATP, and 10 mM HEPES, pH 7.2, adjusted with TEA-OH. Drugs were applied to the cell by a fast perfusion system (Biologic RSC-200; Science Instruments, France) using a large internal diameter (500-μm) triple-barrel pipette assembly. The agonists 500 nM capsaicin or acid, pH 5.5, was applied for 5 s followed by a 30-s wash period. Inhibition of the agonist response was determined after sequential 30-s applications of increasing concentrations of I-RTX or capsazepine with no intervening periods of wash.
For patch-clamp studies, the extracellular solution contained 165 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1.67 mM CaCl2, 17 mM d-glucose, and 10 mM HEPES, pH 7.3 (or pH 6.8 for proton-induced currents), adjusted with NaOH. To buffer acidic solutions (pH 5.5) 10 mM MOPS was included as a substitute for HEPES. In all experiments, unless stated, the holding membrane potential was −60 mV. Current-voltage relationships were constructed from voltage ramps (−80 to +80 mV in 500 ms). To test for heat activation, an eight-barrel heating device (MicroCells) was used. The temperature was changed from 25 to 48°C in approximately 20 s. The temperature of the solution was monitored by a thermoresistor at the tip of the perfusion probe. Recordings were filtered at 2 kHz and digitized at 500 to 1000 Hz using pClamp software (Axon Instruments).
In vitro skin nerve electrophysiological studies were carried out as described by Reeh (1986). Briefly, seven male wild-type (C57/Bl6 × J129 hybrid) were euthanized by cervical dislocation. The saphenous nerve together with the skin innervated by this nerve was dissected free and placed in an organ bath and continuously perfused with synthetic interstitial fluid saturated with 95% O2, 5% CO2 at 34°C. The cut end of the nerve was placed in a separate chamber and split into small filaments that were placed in contact with a recording electrode. Filaments were examined sequentially until action potentials originating from the receptive field of a single C-fiber could be isolated. The conduction velocity and mechanical threshold of each C-fiber was determined. A weighted Plexiglas well was used to isolate the receptive field of the C-fiber and all pharmacological agents were applied to the receptive field via the well for 5 min in each case. Action potentials generated during the baseline, I-RTX or vehicle, and capsaicin application periods were recorded and quantified using a DAPSYS data acquisition system (Johns Hopkins University, Baltimore, MD).
Capsaicin and capsazepine (Sigma-Aldrich, St. Louis, MO) were dissolved in 100% ethanol to yield a stock concentration of 10 mM. Further dilutions were made with 100% ethanol. I-RTX was dissolved in 100% DMSO. Final bath concentrations of DMSO and ethanol were <0.3%.
[3H]RTX Binding.
Human embryonic kidney tsA-201 cells were transfected with rat TRPV1 (Caterina et al., 1997) or human TRPV1 using standard lipid transfection techniques. Three days after transfection, the cells were washed with Dulbecco's phosphate-buffered saline, collected by centrifugation, and frozen at −80°C. Pellets were thawed on ice and then resuspended in 5 ml of cell lysis buffer/T-225 flask (one tablet of protease inhibitor, #1697498; Roche Applied Science, Indianapolis, IN) plus 50 ml of 25 mM HEPES, pH 7.2, by passing through a 25-gauge needle three times followed by 10 strokes in a glass homogenizer. Lysed cells were centrifuged for 10 min at 3,000g, and the pellet was rehomogenized and centrifuged once more for 10 min. at 3,000g. Both supernatants were combined and centrifuged for 30 min at 150,000g. The membrane pellet was resuspended in 1 ml of phosphate-buffered saline per original T-225 flask.
Membranes were diluted to 5 or 100 μg/ml protein and represented a maximum specific [3H]RTX binding concentration (B max) of 0.017 and 0.12 nM for rat and human TRPV1, respectively. TheB max was fixed to the lowest possible value to ensure that inhibitor binding to the RTX site would not cause the free concentration to be substantially different from the total concentration. The free [3H]RTX calculated from the measured total and bound [3H]RTX was used to convert the IC50 values intoK I values (K I = IC50/(1 + [RTX]/K D). The free [3H]RTX was about 100 pM and theK D values for rat and human TRPV1 measured directly with [3H]RTX (data not shown) were approximately 0.05 and 1 nM, respectively. [3H]RTX was incubated overnight in the presence of various concentrations of RTX, I-RTX, or capsazepine. Samples were then filtered on glass fiber filters (#1822 025; Whatman, Maidstone, UK) pretreated with 0.5% polyethylenimine. The filters were washed three times with 10 mM Tris, pH 7.5, and 0.1% Triton X-100. IC50 values were converted toK I values using the free [3H]RTX concentration, and theK D values of 50 and 500 pM for [3H]RTX binding to rat and human TRPV1, respectively.
