


Abstract
D2-like dopamine receptors mediate functional changes via activation of inhibitory G proteins, including those that affect adenylate cyclase activity, and potassium and calcium channels. Although it is assumed that the binding of a drug to a single isoform of a D2-like receptor will cause similar changes in all receptor-mediated functions, it has been demonstrated in brain that the dopamine agonists dihydrexidine (DHX) andN-n-propyl-DHX are “functionally selective”. The current study explores the underlying mechanism using transfected MN9D cells and D2-producing anterior pituitary lactotrophs. Both dopamine and DHX inhibited adenylate cyclase activity in a concentration-dependent manner in both systems, effects blocked by D2, but not D1, antagonists. In the MN9D cells, quinpirole andR-(−)-N-propylnorapomorphine (NPA) also inhibited the K+-stimulated release of [3H]dopamine in a concentration-responsive, antagonist-reversible manner. Conversely, neither DHX, nor its analogs, inhibited K+-stimulated [3H]dopamine release, although they antagonized the effects of quinpirole.S-(+)-NPA actually had the reverse functional selectivity profile from DHX (i.e., it was a full agonist at D2L receptors coupled to inhibition of dopamine release, but a weak partial agonist at D2L receptor-mediated inhibition of adenylate cyclase). In lactotrophs, DHX had little intrinsic activity at D2 receptors coupled to G protein-coupled inwardly rectifying potassium channels, and actually antagonized the effects of dopamine at these D2receptors. Together, these findings provide compelling evidence for agonist-induced functional selectivity with the D2Lreceptor. Although the underlying molecular mechanism is controversial (e.g., “conformational induction” versus “drug-active state selection”), such data are irreconcilable with the widely held view that drugs have “intrinsic efficacy”.
The dopamine receptors, members of the G protein-coupled receptor superfamily, are generally divided into two classes, “D1-like” and “D2-like” (Jaber et al., 1996; Huff, 1997). In mammals, the D1-like family includes receptors cloned from two genes, D1 and D5. Both D1-like receptors are linked to the stimulation of adenylate cyclase and have similar ligand recognition characteristics, yet display markedly different patterns of expression in brain. Three genes that result in the expression of four functional receptors encode the D2-like family (D2, D3, and D4). These include two primary D2 splice variants D2Long (D2L) and D2Short (D2S), as well as the less abundant D3 and D4receptors. In molecular expression systems, these D2-like receptors often inhibit adenylate cyclase, although this effect is less common with D3 receptors. Transduction pathways other than those mediated by cAMP also are known to play a key role in the functional effects mediated by all of the dopamine receptors.
In the central nervous system, dopamine receptors exist on both dopamine neurons (on soma, dendrites, and terminals) and postsynaptically on target cells. Presynaptic receptors (autoreceptors) fall under the D2-like class of dopamine receptors, and serve to autoregulate dopamine synthesis, neuronal impulse activity, and terminal release (Bunney et al., 1973; Walters and Roth, 1976; Aghajanian and Bunney, 1977; Skirboll et al., 1979). Postsynaptic receptors (heteroreceptors) may be either D1-like or D2-like receptors, serving a variety of functions. In addition to multiple molecular isoforms of dopamine receptors, functional diversity is engendered by multiple G protein subunits (Simon et al., 1991; Clapham and Neer, 1993), as well as heterogeneity in the transduction systems affected by G proteins (e.g., numerous forms of adenylate cyclase are known; Sunahara et al., 1996).
Until recently, dogma has held that a single drug binding to a given receptor would have a single type of functional effect (e.g., agonist, partial agonist, or antagonist). The current work is a continuation of studies that may alter this tenet of pharmacology. Nearly a decade ago, we developed dihydrexidine, the first high-affinity, full dopamine D1 receptor agonist (Lovenberg et al., 1989). Our preliminary characterization of this drug revealed, unexpectedly, that DHX had D2 receptor affinity similar to the prototypical D2 receptor agonist quinpirole (Brewster et al., 1990; Mottola et al., 1992). Furthermore, DHX exhibited agonist actions at several functions generally accepted as being mediated by D2-like receptors on target cells. For example, DHX inhibited cAMP synthesis and efflux, inhibited prolactin release, and caused some behavioral changes attributable to D2 receptor agonism (Darney et al., 1991; Mottola et al., 1992). Surprisingly, however, DHX caused little or no inhibition of dopamine release and dopamine cell firing (Mottola et al., 2002), two functions that are generally ascribed to activation of D2 receptors expressed on dopamine neurons (Bowyer and Weiner, 1987). This suggested that DHX may lack agonist activity at D2 receptors coupled to K+ channels (White and Wang, 1984). Yet despite this apparent lack of functional intrinsic activity at autoreceptors, DHX was shown to bind to autoreceptors with identical affinity as to D2-like receptors on striatal target cells (Mottola et al., 2002).
