


Abstract
In cellular models, chronic exposure to μ-opioid agonists converts antagonists into inverse agonists at μ-receptors. Such adaptations could contribute to the development of tolerance and/or dependence. To determine whether δ-receptors respond similarly, or whether this adaptation is unique for μ-receptors, this study examined the effects of prolonged agonist exposure on the intrinsic activity of several δ-opioid ligands in GH3 cells expressing δ-receptors. In opioid naive cells, δ-receptors were constitutively active, and a series of δ-ligands displayed a range of intrinsic activities for G protein activation. Chronic treatment with the full δ-agonist [d-Pen2,5]-enkephalin reduced the acute ability of [d-Pen2,5]-enkephalin to stimulate and the full inverse agonistN,N-diallyl-Tyr-Aib-Aib-Phe-Leu-OH (ICI-174864) to inhibit G protein activation. In contrast, although naloxone and naltriben exhibited weak partial agonism in opioid naive cells, both ligands acted as full inverse agonists to produce concentration-dependent inhibition of guanosine 5′-O-(3-[35S]thio)triphosphate binding after prolonged exposure to [d-Pen2,5]-enkephalin or to the partial agonist morphine. This effect was reversed by a neutral δ-antagonist (N,N-bisallyl)-Tyr-Gly-Gly-ψ-(CH2S)-Phe-Leu-OH (ICI-154129). Finally, as is also characteristic of inverse agonists, naloxone and naltriben demonstrated higher affinities for uncoupled δ-receptors in cells chronically treated with [d-Pen2,5]-enkephalin, relative to opioid naive cells. Therefore, this relatively novel adaptation is shared by both μ- and δ-opioid receptors and therefore may serve as an important common mechanism involved the development of tolerance and/or dependence.
Opioid analgesics are clearly the most efficacious agents currently available for the treatment of moderate-to-severe pain (Reisine and Pasternak, 1996). However, their use for chronic pain management is often limited due to the development of tolerance and/or dependence upon prolonged administration (Nestler et al., 1993). The mechanisms underlying these adaptations to extended opioid exposure are poorly understood. At the cellular level, opioids produce their effects by activation of μ-, δ-, or κ-opioid receptors. All of these receptors are members of the large superfamily of G protein-coupled receptors (GPCRs) that traverse the plasma membrane seven times and activate intracellular G proteins (Law et al., 2000). Many GPCRs exhibit constitutive activity, producing spontaneous regulation of G proteins and effectors in the absence of activation by agonists (Lefkowitz et al., 1993). Both δ- (Merkouris et al., 1997) and μ- (Burford et al., 2000; Liu et al., 2001) opioid receptors display such agonist-independent activity, and alterations in constitutive activity of μ-opioid receptors in response to prolonged opioid exposure have been suggested to contribute to the development of tolerance and/or dependence (Wang et al., 2000;Liu and Prather, 2001).
A two-state receptor model has been proposed to account for constitutive activity in which GPCRs exist in an equilibrium between inactive (R) and active (R*) states (Costa et al., 1992). Agonists stabilize the active R* state and thus display positive intrinsic activity, resulting in an increase in receptor activity. In contrast, inverse agonists stabilize the inactive R state and exhibit negative intrinsic activity, reflected by a reduction in spontaneous, agonist-independent receptor activity. Neutral antagonists have equal preferences for both R and R* states, lack any intrinsic activity, and thus are able to block actions produced by either agonists or inverse agonists. Our laboratory recently demonstrated that chronic exposure of GH3 cells expressing μ-opioid receptors to μ-agonists results in the conversion of neutral antagonists into inverse agonists (Liu and Prather, 2001). Because constitutively active μ-opioid receptors in opioid naive GH3 cells expressing μ-opioid receptors could not be demonstrated with any known inverse agonist, in this original study it was proposed that chronic treatment resulted in a conversion of quiescent receptors into those exhibiting constitutive activity. However, using a novel receptor alkylation technique, it was subsequently shown that μ-opioid receptors in these cells were indeed constitutively active before chronic treatment (Liu et al., 2001). Therefore, combined observations from both studies suggest that chronic agonist treatment might result in subtle alterations in the structure and/or conformation of μ-opioid receptors, revealing negative intrinsic activity of ligands previously demonstrated to posses only neutral antagonist properties. More importantly, such receptor adaptations to prolonged agonist exposure might be unique to μ-opioid receptors, contributing in some manner to tolerance and dependence commonly observed clinically with these drugs.
Selective δ-opioid receptor agonists are being developed as alternatives to μ-opioid analgesics because they may potentially possess fewer side effects (Cowan et al., 1988; Burkey et al., 1998). Acutely, δ- and μ-opioid receptors transduce their intracellular signals by very similar mechanisms, coupling to identical G proteins (Prather et al., 1994; Chakrabarti et al., 1995) and effectors (Piros et al., 1995; Prather et al., 2000). In spite of such similarities, potentially more useful information in the context of developing analgesics with fewer adverse effects might be obtained by comparing the adaptation of these receptors to prolonged agonist exposure. For example, it has been demonstrated that the internalization (Chakrabarti et al., 1997) and desensitization (Kovoor et al., 1997) of μ- and δ-opioid receptors after prolonged agonist exposure can be distinct. Additionally, as previously noted, chronic exposure to μ-opioid agonists results in alterations in the constitutive activity of μ-opioid receptors (Wang et al., 2000; Liu and Prather, 2001). If the development of tolerance and/or dependence in response to prolonged clinical administration of μ-agonists involves changes in constitutive activation of μ-opioid receptors as has been proposed (Wang et al., 1994, 2000; Liu and Prather, 2001), it would be useful to know whether similar adaptations of δ-opioid receptors occur after chronic exposure to δ-agonists. Therefore, the purpose of the present study was to determine the effect of prolonged agonist exposure of GH3 cells expressing δ-opioid receptors (GH3DORT8) on the intrinsic activity of several δ-selective ligands. Evidence is provided that chronic treatment with opioid agonists converts the antagonists naloxone (NAL) and naltriben (NTB), but not other δ-ligands, into inverse agonists at δ-opioid receptors. Therefore, this relatively novel receptor adaptation in response to chronic opioid exposure seems to be shared by both μ- and δ-opioid receptors and therefore may serve as an important common mechanism involved the development of tolerance and/or dependence.
Materials and Methods
Materials.