Capsaicin-Induced Nociceptive Responses in Rats.
Male Sprague-Dawley rats (100–150 g; Bantin and Kingman, Hull, UK) were habituated to individual observation boxes for 1 h before intraplantar injection of capsaicin (13.3 nmol in 50 μl; dissolved in 5% ethanol in phosphate-buffered saline), and the number of flinches was recorded for 5 min. Morphine (1, 3, or 10 mg/kg; dissolved in saline), capsazepine (10, 30, or 60 mg/kg; dissolved in 10% DMSO in saline), or vehicle was administered subcutaneously 30 min before intraplantar injection of capsaicin. In other studies, capsazepine (1.3, 13, or 133 nmol in 10% DMSO in saline), I-RTX (0.001, 0.1, or 1 nmol in 50 μl in 10% DMSO in saline), or vehicle (50 μl of 10% DMSO in saline) was injected into the paw 5 min before capsaicin, and flinching behavior was recorded before and after the administration of capsaicin.
For studies on TRPV1 knockout mice, TRPV1 KO mice were generated as described previously (Caterina et al., 2000) and breeding pairs were supplied by D. Julius (University of California, San Francisco, CA) to establish colonies of homozygous null mutant and wild-type controls (C57BL6/129SVJ hybrids). Male TRPV1 KO and wild-type control mice aged 9 to 13 weeks were habituated to individual observation chambers (diameter 12 cm × height 12 cm) for at least 1 h before intraplantar injection of capsaicin (2.5 μg in 20 μl dissolved in 5% ethanol/phosphate-buffered saline) or I-RTX (10 nmol in 20 μl; dissolved in 10% DMSO in saline). Separate TRPV1 KO and wild-type mice received an intraplantar injection of DMSO vehicle (20 μl of 10% DMSO in saline) as a control for I-RTX. Mice were then returned to the same chamber and the number of flinches and duration of leg raising and licking of the injected paw were recorded for 10 min.
Husbandry and experiments conformed to ethical guidelines for investigation of experimental pain in conscious animals (Zimmermann, 1983) and were in accordance with the UK Scientific Procedures Act (1986) and its associated guidelines. The number of animals and intensity of noxious stimuli were the minimum necessary to demonstrate consistent effects of drug treatments. Animals received drug treatments on one occasion and were humanely killed immediately after testing.
Results
Inhibition of Capsaicin-Induced TRPV1 Currents by I-RTX.
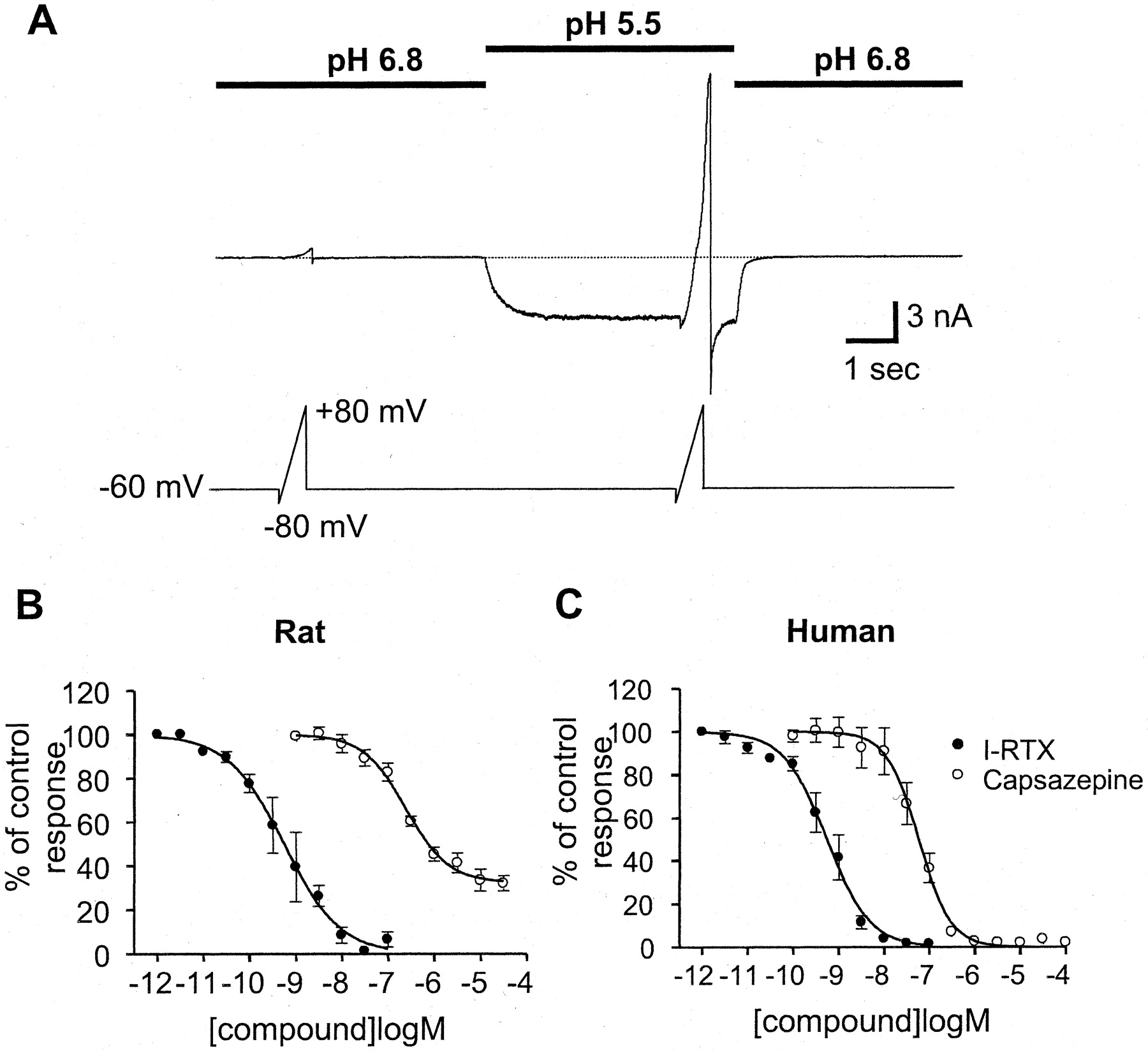
Capsaicin-induced currents were studied using whole cell patch-clamp electrophysiology. Capsaicin activated currents in rat TRPV1 cells with a potency of 447 nM (pEC50 = 6.35 ± 0.03,n = 5), which was comparable with that in human TRPV1 cells of 191 nM (pEC50 = 6.72 ± 0.03 nM,n = 6). The ability of I-RTX to inhibit these currents was compared with capsazepine, a competitive capsaicin antagonist, for both the human and rat channel isoforms of TRPV1 (Fig.1A). I-RTX antagonized the capsaicin-induced human TRPV1 current elicited by a 5-s application of 500 nM capsaicin. A 30-s