Although these findings provide substantial support for the notion that DHX is “functionally selective”, the brain-based functional systems cannot prove this phenomenon unambiguously. The current work uses two different model systems to this end. One system was the MN9D line, cells that, like dopamine neurons, can synthesize dopamine and release it upon depolarization (O'Hara et al., 1996b). These cells do not express endogenous dopamine receptors, but transfected D2L receptors can couple to both inhibition of adenylate cyclase and inhibition of dopamine release (Tang et al., 1994a,b). The second system, pituitary lactotrophs, express only D2L and D2S receptors (Bunzow et al., 1988; Monsma et al., 1989), but not the D3 or D4 isoforms (Sokoloff et al., 1990; Landwehrmeyer et al., 1993; Levant et al., 1993). In lactotrophs, D2 dopamine receptors couple to at least two distinct effector systems: adenylate cyclase (Enjalbert and Bockaert, 1983) and G protein-coupled inwardly rectifying K+-channels (GIRKs) (Einhorn et al., 1991). Thus, in a single cell having a single dopamine receptor isoform, we sought to validate the functional selectivity caused by these drugs in brain tissue (Mottola et al., 2002). The data with these systems convincingly demonstrate that certain drugs, such as dihydrexidine, can have functional effects as diametrically opposed as full agonist in one functional measure, and antagonist in another, even when acting at the same receptor isoform.
Experimental Procedures
Materials
(±)-(DHX) [(6a,12b)-trans-10,11-dihydroxy-5,6,6a,7,8,12b-hexahydrobenzo[a]benzo[a]phenanthridine] and related analogs were synthesized using published methods (Brewster et al., 1990, 1995), and (+)-DHX was resolved as previously reported (Knoerzer et al., 1994). DHX analogs were used as racemic mixtures. Remoxipride was a gift from Astra Läkemüde (Hässle, Mölndal, Sweden). [3H]Spiperone was purchased from Amersham Biosciences (Piscataway, NJ). Quinpirole (LY171555), R-(+)-3-PPP, spiperone, butaclamol,S-(−)-eticlopride, R-(−)-NPA, andS-(+)-NPA were purchased from Sigma/RBI (Natick, MA). Isobutylmethylxanthine (IBMX), cyclic AMP standards, dopamine, pargyline, chlorpromazine, sucrose, tissue culture medium, and other standard laboratory chemicals were purchased from Sigma-Aldrich (St. Louis, MO). HEPES was purchased from Research Organics (Cleveland, OH). Primary antibody for cAMP assays was obtained from Dr. Gary Brooker (Georgetown University, Washington DC). Secondary rabbit anti-goat IgG, covalently linked to magnetic beads, was purchased from Advanced Magnetics (Cambridge, MA).
Cell Culture
MN9Ds.
The establishment of stably transfected MN9D-D2L cells has been described previously (Tang et al., 1994a). The cells were cultured according to these published methods. For membrane preparation, cells were grown in 75-cm2 flasks to approximately 90% confluence, and further processed as described below. Cells used for cyclic AMP accumulation and dopamine release assays were grown on poly-d-lysine-coated six-well plates to 90% confluence. Cells from passages 10 to 20 were used in these experiments.