[3H]Diprenorphine (50 Ci/mmol) was purchased from PerkinElmer Life Sciences (Boston, MA). [8-3H]Adenine (26 Ci/mmol) and guanosine 5′-O-(3-[35S]thio)triphosphate ([35S]GTPγS) (1250 Ci/mmol) were obtained from Amersham Biosciences (Piscataway, NJ). [d-Pen2,5]-enkephalin (DPDPE) was purchased from Phoenix Pharmaceuticals, Inc. (Belmont, CA). Naltrindole (NTD), N-benzylnaltrindole (N-NTD), naltriben, 7-benzylidenenaltrexone (BNTX), ICI-154129, and ICI-174864 were obtained from Tocris Cookson (Ballwin, MO). The National Institute on Drug Abuse (Bethesda, MD) provided morphine. Naloxone, 5′-guanylylimidodiphosphate (GppNHp), GDP, GTPγS, forskolin, and 3-isobutyl-1-methylxanthine were supplied by Sigma-Aldrich (St. Louis, MO). All other reagents were purchased from Fisher Scientific (Pittsburgh, PA).
Cell Culture and Drug Pretreatment.
To create the GH3DORT8 cell line, GH3cells (CCL 82.1) were stably transected with pREP4 plasmids (Invitrogen, Carlsbad, CA) containing cDNA encoding for δ-opioid receptors with a hemagglutinin epitope tag spliced at the N terminus as described previously (Martin et al., 2001). Cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 100 units/ml penicillin, 100 mg/ml streptomycin, and 200 μg/ml hygromycin-B, in a humidified atmosphere of 5% CO2/95% air at 37°C. For receptor binding and [35S]GTPγS binding experiments, cells were seeded into 175-cm3 flasks. When cell growth reached 70% confluence, cells were incubated with various concentrations of morphine, DPDPE, or no drug in fresh culture medium for increasing time periods up to a maximum of 48 h. At the end of drug exposure, cells were detached by incubation with phosphate-buffered saline containing 1 mM EDTA for 5 min and centrifuged at 1000 rpm for 10 min. The cell pellets were then extensively washed three times with 50 volumes of phosphate-buffered saline, pH 7.4, and finally stored at −80°C until use. For adenylyl cyclase assays, cells were seeded into 17-mm (24-well) culture plates at density of 8 × 106 cells/plate and cultured for various time periods in medium containing different concentrations of DPDPE or morphine.
Preparation of Membranes.
Extensively washed, frozen cell pellets were thawed on ice and resuspended in ice-cold homogenization buffer composed of 50 mM HEPES, pH 7.4, 1 mM MgCl2, and 1 mM EGTA. Cells were homogenized with 10 strokes using a glass Dounce homogenizer (Wheaton, Philadelphia, PA). After centrifugation at 40,000g for 10 min (4°C), pellets were resuspended in homogenization buffer, homogenized, and centrifuged again as described. This procedure was repeated twice more. The final pellets were resuspended at 10% of original volume in a 50 mM Tris-HCl buffer, pH 7.4. Protein concentration was determined and aliquots were stored at −80°C.
Opioid Receptor Binding.
For competition binding assays, cell membranes (100 μg of protein) were incubated at room temperature for 90 min in 50 mM Tris-HCl buffer, pH 7.4, in the presence or absence of NaCl (100 mM) and the GTP analog GppNHp (25 μM) with [3H]diprenorphine (1 nM) and increasing concentrations of naltriben or naloxone in final volume of 1 ml. To examine receptor binding after chronic exposure to morphine or DPDPE, membranes prepared from extensively washed pretreated cells were incubated with 1 nM [3H]diprenorphine in the presence or absence of a saturating concentration of DPDPE (10 μM). The remaining specific binding was expressed as a percentage of the specific binding in membranes prepared from control cells that had not been exposed to any opioid (i.e., percentage of control). All receptor binding experiments were performed in triplicate and reactions were terminated by filtration through GF/B fiber filters using a Brandel 24-well cell harvester. Filters were subsequently washed three times with ice-cold binding buffer and bound radioactivity was determined 12 h after the addition of 4 ml of scintillation fluid by counting in a Tri-Carb 2100 TR liquid scintillation counter (Packard Instrument Company, Inc., Meriden, CT).
[35S]GTPγS Binding.
[35S]GTPγS binding was performed as described previously (Liu et al., 2001). Briefly membranes (50 μg/sample) were incubated with [35S]GTPγS (0.1 nM) in a binding buffer composed of 20 mM HEPES, pH 7.4, 10 mM MgCl2, 100 mM KCl, and 10 μM GDP at 30°C for 1 h in the presence of increasing concentrations of naloxone or naltriben, in a final volume of 1 ml. Nonspecific binding was determined in the presence of nonradioactive GTPγS (10 μM). Reactions were terminated by rapid filtration and bound radioactivity was determined by liquid scintillation counting as described above.
Adenylyl Cyclase Assay.
The effect of the absence or presence of opioids on the conversion of [3H]adenine-labeled ATP pools to cAMP in whole, intact cells was measured as described previously (Liu and Prather, 2001). Briefly, cells were seeded into 24-well plates and cultured for 24 h in the presence or absence of morphine (20 μM) or DPDPE (1 μM). At the end of agonist exposure, media were removed and cells were washed three times with serum-free medium. After washes, an incubation mixture (at 37°C) of Dulbecco's modified Eagle's medium containing the same concentration of the opioid used for pretreatment, 0.9% NaCl, 500 μM 3-isobutyl-1-methylxanthine, and 1.25 μCi/well of [3H]adenine was added for 1 to 2 h. After incubation, the mixture was removed and cells were washed three times with serum-free medium. Each plate was then floated in an ice-water bath for 5 min. During this time, an assay mixture of ice-cold Krebs-Ringer-HEPES buffer, pH 7.4, containing 500 μM 3-isobutyl-1-methylxanthine, 10 μM forskolin, and the appropriate concentration of the opioid ligand to be tested was added. Plates were then placed on a water bath at 37°C for 15 min. The reaction was terminated by the addition of 50 μl of 2.2 N HCl, cAMP was separated by using Alumina column chromatography, and radioactivity was determined by liquid scintillation counting as described above.
Data Analysis and Statistics.
Unless otherwise stated, data reported represent the mean ± standard error of the mean for at least three separate experiments that were each performed in triplicate. The IC50 andImax values were obtained from full concentration-effect curves subjected to sigmoidal curve fitting using the nonlinear regression analysis function of GraphPad Prism, version 2.0b, for Macintosh (GraphPad Software, San Diego, CA). For statistical comparisons involving three or more groups, differences between means were determined by a one-way analysis of variance (ANOVA) followed by post hoc comparisons using Dunnett's or Tukey's test. When only two groups were compared, differences between the means were determined by the nonpaired Student's t test.
Results
δ-Opioid Receptors Are Constitutively Active in Opioid Naive GH3DORT8 Cells.