application of 100 nM I-RTX was shown to be sufficient to reduce the capsaicin-induced current to just 2.3 ± 1.2% of control (n = 7). Membrane responses to voltage ramps (−80 to +80 mV) indicate that application of I-RTX inhibited human TRPV1 with no shift in the reversal potential or change in the outward rectification properties of the capsaicin-induced current. I-RTX did not exert any intrinsic agonist activity at the maximum concentration used in this study (10 μM; n = 3). I-RTX did not induce any inward current at a holding potential of −60 mV nor did it induce any outward current at +80 mV using voltage ramps (Fig. 1C). Cumulative concentration-response curves for both rat and human TRPV1 were constructed to compare the inhibitory action of I-RTX with capsazepine. Inhibition of the capsaicin response (500 nM) was determined after sequential 30-s applications of increasing concentrations of I-RTX or capsazepine with no intervening periods of wash. The amplitude of currents in untreated cells was unaffected by repetitive administration of capsaicin using this protocol (e.g., for rat TRPV1 application 2 = 105 ± 2.6%, n = 7; application 7 = 103.6 ± 3.5%, n = 7, application 14 = 97.7 ± 5.1% of control, n= 5). When coapplied with capsaicin, I-RTX produced a high-affinity block of both rat and human TRPV1 receptors with calculated IC50 values of 0.7 and 5.4 nM, respectively (Table 1). In both species, the affinity of I-RTX was significantly higher than that of capsazepine, with an 800-fold difference observed for the rat receptor (capsazepine IC50 = 562 nM; Fig. 1, D and E). Application of vehicle alone had no effect on currents activated by capsaicin, e.g., for rat TRPV1 the current in response to capsaicin (500 nM) in 0.1% ethanol was 108.1 ± 5.4% (n = 5), and for 0.1% DMSO was 95.1 ± 1.2% (n = 3), that of control.

Activation of TRPV1 (VR1) receptors by capsaicin and antagonism of currents by capsazepine and I-RTX measured using whole cell voltage-clamp electrophysiology. A, chemical structure of capsaicin, capsazepine, and iodo-resiniferatoxin. B, whole cell patch-clamp recording of CHO cell expressing human TRPV1, showing current activated by 500 nM capsaicin and its block by 100 nM I-RTX. Lower trace represents holding membrane potential and time course of voltage-ramp protocols. C, current-voltage relationship from a representative cell expressing human TRPV1 receptors during control conditions (Con), exposure to capsaicin (1 μM; Cap), and with I-RTX alone (10 μM; I-RTX). Membrane potential, −60 mV. Inset shows a blow up of the currents activated under control conditions and during the presence of I-RTX demonstrating that there was no intrinsic activity of this antagonist. D, concentration-response curves to I-RTX (●) and capsazepine (○) on rat TRPV1 using 500 nM capsaicin as the agonist. E, concentration-response curves to I-RTX (●) and capsazepine (○) on human TRPV1 using 500 nM capsaicin as the agonist.