Preparation and Electrophysiology of Primary Cultures of Rat Pituitary Lactotrophs
For each preparation, one to two adult female Sprague-Dawley rats (determined to be in proestrus phase by vaginal smear) were sedated with a 5:5:1 ketamine/acepromazine/xylazine i.m. injection, and humanely decapitated. Anterior pituitary lobes were extracted from isolated brains, minced, and rinsed in sterile Hanks' calcium- and magnesium-free balanced salt solution (HCMF) and incubated in HCMF containing 1 mg/ml each of collagenase (ca. 200 dU/mg) and DNase I at 37°C for 1 h. Tissue then was triturated with a silicon-treated, fire-polished Pasteur pipette and gravity filtered through sterile nylon mesh (20 μm) in a Swinnex holder. Cells were washed twice in HCMF, passed through 10-μm mesh, and suspended in Dulbecco's modified Eagle's medium containing 0.1% bovine serum albumin for use in the reverse hemolytic plaque assay (see below). Alternatively, cells were enriched for lactotrophs by passing through a discontinuous Percoll gradient. For cyclase assays, 50-μl aliquots of cells at a density of ca. 106 cells/ml were plated in 96-well sterile culture plates treated with 0.25 mg/ml poly-l-lysine and incubated at 37°C overnight. For electrophysiology, cells were plated on plastic coverslips coated with 0.25 mg/ml poly-l-lysine at a density of ca. 104 cells/ml and incubated for up to 3 days.
In addition to lactotroph enrichment by discontinuous Percoll gradient, some electrophysiology experiments used the reverse hemolytic plaque assay for unambiguous identification of lactotrophs. The method (Smith et al., 1986) uses an immunologically triggered complement lysis of bovine red blood cells in the periphery of the antigen-secreting cells. Both whole-cell and perforated patch recording methods were used to measure agonist-activated membrane currents, and followed the protocols outlined in Einhorn et al. (1991).
Membrane Preparation from Cultured MN9D Cells
Cells were rinsed once with 10 volumes per flask ice-cold distilled water, followed by incubation for 10 min at 4°C in cold distilled water. Flasks were scraped and rinsed once more with 5 volumes of distilled water, and the lysates transferred to a glass/Teflon Potter-Elvehjem tissue grinder. While on ice, the cells were homogenized using 10 strokes of the tissue grinder. Homogenates were then centrifuged at 50,000g at 4°C for 25 min, and the resulting pellet was resuspended in 1 volume of ice-cold homogenizing buffer (0.32 M sucrose and 2.5 mM Tris, pH 6.9) using 10 strokes of the homogenizer. Centrifugation and pellet disruption were repeated as described above, except that the final pellet was resuspended in 1 volume (1 ml/flask) of ice-cold storage buffer (0.32 M sucrose, pH 6.9). Membrane aliquots were stored at −70°C until use, when protein concentrations (Lewis et al., 1998) were adjusted to 2 mg protein/ml assay buffer.
Radioreceptor Binding in Cultured Cell Membranes
Competition binding studies were done according to Tang et al. (1994a), with the following modifications: Experiments with dopamine also contained 10 μM pargyline. After incubation, assays were terminated according to Watts et al. (1993) and analyzed using nonlinear regression with a sigmoid dose-response model (GraphPad Software, San Diego, CA).
cAMP Accumulation Assays in MN9D Cells
Cells grown in six-well culture plates were incubated for 5 min with serum-free medium + 500 μM IBMX at 37°C for 5 min. The cells were then aspirated, and incubated with 10 μM forskolin and varying concentrations of agonist and/or antagonist for 10 min. Cells were rinsed with the serum-free medium with IBMX, and then lysed by the addition of 0.1 M HCl. The wells were scraped to remove cells, and the soluble cAMP, collected, and centrifuged at 5000g for 5 min. An aliquot of the supernatant then was used for determination of cAMP levels by radioimmunoassay.
Measurement of Adenylate Cyclase Activity in Intact Lactotrophs
Two methods were used to elevate cAMP levels ca. 50- to 100-fold higher than basal levels: 1) forskolin (FSK; 100 μM) and 2) vasoactive intestinal peptide (VIP; 1 μM). Test drugs were dissolved in a vehicle consisting of warm Dulbecco's modified Eagle's salts, 10 μM ascorbic acid, and either FSK or VIP. In VIP studies, 0.5% bovine serum albumin and 500 μM IBMX were included. Cells were incubated at 37°C with either vehicle, or dopamine antagonist, for 10 min, after which vehicle or dopamine agonist was added to the medium for 15 min. Cells then were lysed with 200 μl of ice-cold 0.1 M HCl, collected, and wells rinsed once with an additional 200 μl of HCl. Lysates were spun at 14,000 rpm for 5 min, and the supernatants frozen at −20°C. The concentration of cAMP in each sample was determined via radioimmunoassay of acetylated samples following published protocols (Watts et al., 1993; Lewis et al., 1998).