Constitutive activity of δ-opioid receptors has been well characterized by use of the inverse agonist ICI-174864 (Merkouris et al., 1997). In the absence of agonists (i.e., under basal conditions), constitutively active GPCRs stimulate G proteins, resulting in an increase in the binding of the hydrolysis-resistant GTP analog [35S]GTPγS to G protein α-subunits. Inverse agonists possess negative intrinsic activity and stabilize the inactive state of the receptor, resulting in a decrease in basal [35S]GTPγS binding in systems containing constitutively active receptors. Because the purpose of the present study was to examine the effect of chronic agonist exposure on the intrinsic activity of δ-selective ligands in GH3DORT8 cells, we first determined whether δ-opioid receptors in this cell line constitutively activated G proteins (Fig. 1). As anticipated, the well characterized δ-opioid agonist DPDPE (100 nM) produced a significant increase of 71.8 ± 2.6% (P < 0.01) in [35S]GTPγS binding to GH3DORT8 membranes (Fig. 1, left). In contrast, the established δ-opioid inverse agonist ICI-174864 (1 μM) produced a −29.7 ± 3.9% reduction of [35S]GTPγS binding (P < 0.01) (Fig. 1, middle). A second δ-opioid antagonist, ICI-154129 (10 μM), showed no significant effect on [35S]GTPγS binding when tested alone, demonstrating neutral antagonist activity (Fig. 1, right). Importantly, both the stimulatory and inhibitory actions of DPDPE and ICI-174864 on [35S]GTPγS binding, respectively, were blocked by coadministration with the neutral δ-opioid antagonist ICI-154129 (10 μM) (P < 0.05). These data suggest that δ-opioid receptors in GH3DORT8 cells produce constitutive activation of G proteins. In addition, we have previously demonstrated that δ-opioid receptors in GH3DORT8 cells also constitutively inhibit adenylyl cyclase activity (Martin et al., 2001). In that study, it was shown that although DPDPE maximally reduced intracellular cAMP levels by over 70%, ICI-174864 (1 μM) produced a 21.5% stimulation of adenylyl cyclase activity above control levels when tested alone.

[35S]GTPγS binding to opioid naive GH3DORT8 membranes in response to the agonist DPDPE, the inverse agonist ICI-174864, or the neutral antagonist ICI-154129. Membranes were prepared from opioid naive GH3DORT8 cells, as described under Materials and Methods. [35S]GTPγS (0.1 nM) binding to membranes (50 μg) in response to 100 nM DPDPE (left) or 1 μM ICI-174864 (middle) was determined in the absence (−) or presence (+) of 10 μM ICI-154129. The effect of ICI-154129 (10 μM) alone on [35S]GTPγS binding was also tested (right). Nonspecific binding was defined by the inclusion of 10 μM GTPγS. Data are presented as the percentage of [35S]GTPγS binding in the presence of the indicated drug concentration, compared with basal binding in the absence of any test ligand (i.e., % of control). The control value used for all calculations (i.e., basal [35S]GTPγS binding) was 64.0 ± 7.3 fmol/mg of protein. Values represent the mean ± S.E.M. of three independent experiments performed in triplicate. ★, significantly different from [35S]GTPγS binding in the presence of DPDPE or ICI-174864 alone (P < 0.05; Student's t test). +, significantly different from basal [35S]GTPγS binding (P < 0.05; one-way ANOVA plus Dunnett's post hoc test).
Chronic Exposure of GH3DORT8 Cells to Full Agonist DPDPE, but Not to Partial Agonist Morphine Produces Desensitization of δ-Opioid Receptor-Induced Inhibition of Adenylyl Cyclase Activity.
We have previously demonstrated (Martin et al., 2001) that DPDPE binds to stably transected δ-opioid receptors in GH3DORT8 cell membranes with a high affinity of 6.08 ± 1.1 nM (Table 1). In the present study, as expected, it was determined that the affinity of the relatively μ-selective ligand morphine for δ-opioid receptors in this cell line (116.8 ± 15.4 nM) was considerably less than that of DPDPE. Both morphine and DPDPE produced similar efficacious inhibition of the adenylyl cyclase activity, withImax values of 72.7 ± 0.67 and 71.3 ± 1.2%, respectively (Table 1). However, much less DPDPE (IC50 = 0.43 ± 0.04 nM) was required to produce a half-maximal reduction in intracellular cAMP levels relative to morphine (IC50 = 272.1 ± 21.3 nM) (P < 0.01). Based on comparison of theKi and IC50values for these drugs, it would be predicted that the maximal effects of DPDPE on intracellular cAMP accumulation require occupancy of only a fraction of δ-opioid receptors, whereas near full receptor occupancy would be needed for morphine to produce a similar level of efficacy. Therefore, at δ-opioid receptors expressed in GH3DORT8 cells, morphine acts as a partial agonist relative to the full agonist DPDPE to inhibit adenylyl cyclase activity.
Affinity (Ki) values and inhibition of adenylyl cyclase activity by morphine and DPDPE in opioid naı̈ve GH3DORT8 cells and in cells chronically treated with morphine or DPDPE
Next, the effect of chronic exposure of cells to DPDPE or morphine on the ability of δ-opioid receptors to inhibit adenylyl cyclase activity was examined. GH3DORT8 cells were treated for 24 h with a concentration of morphine (20 μM) or DPDPE (1 μM) predicted to produce total receptor occupancy (i.e., >150 times Ki; Table 1). After extensive washing to remove residual chronic drugs, the ability of increasing concentrations of DPDPE (10−12-10−6 M) to inhibit 10 μM forskolin-stimulated cAMP levels was examined (Fig2; Table 1). In opioid naive cells, 0.43 ± 0.04 nM DPDPE was required to produce half-maximal inhibition of adenylyl cyclase activity. After prolonged exposure to morphine, neither the potency (IC50 = 1.69 ± 0.13 nM) nor the efficacy (Imax = 73.3 ± 1.7%) of DPDPE was significantly different from that observed in opioid naive cells. However, in cells treated 24 h with DPDPE, the IC50 for DPDPE was increased over 25-fold to 11.3 ± 3.2 nM (P < 0.05) and the maximal inhibition was significantly reduced to only 59.3 ± 1.9% (P < 0.05). Finally, the desensitization produced by DPDPE was also determined to be concentration- and time-dependent, with maximal effects obtained with 100 nM drug pretreatment and at 24 h of drug exposure, respectively (data not shown). Thus, chronic exposure to receptor-saturating concentrations of the full agonist DPDPE, but not to the partial agonist morphine results in a desensitization of the ability of δ-opioid receptors to acutely inhibit adenylyl cyclase activity.