Comparison between the pharmacology of capsazepine and I-RTX on rat and human TRPV1 (VR1) receptors
I-RTX Inhibition of Proton-Induced TRPV1 Responses.
Both rat and human TRPV1 receptors were activated by acidic solutions. A 5-s application of pH 5.5 was used to repetitively activate a proton-induced current every 30 s with minimal run-down. A control wash of pH 6.8 was used routinely to desensitize endogenous proton-activated currents present in tsA-201 cells. Identical protocols were consequently used for both rat and human TRPV1 isoforms (Fig.2A). As shown in Fig. 2, B and C, I-RTX completely inhibited the proton-induced current for both rat and human TRPV1, with an identical affinity (IC50 = 550 versus 550 pM, respectively). In a similar manner to capsaicin-induced responses, capsazepine inhibited the proton-activated human and rat TRPV1 currents with approximately 100- to 1000-fold lower affinity compared with I-RTX (IC50 = 246 and 58 nM, respectively). However, capsazepine also differed in its ability to inhibit proton-induced rat TRPV1 currents. At a saturating concentration of capsazepine (30 μM) over 30% of the total current remained unblocked (67.9 ± 3.3% inhibition, n = 4; Fig. 2C). Capsazepine was also an incomplete antagonist of currents activated by pH in transiently transfected rat TRPV1 CHO cells. In five cells the current activated by pH 5.5 (1455 ± 553 pA) was reduced by 83 ± 3% (227 ± 81 pA) with capsazepine (30 μM). No pH-activated currents were observed in untransfected or control plasmid transfected CHO cells (c.f. tsA-201 cells; see Materials and Methods). Under identical recording conditions capsazepine completely blocked proton-induced currents in CHO cells stably transfected with human TRPV1 (Fig. 2C), or proton-induced currents mediated by human TRPV1 receptors that had been expressed in tsA-201 cells (97.0 ± 1.5% at 10 μM, n = 3). Application of vehicle alone had no effect on currents activated by pH, e.g., for rat TRPV1 the current in response to pH 5.5 in 0.1% ethanol was 105.7 ± 3.9% (n = 3), and for 0.1% DMSO was 99.7 ± 1.7% (n = 3) that of control.

Activation of TRPV1 receptors by protons and antagonism of currents by capsazepine and I-RTX measured using whole cell voltage-clamp electrophysiology. A, membrane current in a representative cell expressing human TRPV1 receptors during transitions between extracellular solutions buffered at pH 6.8 and 5.5. A voltage ramp (−80 to +80 mV) was applied to the cell before and during the application of the proton challenge to measure the degree of outward rectification of the current (voltage protocol indicated by lower trace). B, concentration-response curves to I-RTX (●) and capsazepine (○) on rat TRPV1 using pH 5.5 as the agonist. C, concentration-response curves to I-RTX (●) and capsazepine (○) on human TRPV1 using pH 5.5 as the agonist.
I-RTX Inhibits Heat-Induced Currents.
Application of a heat ramp to cells expressing human TRPV1 receptors induced a rapidly activating inward current at elevated temperatures. As was seen with other modes of channel activation, application of I-RTX completely antagonized these heat-activated currents (97.5 ± 1.9% inhibition at 1 μM, n = 3; Fig.3).

Whole cell currents activated by temperature ramps from 25 to 48°C. Repetitive heat transients elicit stable temperature-activated currents that can be completely antagonized by 1 μM I-RTX. The percentage of block of the heat-activated current was 97.5 ± 1.9% with 1 μM I-RTX.
I-RTX Inhibits [3H]RTX Binding.