Dopamine Release Assays
These studies were done essentially as described by O'Hara et al. (1996b) with the following modifications: Cells were incubated in buffer at 37°C for 5 min, as described by Mottola et al. (1992). After this, 25 nM [3H]dopamine was added for 5 min, aspirated, the cells washed three times with KRS, and then incubated with 1 ml of KRS or high K+ buffer (equivalent to KRS except without NaCl and containing 97.5 mM KCl), with or without test drugs. Samples were collected, centrifuged for 5 min at 1000g, and the radioactivity quantified by liquid scintillation spectroscopy. Stimulated [3H]dopamine release was defined as the difference in the amounts of [3H]dopamine released between KRS-treated and high K+buffer-treated cells.
Results
Ligand Affinity for Transfected D2 Receptors.
Several compounds of the hexahydrobenzo[a]phenanthridine family compete for [3H]spiperone-labeled binding sites in D2L receptor-transfected MN9D cells. Figure 1 displays representative competition curves for DHX and the two reference agonists quinpirole and dopamine. DHX and several of its analogs have affinities (K0.5) in the low micromolar range, similar to those of the two full agonists, quinpirole andR-(−)-NPA, and the endogenous ligand dopamine (Table1). The indirect Hill slopes for DHX and its analogs are less than unity (range of −0.65 to −0.85), and are similar to those obtained for quinpirole and dopamine.

Competition of DHX and reference compounds for [3H]spiperone binding to D2L receptors in MN9D cells. Cell membranes were incubated with 0.07 nM [3H]spiperone. Data are plotted as percentage of specific binding (total − nonspecific), where nonspecific binding is defined by 1 μM chlorpromazine. Each value represents the mean ± S.E.M. of data points, run in triplicate, from a single assay.
Affinity of dopamine receptor agonists for [3H]spiperone binding to D2L receptors transfected in MN9D cells
Agonists Inhibit cAMP Accumulation in MN9D-Transfected Cells.
D2L receptors expressed in MN9D cells are linked to inhibition of adenylate cyclase and inhibition of potassium-stimulated [3H]dopamine release. Figure 2A illustrates the effects of 10 μM test compounds on adenylate cyclase inhibition. Dopamine, quinpirole, and R-(−)-NPA produce greater than 50% inhibition of forskolin-stimulated cAMP accumulation. Likewise, DHX andN-Pr-DHX produced inhibition of adenylate cyclase comparable with that observed with dopamine and quinpirole. It is noteworthy thatS-(+)-NPA had lower intrinsic activity thanR-(−)-NPA, causing significantly less inhibition of adenylate cyclase. The inhibition of cyclase activity was blocked by both the nonselective DA antagonist butaclamol (data not shown), as well as the D2-selective antagonist spiperone (dopamine and DHX, Fig. 2A; N-Pr-DHX, data not shown).

Intrinsic activity of dopamine receptor agonists for inhibition of cAMP accumulation and [3H]dopamine release in D2L receptor-transfected MN9D cells. A, agonist inhibition of cAMP accumulation was measured in whole cells incubated with 10 μM forskolin to stimulate cAMP synthesis. B, agonist inhibition of dopamine release was measured in intact cells that were preloaded with 25 nM [3H]dopamine and stimulated with 100 mM potassium as described under Experimental Procedures. Data are expressed as percentage of stimulation by forskolin (for cAMP accumulation) or K+ depolarization (for [3H]dopamine release) in the absence of test compound. All compounds were tested at 10 μM (n = 3–5 independent experiments for each compound and functional measure). Full concentration-response curves for selected agonists are shown in Figs.3. Both agonist-induced inhibition of cAMP accumulation and dopamine release were blocked by addition of 10 μM spiperone. ∗, significantly different versus control (P < 0.05) by one-way analysis of variance and post hoc Tukey-Kramer multiple comparison test.