Effect of chronic pretreatment with either the full agonist DPDPE or the partial agonist morphine on δ-opioid receptor inhibition of adenylyl cyclase activity in whole GH3DORT8 cells. Whole cell adenylyl cyclase assays were conducted as described under Materials and Methods. The ability of increasing concentrations of DPDPE to inhibit 10 μM forskolin-simulated cAMP levels after no chronic drug exposure (filled squares), 24 h of 20 μM morphine exposure (filled triangles), or 24 h of 1 μM DPDPE exposure (filled circles) was examined. Data are presented as the percentage of cAMP levels in the presence of the indicated drug concentrations, compared with that observed in the absence of any drug (i.e., % of control). Control values were 54.7 ± 10.8 fmol/mg/min for no chronic treatment, 60.7 ± 11.5 fmol/mg/min for chronic morphine treatment, and 89.5 ± 15.8 fmol/mg/min for chronic DPDPE treatment. Data represent the mean ± S.E.M. for three independent experiments performed in triplicate. The IC50 and Imax values determined for DPDPE under each condition are presented in Table 1.
Chronic Exposure of GH3DORT8 Cells to Either DPDPE or Morphine Produces Down-Regulation of δ-Opioid Receptors.
The effect of chronic DPDPE or morphine exposure on δ-opioid receptor density was also determined by measuring the amount of binding remaining of the nonselective opioid antagonist [3H]diprenorphine in membranes prepared from pretreated cells after extensive washing to remove residual agonist (Fig 3). It has been previously determined (Martin et al., 2001) by saturation binding with [3H]diprenorphine that GH3DORT8 cells express 2.16 ± 0.38 pmol/mg of δ-opioid receptors (Kd = 1.14 ± 0.22 nM). To ensure that maximal effects of chronic drug exposure on receptor binding were obtained, cells were incubated with the indicated concentrations of opioids for 48 h. Treatment of cells with increasing concentrations of DPDPE or morphine significantly decreased [3H]diprenorphine binding to membranes prepared from these cells by 76.8 ± 6.8 and 37.3 ± 3.2%, respectively. In addition to being less efficacious in reducing [3H]diprenorphine binding (P < 0.01), a much greater amount of the partial agonist morphine (IC50 = 1.4 ± 0.32 μM) relative to the full agonist DPDPE (IC50 = 0.62 ± 0.041 nM) was required to produce half-maximal down-regulation of δ-opioid receptors in GH3DORT8 cells (P < 0.01). These data demonstrate that chronic pretreatment with the full agonist DPDPE produces profound down-regulation of δ-opioid receptors, whereas prolonged exposure to the partial opioid agonist morphine, surprisingly, also significantly reduces receptor number. Our observations with morphine are in agreement with a previous study byZaki et al. (2001); however, other studies have demonstrated that chronic exposure to morphine does not result in a decrease in δ-opioid receptor number (Keith et al., 1996; Remmers et al., 1998).

Effect of chronic pretreatment of GH3DORT8 cells with increasing concentrations of either the full agonist DPDPE or the partial agonist morphine on δ-opioid receptor binding. GH3DORT8 cells were cultured for 48 h in the presence of increasing concentrations of morphine (filled triangles) or DPDPE (filled circles). After extensive washing to remove residual agonist, membranes were prepared and receptor binding experiments performed as described under Materials and Methods. Nonspecific binding was defined by inclusion of 10 μM naloxone in the binding buffer. Data are presented as the percentage of [3H]diprenorphine (1 nM) binding remaining in membranes prepared form cells chronically exposed to the indicated opioid concentration, compared with binding observed in cells that received no chronic treatment (i.e., % of control). The control receptor binding value used for all calculations was 1.80 ± 0.14 pmol/mg of protein. Data represent the mean ± S.E.M. for three independent experiments performed in triplicate.
Prolonged Exposure of GH3DORT8 Cells to Full Opioid Agonist DPDPE Converts Naloxone and Naltriben into Inverse Agonists at δ-Opioid Receptors, as Measured by [35S]GTPγS Binding.
To determine the effect of chronic opioid pretreatment on the intrinsic activity of selected δ-opioid ligands, a series of eight drugs with varying intrinsic activities in opioid naive GH3DORT8 cells were evaluated (Fig.4, closed columns). Based on previously reported affinities of the test ligands for δ-opioid receptors (Shaw et al., 1982; Cotton et al., 1984; Portoghese et al., 1990, 1992;Korlipara et al., 1994; Tables 1 and 2), the concentrations used for the initial screen were selected to produce near full receptor occupancy, resulting in maximal effects. In membranes prepared from opioid naive cells, DPDPE (100 nM) exhibited full agonist activity, producing a 71.8 ± 2.6% increase in [35S]GTPγS binding. NTD (1 μM) and N-NTD (1 μM), previously described as potent and selective δ-opioid antagonists (Portoghese et al., 1990; Korlipara et al., 1994), acted as partial agonists, inducing increases of 34.8 ± 3.6 and 35.5 ± 6.1% in [35S]GTPγS binding, respectively. NAL (10 μM) and NTB (1 μM) exhibited weak partial agonist activity, producing respective increases of only 19.8 ± 2.8 and 19.3 ± 3.0% in [35S]GTPγS binding. Another well characterized δ-opioid antagonist, ICI-154129 (10 μM) (Shaw et al., 1982), showed no significant effect on [35S]GTPγS binding and thus possessed neutral antagonist properties in this assay. As observed in previous studies (Merkouris et al., 1997; Neilan et al., 1999), BNTX (1 μM) and ICI-174864 (10 μM) behaved as inverse agonists, producing decreases of −12.1 ± 2.0 and −29.7 ± 3.9% in [35S]GTPγS binding, respectively. These observations demonstrate that the ligands tested exhibit a range of intrinsic activities at δ-opioid receptors expressed in opioid naive GH3DORT8 cells, from full agonists (i.e., DPDPE) to full inverse agonists (i.e., ICI-174864).