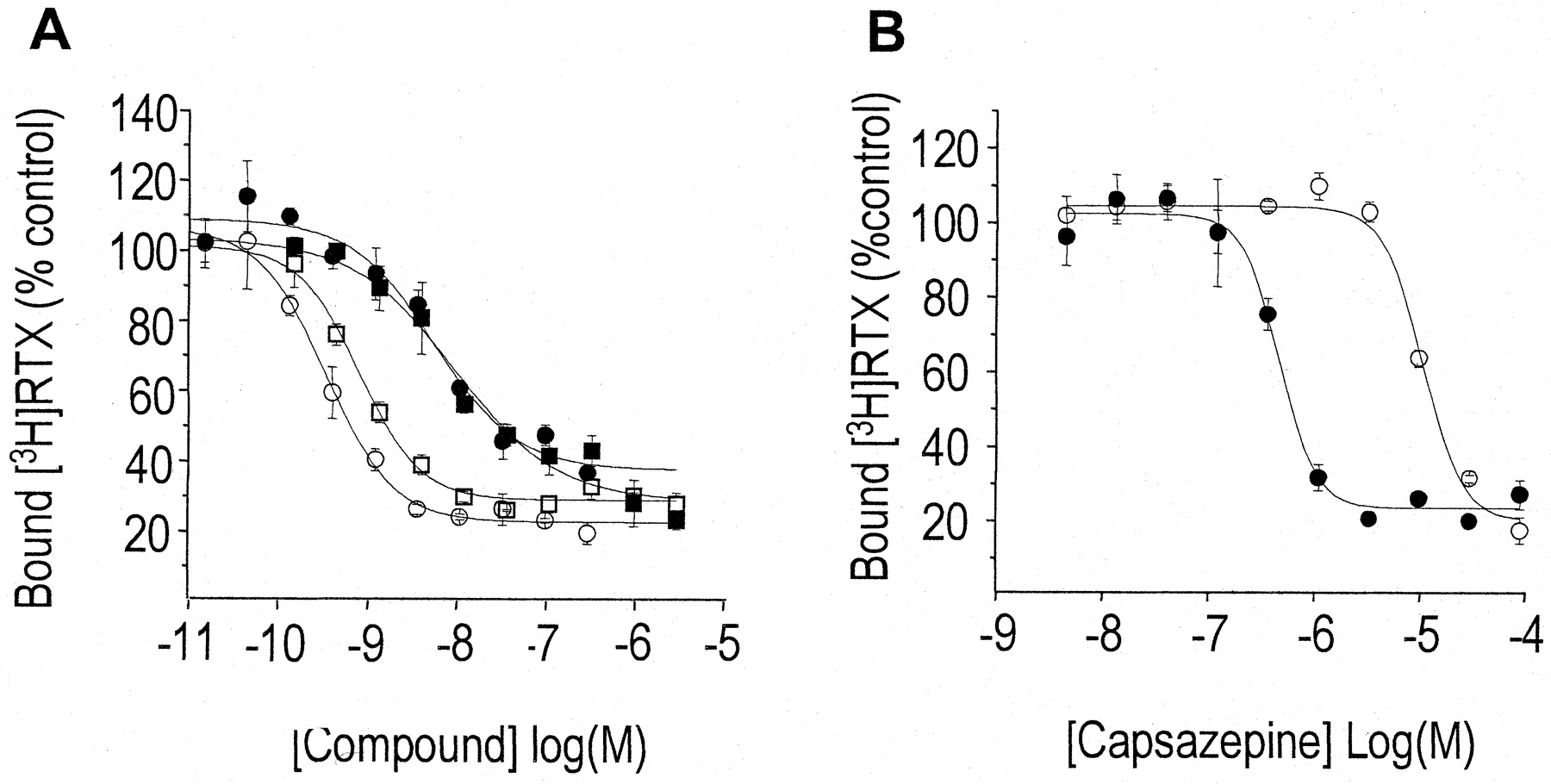
[3H]RTX was used to directly measure the equilibrium binding affinity of I-RTX to both rat and human TRPV1 transiently expressed in tsA-201 cells (Fig.4A). RTX itself inhibited [3H]RTX binding to rat TRPV1 (K I = 0.158 ± 0.051 nM,n = 3) with 20-fold higher affinity than human TRPV1(K I = 3.67 ± 1.5 nM,n = 3). I-RTX inhibited binding with 2-fold lower affinity to both rat (K I = 0.386 ± 0.025 nM, n = 3) and human (K I = 6.70 ± 1.95 nM,n = 3) TRPV1 but displayed the same difference in relative affinity between species as RTX. Capsazepine was less effective at displacing [3H]RTX binding. In contrast to both RTX and I-RTX, capsazepine had a lower affinity for rat TRPV1 (K I = 4300 ± 1000 nM,n = 4; Fig. 4B) relative to human TRPV1 (K I = 583 ± 280 nM,n = 4).

Inhibition of [3H]RTX binding to rat and human TRPV1. Membranes were prepared from tsA-201 cells expressing either rat or human TRPV1. A, [3H]RTX was incubated with rat (open symbols) or human (closed symbols) TRPV1 with 60 or 120 pM [3H]RTX, respectively. The indicated concentration of RTX (circles) or I-RTX (squares) was added, and samples were incubated overnight at room temperature. The bound [3H]RTX was determined by filtration on glass fiber filters. The data represent the average and standard deviation of three separate filtrations. Results are summarized in Table 1. B, same as in A except the indicated concentrations of capsazepine were used to inhibit binding to rat (open circles) or human (closed circles) TRPV1.