Dopamine Release in D2-Transfected MN9D Cells.
The functional effects of the DHX analogs on dopamine release are markedly different from their effects on adenylate cyclase (Fig. 2B). None of the DHX analogs produce measurable inhibition of dopamine release, whereas quinpirole and both enantiomers of NPA produce ca. 50% inhibition (with dopamine not being tested because of the type of assay used). Butaclamol (data not shown) and spiperone reversed the inhibition of dopamine release exhibited by R-(−)-NPA and quinpirole; similar antagonist blockade (using eticlopride) was observed forS-(+)-NPA-induced inhibition of release (data not shown). Notably, the absence of effects for DHX and N-Pr-DHX at dopamine release stands in remarkable contrast to the inhibition of adenylate cyclase activity by these two drugs.
The selective activity of DHX and its analogs for inhibition of adenylate cyclase is illustrated further in Fig.3A that shows full concentration-response curves for selected agonists at both functional measures. DHX andN-Pr-DHX display dose-dependent inhibition of forskolin-stimulated cAMP accumulation with a potency and a maximal effect similar to that achieved by the prototypical full D2 receptor agonist quinpirole (Fig. 3A, left; Table 2). In contrast, the concentration-response functions for DHX and N-Pr-DHX at [3H]dopamine release stimulated by potassium-induced depolarization are essentially flat (Fig. 3A, right; Table 2), whereas quinpirole displays concentration-dependent inhibition of release with a potency similar to that shown for adenylate cyclase inhibition. Interestingly, the two enantiomers of NPA actually reveal the opposite pattern of functional discrimination (Fig.3B). S-(+)-NPA displays near maximal inhibition of the [3H]dopamine release, yet produces only modest inhibition of adenylate cyclase. R-(−)-NPA inhibits both dopamine release and adenylate cyclase accumulation in a manner similar to the full agonist quinpirole, albeit with somewhat lower potency (Table 2).

Functional effects of DHX and N-Pr-DHX at D2L receptor-transfected MN9D cells. Agonist inhibition of cAMP accumulation was measured in intact cells incubated with 10 μM forskolin to stimulate cAMP synthesis. Agonist inhibition of dopamine release was measured in intact cells that were preloaded with 25 nM [3H]dopamine and stimulated with 100 mM potassium. Data are expressed as percentage of stimulation by forskolin (for cAMP accumulation) or K+ depolarization (for [3H]dopamine release) in the absence of drug. A, effects of quinpirole, DHX, and N-propyl-DHX. B, effects of quinpirole, R-(−)-NPA, and S-(+)-NPA. Each value represents the mean ± S.E.M. for a single representative assay conducted in triplicate (cAMP) or duplicate ([3H]dopamine release). Assays were repeated three to five times for both functional measures.
Agonist potency for D2L receptor-mediated functions in MN9D cells
Additional experiments were performed to determine whether DHX and its analogs possess antagonistic properties toward the dopamine release inhibitory effects that are exhibited by typical D2 agonists. Figure4 shows that DHX dose dependently reversed the inhibition of stimulated [3H]dopamine release caused by quinpirole; similar antagonistic effects were obtained for several other DHX analogs (data not shown).

Antagonist effects of DHX on quinpirole-induced inhibition of dopamine release in D2L receptor-transfected MN9D cells. Quinpirole inhibition of dopamine release was measured in intact cells preloaded with 25 nM [3H]dopamine and subsequently depolarized by exposure to 100 mM K+. Cells were incubated with 10 μM quinpirole in the presence or absence of varying concentrations of either DHX or butaclamol. Similar results, with average reversals of 50 to 70% of quinpirole inhibition, are exhibited by N-Pr-DHX, 4-Me-DHX, and 4-Me-N-Pr-DHX (data not shown). Each value represents the mean ± S.E.M. of data points, run in duplicate, from a single assay.