[35S]GTPγS binding in response to a series of δ-opioid-selective ligands in membranes prepared from opioid naive or chronic DPDPE-pretreated GH3DORT8 cells. GH3DORT8 cells received no chronic pretreatment (filled columns) or were cultured for 48 h in the presence of the 1 μM DPDPE (open columns). After extensive washing to remove residual agonist, membranes were prepared and [35S]GTPγS binding experiments performed as described under Materials and Methods. [35S]GTPγS (0.1 nM) binding to membranes (50 μg) in response to 1 μM DPDPE (DP), 1 μM NTD, 1 μM N-NTD, 10 μM NAL, 1 μM NTB, 10 μM ICI-154129 (ICI-154), 1 μM BNTX, or 10 μM ICI-174864 (ICI-174) was examined. Nonspecific binding was defined by the inclusion of 10 μM GTPγS. Data are presented as the percentage of [35S]GTPγS binding in the presence of the indicated drug concentration, compared with basal binding in the absence of any test ligand (i.e., % of control). The control values used for all calculations (i.e., basal [35S]GTPγS binding) were 66.1 ± 2.0 fmol/mg of protein for no chronic pretreatment and 38.0 ± 1.0 fmol/mg of protein for chronic DPDPE pretreatment. Values represent the mean ± S.E.M. of four to six independent experiments performed in triplicate. ★, significantly different from [35S]GTPγS binding in the presence of the indicated test ligand in membranes prepared from cells receiving no pretreatment (P < 0.05; Student's t test). +, significantly different from basal [35S]GTPγS binding (P < 0.05; one-way ANOVA plus Dunnett's post hoc test).
Affinity (Ki) values for the inhibition of [3H]diprenorphine binding by naloxone or naltriben in the absence or presence of GppNHp/NaCl to membranes prepared from opioid naı̈ve, morphine, or DPDPE-pretreated GH3DORT8 cells
To screen for potential alterations in intrinsic activity after prolonged opioid exposure, [35S]GTPγS binding in response to all eight δ-opioid ligands was examined in membranes prepared from GH3DORT8 cells chronically treated with the full agonist DPDPE (1 μM, 48 h) (Fig. 4, open columns). Chronic DPDPE was selected for the screen because initial experiments had determined that prolonged exposure of cells to DPDPE produced significantly greater desensitization and down-regulation relative to morphine. Chronic exposure of GH3DORT8 cells to DPDPE significantly reduced both the stimulatory effects of the full agonist DPDPE (1 μM) (from 71.8 ± 2.6 to 24.8 ± 2.6%), as well as the inhibitory effects of the inverse agonists ICI-174864 (10 μM) (from −29.7 ± 3.9 to −12.1 ± 2.0%) and BNTX (1 μM) (from −12.1 ± 2.3 to 7.1 ± 5.8%) on [35S]GTPγS binding. Interestingly, the intrinsic activity of the partial agonists NTD (1 μM) (from 35.5 ± 6.1 to 22.9 ± 4.3%) and N-NTD (1 μM) (from 34.8 ± 3.6 to 31.8 ± 4.2%), as well as the neutral antagonist ICI-154129 (10 μM) (from −1.9 ± 2.9 to −3.6 ± 2.5%), was not significantly altered by prolonged exposure to DPDPE. Most importantly, after chronic DPDPE treatment, the intrinsic activity of naloxone (10 μM) and naltriben (1 μM) was converted from weak partial agonism (19.8 ± 2.8 and 18.2 ± 3.9%, respectively) in opioid naive cells, to full inverse agonism (−16.1 ± 1.7 and –26.9 ± 3.1%, respectively). Importantly, the efficacy of inhibition of [35S]GTPγS binding by these drugs after chronic opioid exposure was equivalent to that produced by the accepted inverse agonist ICI-174864 (10 μM).
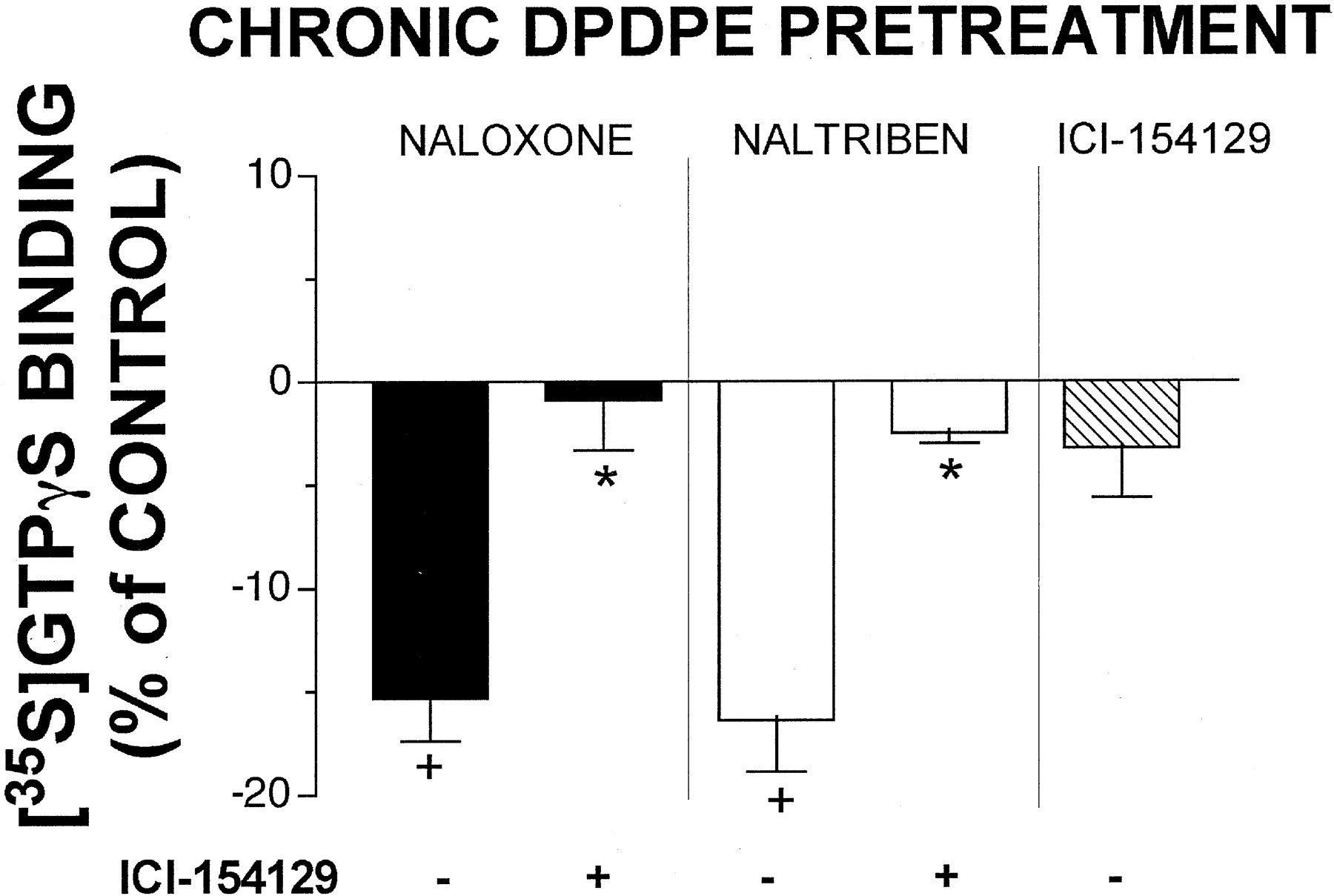
ICI-154129 was concluded to be a neutral δ-opioid antagonist based on absence of any effects on [35S]GTPγS binding in membranes prepared from either opioid naive cells or cells treated chronically with DPDPE (Fig. 4). Therefore, to confirm that the reduction in [35S]GTPγS binding after chronic DPDPE treatment by naloxone and naltriben specifically involved δ-opioid receptors, ICI-154129 was tested for its ability to block their apparent inverse agonism (Fig. 5). Coincubation with ICI-154129 (10 μM) significantly reversed the ability of both naloxone (10 μM) (Fig. 5, left) and naltriben (1 μM) (Fig. 5, middle) to decrease [35S]GTPγS binding in GH3DORT8 cells chronically exposed to DPDPE (P < 0.05). It is also important to point out that the lack of any effect on [35S]GTPγS binding by ICI-154129 in cells chronically treated with DPDPE (Fig. 5, right) strongly implies that the apparent inverse agonist effects of naloxone and naltriben in DPDPE pretreated cells was not simply due to antagonism of the stimulation of [35S]GTPγS binding produced by residual chronic opioid present in the incubation medium. If this were the case, then the antagonist ICI-154129 should have also demonstrated a similar “pseudo” inverse agonist effect of reducing residual agonist-induced [35S]GTPγS binding after chronic opioid exposure.