Inhibition of Capsaicin-Induced Paw Flinching.
Intraplantar injection of capsaicin (4 μg, equivalent to 13 nmol) in rats evoked short-lasting (<5 min) flinching of the injected paw. Pretreatment with morphine (1–10 mg/kg s.c., 30 min previously) dose-dependently and completely abolished flinching behavior induced by capsaicin (Fig.5A). Systemic administration of capsazepine (10–60 mg/kg s.c., 30 min previously) reduced capsaicin-evoked flinching response but the effect was not dose-related (Fig. 5B). Similarly, intraplantar injection of capsazepine (13 and 133 nmol) 5 min before capsaicin attenuated capsaicin-induced flinching (Fig. 6A). The difference in number of flinches to capsaicin administration alone seen in Fig. 5 versus Fig. 6may relate to different levels of stress associated with dosing because the pretreatment time for intraplantar injections (5 min) was shorter than that for the subcutaneous route (30 min). No flinching behavior was observed after injection of capsazepine alone (data not shown). In contrast, intraplantar injection of I-RTX (0.001, 0.1, or 1 nmol) had no effect on capsaicin-induced flinching (Fig. 6B). Intraplantar injection of high doses of I-RTX (1 nmol; Fig. 6C) elicited a flinching response of similar magnitude to that evoked by capsaicin.

Inhibition of capsaicin (4 μg ipl) induced flinching in rats by systemic administration of morphine 1, 3, or 10 mg/kg s.c. 30-min pretreatment time (A) and capsazepine 10, 30, or 60 mg/kg s.c. 30-min pretreatment time (B). Data are expressed as the mean ± S.E.M. of the number of flinches in 0- to 5-min observation period postcapsaicin. ★, p < 0.05 relative to vehicle.

Effect of intraplantar injection of capsazepine, 1.3 to 133 nmol ipl (5-min pretreatment) (A) and I-RTX 0.001 to 1 nmol ipl (5-min pretreatment) (B) on capsaicin (4 μg ipl)-induced flinching. C, flinching induced by I-RTX administration alone (0.001–1 nmol). Data are expressed as the mean ± S.E.M. of the number of flinches in 0- to 5-min observation period after test compound. ★,p < 0.05 relative to vehicle.
Inhibition of Capsaicin-Induced C-Fiber Activation.
To test directly whether I-RTX could block capsaicin-induced C-fiber activation we used an in vitro isolated skin-nerve electrophysiological recording preparation. A total of 13 C-fibers were recorded from eight wild-type mice (see Materials and Methods). The mean conduction velocity of the C-fibers was 0.68 ± 0.05 m/s and the median mechanical threshold was 7.48 mN (range 3.4 to 47.7). All C-fibers were mechanically sensitive and most C-fibers had little or no ongoing activity in the absence of any external stimuli (Fig.7, A and B). In vehicle-treated C-fibers capsaicin caused a significant increase in the number of action potentials (Fig. 7A) with a latency of C-fiber response to capsaicin application of 34 ± 16 s. After the application of either I-RTX (10 μM; n = 8) or vehicle (n = 5), there was no significant increase in the number of action potentials generated compared with baseline response (Fig. 7C). In the presence of I-RTX (10 μM), the effect of subsequent application of 3 μM capsaicin was inhibited (Fig. 7C; p < 0.05; analysis of variance with Newman-Keuls post hoc analysis).

Inhibition of C-fiber activity in an in vitro skin-nerve preparation by I-RTX in wild-type mice. A, event marker traces depicting representative recordings of single C-fiber responses to a 28 mN von Frey (VF) fiber stimulation of the cutaneous receptive field and subsequent 300-s time bins during baseline, vehicle, or capsaicin (3 μM) application (see Materials and Methods). Inset, C-fiber action potential waveform. Although there were few action potentials during baseline recording or vehicle application, the C-fiber responded robustly during capsaicin application. B, mechanically sensitive C-fiber generated few action potentials during baseline recording or during the application of either 10 μM I-RTX alone or I-RTX in the presence of 3 μM capsaicin. C, response to capsaicin was significantly greater in C-fibers treated with capsaicin compared with C-fibers treated with I-RTX or capsaicin in the presence of I-RTX. There was no significant difference between responses during vehicle application and responses during application of I-RTX alone or I-RTX in the presence of capsaicin (★, p < 0.05; analysis of variance with Newman-Keuls post hoc analysis; data are expressed as the mean ± S.E.M.).
Activity of I-RTX in TRPV1 Knockout Mice.
To test the relationship between the spontaneous activity induced by I-RTX alone, its effects were also examined in TRPV1 knockout mice. In wild-type mice capsaicin caused a robust increase in paw-flinching behavior and this effect was abolished in TRPV1 knockout mice. In TRPV1 knockout mice the paw-flinching behavior induced by capsaicin (2.5 μg ipl, equivalent to 8.2 nmol) was significantly reduced (2 ± 1 flinches over 10-min period) compared with wild-type mice (47 ± 12 flinches over 10 min). Similarly, the duration of leg raising and kicking of the injected paw was abolished in TRPV1 knockout mice (duration 2 ± 1 s for TRPV1 knockout and 16 ± 4 s for wild-type mice). As previously found in rats, intraplantar administration of I-RTX alone caused nocifensive behaviors (flinching and licking) in wild-type mice and an identical effect was seen in TRPV1 knockout mice (Fig. 8).

Effect of intraplantar I-RTX on nocifensive behavior in TRPV1 KO mice. I-RTX was applied at a dose of 10 nmol dissolved in 10% DMSO/90% saline (dose volume 20 μl). Leg raising (A) and paw-flinching behaviors (B) were observed for 10 min after I-RTX or vehicle administration. Injection of vehicle alone did not induce flinching or leg raising/licking behaviors. In contrast to acute administration of capsaicin in rats (Fig. 5) the paw-flinching and leg raising/licking behavior in wild-type and TRPV1 KO mice in response to application of I-RTX was slower in onset (typically occurring 3–10 min postadministration) and was submaximal in its intensity. Under identical conditions, intraplantar injection of capsaicin induced flinching and leg raising/licking of the injected paw in wild-type mice [47 ± 12 flinches and 16 ± 4 s duration of licking over 10 min in wild-type (WT) mice] but these behaviors were abolished in TRPV1 KO mice (2 ± 1 flinches and 2 ± 1-s duration of licking over 10 min in VR1 KO mice). Data are expressed as the mean ± S.E.M.
Discussion
In this study, we have characterized the pharmacological properties of iodo-resiniferatoxin, a high-affinity rat TRPV1 receptor antagonist (Wahl et al., 2001) that also blocks responses to capsaicin, protons, and heat on human TRPV1 receptors. Furthermore, we show in electrophysiological assays that I-RTX has a 10-fold lower affinity for responses to capsaicin on human TRPV1 receptors compared with other methods of activating these currents. Of particular note is the finding that the pharmacology of human and rat TRPV1 receptors differ, such that under specific conditions capsazepine is an incomplete antagonist of responses mediated by acidic pH in rat TRPV1 receptors. These results are important because in tissues or assays where there is a high channel density capsazepine may be ineffective as an antagonist on TRPV1 currents activated by protons despite the fact that TRPV1 receptors are involved in the functional response. Furthermore, we show that despite the high affinity of I-RTX for rat TRPV1 receptors this compound is ineffective at blocking responses to intraplantar capsaicin administration in rats. We consider this most likely a consequence of RTX being a high molecular weight lipophilic molecule with poor aqueous solubility (see Materials and Methods). At high doses, equivalent to concentrations that do not exhibit any intrinsic agonist activity on TRPV1 receptors in vitro, I-RTX alone induced spontaneous paw-flinching behavior. This effect was consistent with a nonspecific pharmacological effect because this activity was also apparent in TRPV1 knockout mice.
Previous studies using luminescence-based assays have suggested that the pharmacology of human and rat receptors is different (McIntyre et al., 2001) and that the pharmacology of cloned versus native receptors may differ (Shin et al., 2001). Although the latter possibility cannot be excluded, the results of the present study confirm that there are subtle differences between rat and human TRPV1 receptors that may reflect the underlying differences in the structure of these two receptors between species.
Neurons express several classes of cation channels that are gated by protons, including members of the TRP and ASIC family (Harteneck et al., 2000). Previous studies have shown that some cell lines, in particular human embryonic kidney cells, express endogenous proton-activated currents that belong to the ASIC family (Hayes et al., 2000; Gunthorpe et al., 2001). Indeed, in the present study it was necessary to isolate proton-activated currents mediated by TRPV1 receptors in tsA-201 cells by desensitizing native currents with solution buffered at pH 6.8 (Vellani et al., 2001). It is unlikely that the differences in pharmacology of pH responses between human and rat receptors seen in the present study resulted simply from cell-specific changes in the expression of endogenous proton-activated currents. This is because 1) I-RTX completely blocked the proton responses of both rat and human TRPV1 receptors, and 2) human TRPV1 currents activated by protons were completely blocked by capsazepine irrespective of whether the channels were stably expressed in CHO cells or transiently expressed in the tsA-201 cell line (whereas capsazepine was only an incomplete antagonist of proton-activated rat TRPV1 receptors in the same cells).