Blockade of naloxone and naltriben inhibition of [35S]GTPγS binding to membranes prepared from DPDPE-pretreated cells by ICI-154129. Membranes were prepared from extensively washed GH3DORT8 cells pretreated for 48 h with 1 μM DPDPE, as described under Materials and Methods. [35S]GTPγS binding to membranes in response to 10 μM naloxone (left) or 1 μM naltriben (middle) was determined in the absence (−) or presence (+) of 10 μM ICI-154129. The effect of ICI-154129 (10 μM) alone on [35S]GTPγS binding was also tested (right). Nonspecific binding was defined by the inclusion of 10 μM GTPγS. Data are presented as the percentage of [35S]GTPγS binding in the presence of the indicated drug concentration, compared with basal binding in the absence of any test ligand (i.e., % of control). The control value used for all calculations (i.e., basal [35S]GTPγS binding) was 30.1 ± 1.0 fmol/mg of protein. Values represent the mean ± S.E.M. of four or five independent experiments performed in triplicate. ★, significantly different from [35S]GTPγS binding in the presence of DPDPE or naltriben alone (P < 0.05; Student's t test). +, significantly different from basal [35S]GTPγS binding (P < 0.05; one-way ANOVA plus Dunnett's post hoc test).
Apparent Inverse Agonism of Naloxone and Naltriben after Chronic DPDPE Pretreatment Is Concentration-Dependent and Also Occurs after Prolonged Exposure to Partial Agonist Morphine.
As presented in Fig. 6A, naloxone demonstrated slight (Emax = 19.8 ± 2.8%) but significant stimulation of [35S]GTPγS binding to membranes prepared from opioid naive cells (P < 0.01). In marked contrast, naloxone produced significant (P < 0.01), concentration-dependent inhibition of [35S]GTPγS binding in membranes prepared from cells chronically treated not only with DPDPE (IC50 = 23.5 ± 2.1 nM;Imax = 16.1 ± 1.7%) but also with morphine (IC50 = 11.2 ± 3.5 nM;Imax = 19.4 ± 1.0%). Similarly, naltriben produced a concentration-dependent inhibition of [35S]GTPγS binding with IC50 values of 1.6 ± 0.42 and 2.6 ± 0.31 nM in membranes prepared from cells chronically treated with morphine or DPDPE, respectively (Fig. 6B). However, unlike that observed for naloxone, naltriben produced significantly (P < 0.01) greater maximal inhibition in cells chronically treated with morphine (Imax = 41.4 ± 4.7%), relative to DPDPE (Imax = 26.9 ± 3.1%). Naltriben also showed slight (from 14.2 to 19.3%), but significant (P < 0.05) stimulation of [35S]GTPγS binding to membranes prepared from opioid naive cells. Interestingly, this effect was not concentration-dependent and similar modest levels of stimulation occurred when concentrations of naltriben as low as 10−14 M were tested. It is possible that this method lacks sufficient sensitivity to consistently detect the weak partial agonist activity exhibited by naltriben in opioid naive cells. In any case, it is important to note that the IC50 values for inhibition of [35S]GTPγS binding determined for both naloxone and naltriben in chronically treated cells are similar to their affinity (Ki) for δ-opioid receptors (Table 2).

Concentration-effect of naloxone and naltriben on [35S]GTPγS binding to membranes prepared from opioid naive, morphine-, or DPDPE-pretreated GH3DORT8 cells. GH3DORT8 cells were treated with no opioid (filled squares), 20 μM morphine (filled triangles), or 1 μM DPDPE (filled circles) for 48 h. Membranes were prepared from extensively washed pretreated cells, as described under Materials and Methods. [35S]GTPγS binding to membranes in response to naloxone (A) or naltriben (B) was determined with 0.1 nM [35S]GTPγS in the presence of increasing concentrations of naloxone (10−10-10−5 M) or naltriben (10−11-10−6 M). Nonspecific binding was defined by the inclusion of 10 μM GTPγS. Data are presented as the percentage of [35S]GTPγS binding in the presence of the indicated drug concentration, compared with basal binding in the absence of any test ligand (i.e., % of control). The control values used for all calculations (i.e., basal [35S]GTPγS binding) were 49.8 ± 4.0 fmol/mg of protein for no chronic pretreatment, 44.5 ± 1.2 fmol/mg of protein for chronic morphine pretreatment, and 31.5 ± 2.2 fmol/mg of protein for chronic DPDPE pretreatment. Values represent the mean ± S.E.M. of four to six independent experiments performed in triplicate. ★ and ★★, significantly different from control (i.e., no opioid) (★,P < 0.05; ★★, P < 0.01; one-way ANOVA plus Dunnett's post hoc test). a–c, significantly different from maximal inhibition determined for no chronic pretreatment (a), chronic morphine pretreatment (b), or chronic DPDPE pretreatment (c) (P < 0.01; one-way ANOVA plus Tukey's post hoc test).
Chronic Exposure of GH3DORT8 Cells to DPDPE, but Not to Morphine Increases Affinity of Naloxone and Natriben for Uncoupled Form of δ-Opioid Receptors.