There are conflicting reports regarding whether capsazepine is able to fully block responses to acidic pH in native tissues. This may in part relate to the expression of additional proton gated ion channel subtypes in such cells (see above) or alternatively that capsazepine is an incomplete antagonist on rat TRPV1 (present study; McIntyre et al., 2001). For example, Reeh and colleagues (Habelt et al., 2000) have shown that nociceptive pH responses on heat-sensitive C-fibers of rat saphenous nerve are not blocked by capsazepine, as is also observed in rat trigeminal ganglia (Martenson et al., 1997), whereas in other tissues (Santicioli et al., 1993) or species such as guinea pig, capsazepine has been reported to be an effective antagonist of pH responses on sensory nerves (Franco-Cereceda and Lundberg, 1992). There are also reports that sustained pH currents recorded from dorsal root ganglia neurons are not fully antagonized by capsazepine (Vyklicky et al., 1998). Nonetheless, capsazepine has been used to investigate the functional significance of vanilloid receptors in inflammation and pain (for review, see Szallasi and Blumberg, 1999). In particular it has been shown that capsazepine can block thermal hyperalgesia (Kwak et al., 1998), a phenotype similar to that described for TRPV1 knockout mice (Caterina et al., 2000; Davis et al., 2000). These data confirm an important role for TRPV1 receptors in some forms of pain and inflammation. In the present study capsazepine, when applied either subcutaneously or intraplantar, was effective at attenuating the nocifensive behavior induced by intraplantar administration of capsaicin.
I-RTX has been recently reported to be a high-affinity antagonist of TRPV1 receptors (Wahl et al., 2001). The antagonist properties of 4-hydroxy-5-iodo-3-methoxyphenylacetate I-RTX has been confirmed in the present study and its pharmacology extended to include capsaicin and pH activation of rat and human TRPV1 receptors. A recent study (McDonnell et al., 2002) also shows that iodination at a different position on the molecule (4-hydroxy-2-iodo-5-methoxyphenylacetate) confers partial agonist-like activity to RTX. Notably, in the previous study by Wahl et al., (2001) it was also reported that I-RTX has analgesic activity against peripheral administration of capsaicin when applied intrathecally. To determine whether I-RTX was directly effective against peripheral TRPV1 receptors in the present study we examined whether intraplantar administration of this antagonist could directly block the nociceptive responses to capsaicin. I-RTX was not given systemically like capsazepine because it is likely to be highly plasma protein-bound and therefore unlikely to be accessible from the plasma to interact with the receptor in the skin. The quantity of material required for such study is also impractical given the limited availability of the parent toxin RTX. Nevertheless despite its high affinity in vitro, I-RTX (up to 1 nmol/paw, equivalent to that used intrathecally by Wahl et al., 2001) did not block the aversive behavior induced by a submaximally effective dose of capsaicin. Given these data we tested directly whether I-RTX could block C-fiber activation by capsaicin using an in vitro isolated skin-nerve preparation. Under these conditions I-RTX completely antagonized the effects of capsaicin. Thus, the most parsimonious explanation for the lack of activity of I-RTX after intraplantar administration is limited access to the receptive field in the skin due to its poor physicochemical properties (see Materials and Methods). Interestingly, it was found that at a high dose intraplantar I-RTX also induced spontaneous aversive behavior (leg raising and paw flinching) in the absence of capsaicin. To test whether this was an effect mediated by TRPV1 receptors we examined its activity in TRPV1 knockout mice. In both wild-type and TRPV1 knockout mice a similar aversive behavior to intraplantar I-RTX was observed. Because I-RTX did not have agonist activity on recombinant TRPV1 receptors, or native receptors in the skin-nerve preparation, coupled with the fact that the nociceptive effect of I-RTX was still observed in TRPV1 knockout mice. Therefore, we conclude that this activity is due to effects on other receptors or pathways that do not involve TRPV1.
These data clearly demonstrate that I-RTX has limited utility for behavioral studies and that antagonists with better pharmaceutical properties are required to fully explore the functional relevance of TRPV1 receptors in vivo. We show that, in contrast to capsazepine, I-RTX is a nanomolar-affinity full antagonist on rat and human TRPV1 using either capsaicin, pH, or heat as the agonist. These indicate that I-RTX will be a useful tool for further characterization of the functional properties of cloned TRPV1 receptors and that of vanilloid receptor subtypes in isolated tissues.
Footnotes
- Received June 18, 2002.
- Accepted August 15, 2002.
DOI: 10.1124/jpet.102.040394
Abbreviations
- TRPV1
- VR1, transient receptor potential vanilloid receptor
- RTX
- resiniferatoxin
- I-RTX
- iodo-resiniferatoxin
- KO
- knockout
- CHO
- Chinese hamster ovary
- MOPS
- 3-(N-morpholino)propanesulfonic acid
- DMSO
- dimethyl sulfoxide
- ipl
- intraplantar
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}