According to the two-state model of receptor activation, GPCRs exist in an equilibrium between active (R*, coupled) and inactive (R, uncoupled) states (Costa et al., 1992). Because inverse agonists preferentially stabilize the inactive (R) conformation of the receptor, they demonstrate higher affinity for the uncoupled form of the receptor. Based on the observation that naloxone and naltriben act as inverse agonists to decrease basal [35S]GTPγS binding only in membranes prepared from cells chronically exposed to opioids, it would be expected that these ligands would have a higher affinity for the uncoupled (R) state of the δ-opioid receptor under these conditions. Therefore, competition binding assays with [3H]diprenorphine were performed in opioid naive and chronically treated membranes in the presence or absence of NaCl and GppNHp (i.e., conditions known to produce uncoupling of GPCRs from G proteins; Childers and Snyder, 1980; Appelmans et al., 1986) (Fig. 7; Table 2). We have previously demonstrated that this method accurately reflects the binding characteristics for both agonists and inverse agonists in GH3DORT8 cells (Martin et al., 2002). For example, the presence of GppNHp/NaCl in the binding buffer resulted in a 28-fold reduction in the affinity of the agonist DPDPE for δ-opioid receptors, whereas these conditions enhanced the affinity of the inverse agonist ICI-174864 over 8-fold (Martin et al., 2002). As predicted for an antagonist, in membranes prepared from cells not exposed to opioids, naltriben showed equivalent affinity for the coupled (Ki = 0.23 ± 0.04 nM) and uncoupled (Ki = 0.15 ± 0.03 nM) states of the receptors (Fig. 7A). Unexpectedly, naltriben also demonstrated similar affinity in the absence (Ki = 0.19 ± 0.02 nM) and presence (Ki = 0.11 ± 0.03 nM) of GppNHp/NaCl in cells chronically exposed to morphine (Fig. 7B). However, as is characteristic of an inverse agonist, in membranes prepared from cells pretreated with DPDPE, the affinity of naltriben was more than 8-fold greater for the uncoupled (Ki = 0.036 ± 0.01 nM) relative to the coupled (Ki = 0.22 ± 0.07 nM) state of the receptor (P < 0.05) (Fig. 7C). Similar to naltriben, naloxone also demonstrated a higher affinity for the uncoupled form of δ-opioid receptors in membranes prepared from cells chronically treated with DPDPE (Ki = 5.3 ± 1.4 nM), relative to that observed in morphine-treated (Ki= 11.3 ± 2.4 nM) or in opioid naive cells (Ki = 12.4 ± 0.97 nM) (Table 2). Interestingly, but distinct from naltriben, naloxone also showed significantly (P < 0.01) higher affinity for δ-opioid receptors in the presence of GppNHp/NaCl, independent of any treatment condition (i.e., in opioid naive, morphine-, and DPDPE-treated cells).

Effect of GppNHp/NaCl on the competitive inhibition of [3H]diprenorphine binding by naltriben in membranes prepared from opioid naive, morphine-, or DPDPE-pretreated GH3DORT8 cells. GH3DORT8 cells were cultured for 48 h in the presence of no opioid (A), 20 μM morphine (B), or 1 μM DPDPE (C). After extensive washing to remove residual agonist, membranes were prepared and receptor binding experiments performed as described under Materials and Methods. Assays were conducted in the absence (filled symbols) or presence (open symbols) of 25 μM GppNHp and 100 mM NaCl in the binding buffer. Data are presented as the percentage of [3H]diprenorphine binding in the presence of the indicated drug concentration, compared with binding in the absence of any competing ligand (i.e., % of control). Control values for competition in opioid naive membranes were 1.90 ± 0.10 pmol/mg (−GppNHp/NaCl) and 1.62 ± 0.08 pmol/mg (+GppNHp/NaCl). Control values for competition in morphine-pretreated membranes were 1.54 ± 0.10 pmol/mg (−GppNHp/NaCl) and 1.44 ± 0.14 pmol/mg (+GppNHp/NaCl). Control values for competition in DPDPE-pretreated membranes were 0.81 ± 0.13 pmol/mg (−GppNHp/NaCl) and 0.80 ± 0.12 pmol/mg (+GppNHp/NaCl). Data represent the mean ± S.E.M. for three to five experiments performed in duplicate. The affinity (Ki) values calculated for each binding condition are presented in Table2.
Discussion
In cellular models, chronic exposure to μ- and δ-opioid agonists results in several well characterized adaptive responses, including receptor desensitization (Kovoor et al., 1997), receptor down-regulation (Chakrabarti et al., 1997), receptor internalization (Keith et al., 1996), and adenylyl cyclase supersensitization (Sharma et al., 1975). These processes have been proposed to play a role in the development of tolerance and/or dependence that occurs upon prolonged opioid administration (Collier, 1984). However, both μ- (Burford et al., 2000; Liu et al., 2001) and δ- (Merkouris et al., 1997) opioid receptors also exhibit constitutive activity, and we previously demonstrated that chronic exposure to μ-agonists converts antagonists into inverse agonists at μ-opioid receptors (Liu and Prather, 2001). Therefore, it is possible that an additional mechanism contributing to the development of opioid tolerance and dependence might be produced by alterations in constitutive activity of these receptors. Because δ-opioid agonists are being developed as potential alternative analgesics with fewer side effects than drugs acting at μ-opioid receptors (Cowan et al., 1988; Burkey et al., 1998), this study determined whether prolonged exposure to δ-agonists would produce similar adaptations in δ-opioid receptor signal transduction as has been shown previously for μ-opioid receptors.
This was accomplished by comparing the intrinsic activity of several δ-selective ligands to activate G proteins in GH3DORT8 cells before and after chronic exposure to opioid agonists. The most significant finding of this article was that although naloxone and naltriben exhibited weak partial agonism in opioid naive cells, both ligands acted as full inverse agonists after chronic exposure to maximally effective concentrations of DPDPE or morphine. This is demonstrated by the following observations. First, naloxone and naltriben produced concentration-dependent inhibition of [35S]GTPγS binding only in membranes prepared from chronically treated cells. Second, the IC50values for G protein inhibition for both drugs were similar to their affinity (Ki) for δ-opioid receptors. Third, this inhibitory effect was reversed by a neutral δ-opioid antagonist, ICI-154129. Finally, as is also characteristic of inverse agonists, naloxone and naltriben demonstrated higher affinities for the uncoupled form of δ-opioid receptors in membranes prepared from cells chronically treated with DPDPE, relative to that observed in opioid naive cells. Therefore, as far as this specific adaptation is concerned, both μ- and δ-opioid receptors respond similarly to prolonged opioid exposure.
The mechanisms underlying this novel adaptive response to prolonged opioid exposure are unknown; however, data provided in the present study lend insight into two alternative possibilities. First, it is reasonable to propose that the observation of inverse agonism after chronic opioid exposure by drugs previously demonstrated to exhibit only antagonist or partial agonist activity reflects an overall increase in constitutive activity of δ-opioid receptors. This would necessitate that even though fewer receptors are present, the measure of total constitutive activity is enhanced. Therefore, the apparent “conversion” to inverse agonism of some drugs might simply be due to the fact that chronic opioid treatment makes measurement of the functional consequence of this alteration easier to observe. Indeed, several studies have demonstrated that chronic exposure to morphine results in an apparent enhancement of the constitutive activity of μ-opioid receptors (Wang et al., 1994, 2000). If this hypothesis is correct then in addition to the appearance of inverse agonism by naloxone and naltriben, it would be predicted that the efficacy of the full inverse agonist ICI-174864 would also increase proportionally after chronic agonist exposure. In marked contrast, although ICI-174864 demonstrated full inverse agonism in opioid naive cells, chronic DPDPE pretreatment significantly reduced this negative intrinsic activity by over half. Consequently, although this mechanism may play some role in the conversion of antagonists into inverse agonists after chronic opioid pretreatment, this simple hypothesis is insufficient to explain all of the present observations.
Second, chronic opioid agonist exposure has been shown to uncouple GPCRs from G proteins and their subsequent downstream signaling processes (Tao et al., 1993). Additionally, inverse agonists have a higher affinity for, and stabilize the inactive (R), uncoupled state of GPCRs (Costa et al., 1992). Therefore, it is also possible that the manifestation of inverse agonism observed after chronic agonist treatment is due to a preferential enhancement (or enrichment) in inverse agonist binding in response to receptor uncoupling. Interestingly, our data seem to support part, but not all, of this hypothesis. For example, chronic morphine pretreatment did not produce significant desensitization of δ-opioid receptor inhibition of adenylyl cyclase activity. This implies that prolonged morphine exposure apparently did not produce an uncoupling of δ-opioid receptors from G proteins and other downstream signaling processes. Therefore, it would be predicted that the affinity of inverse agonists for the uncoupled form of δ-opioid receptors would not change after this drug treatment. Indeed, it was demonstrated that the affinity of neither naloxone nor naltriben for the uncoupled state of δ-opioid receptors was altered after prolonged morphine exposure. Applying similar reasoning, because chronic morphine pretreatment did not produce receptor uncoupling or enhance the binding of the putative inverse agonists for uncoupled δ-opioid receptors, it would also be predicted that prolonged morphine exposure would not alter the intrinsic activity of naloxone or naltriben. In contrast, chronic pretreatment with either DPDPE or morphine converted these ligands into inverse agonists. Therefore, the ability of naloxone and naltriben to act functionally as inverse agonists after prolonged opioid agonist exposure cannot be explained solely by an enhancement of their affinity for uncoupled δ-opioid receptors in response to chronic treatment.
Regardless of the potential mechanisms underlying the present observations, two important implications can be inferred from data presented in the present study. First, the consequence of chronic agonist exposure on the intrinsic activity of drugs is highly dependent on the specific ligand being examined. For example, chronic opioid agonist treatment decreased the intrinsic activity of some ligands (DPDPE, ICI-174864, and BNTX), converted the intrinsic activity of some ligands from weak partial agonists into inverse agonists (naloxone and naltriben), or had no effect at all on the intrinsic activity of other ligands (N-NTD, NTD, and ICI-154129). This might be explained by observations that different ligands have been shown to bind to δ-opioid receptors in distinct manners (Befort et al., 1996). Therefore, if chronic agonist treatment produced subtle changes in receptor conformation, the quality and magnitude of alterations in the intrinsic activity produced by a drug after prolonged exposure would be determined by the unique manner in which that drug binds to the receptor.
Second, our data also suggest that chronic exposure to different agonists produces distinct cellular adaptations. For example, after both chronic DPDPE and morphine pretreatment, naloxone and naltriben displayed inverse agonist activity when measured by G protein activation. However, only chronic DPDPE resulted in an increase in the affinity of the ligands for the uncoupled form of δ-opioid receptors. It was recently shown that chronic exposure to [d-Ala2,N-MePhe4,Gly-ol5]-enkephalin or morphine produced distinctly different phosphorylated forms of μ-opioid receptors (Chakrabarti et al., 1998). Therefore, prolonged exposure to individual opioid ligands may produce uniquely phosphorylated forms of δ-opioid receptors, resulting in different active conformational states with distinct signaling properties.
Similar enhancement of the inverse agonist efficacy has been observed for ligands acting at β2-adrenergic receptors after chronic exposure to β-agonists (Chidiac et al., 1996). It was suggested in this study that in addition to the inactive (R) and active (R*) states of the β2-adrenergic receptor, chronic agonist exposure results in the formation of a distinct active conformation of the desensitized receptor (Rd*). Therefore, it was proposed that differences in the responsiveness between control and chronically treated membranes to ligands might reflect differences in the relative affinities of these drugs for the active versus the inactive state of each conformation of the receptor. This provides a general mechanism for agonist-induced modulation of inverse agonist efficacy, irrespective of the GPCR system being examined.
In summary, we have presented data demonstrating that the intrinsic activity of δ-, in addition to μ-opioid ligands, can be altered after chronic exposure to opioid agonists. This novel receptor adaptation might represent a common mechanism involved in the development of tolerance and/or dependence, and thus serve as an important target for potential future clinical interventions.
Footnotes
-
This work was supported in part by National Institute on Drug Abuse Grant DA10936 (to P.L.P.).
-
DOI: 10.1124/jpet.102.035964
- Abbreviations:
- GPCR
- G protein-coupled receptor
- GH3DORT8
- GH3 cells transfected with δ-opioid receptors
- NAL
- naloxone
- NTB
- naltriben
- [35S]GTPγS
- guanosine 5′-O-(3-[35S]thio)triphosphate
- DPDPE
- [d-Pen2,5]-enkephalin
- NTD
- naltrindole
- N-NTD
- N-benzylnaltrindole
- BNTX
- 7-benzylidenenaltrexone
- GppNHp
- 5′-guanylylimidodiphosphate
- ANOVA
- analysis of variance
- ICI-174864
- N,N-diallyl-Tyr-Aib-Aib-Phe-Leu-OH
- ICI-154129
- (N,N-bisallyl)-Tyr-Gly-Gly-ψ-(CH2S)-Phe-Leu-OH
- Received March 11, 2002.
- Accepted May 10, 2002.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}