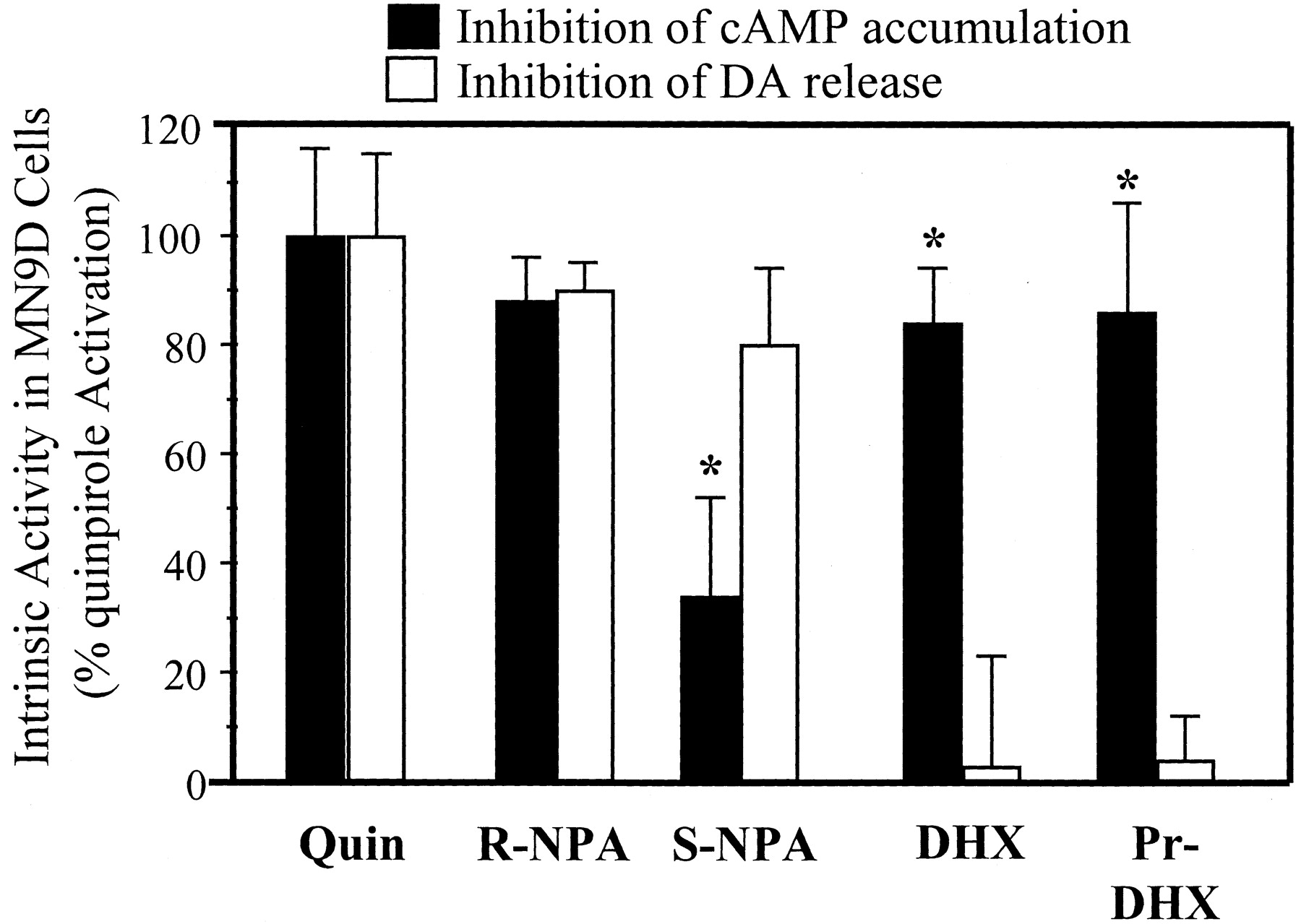
A summary of the observed agonist-specific patterns of effector activation is presented in Fig. 5. To provide a basis for comparison, the effects of all tested compounds are expressed relative to the effects of the full D2agonist quinpirole (10 μM). This method of presentation illustrates clearly that DHX and its analogs produce selective activation of D2L receptors linked to adenylate cyclase, whereas S-(+)-NPA favors the pathway linked to dopamine release.

Intrinsic activity of NPA, DHX, andN-Pr-DHX on two separate functional measures in D2L receptor-transfected MN9D cells. The maximal inhibitory effects of quinpirole (10 μM) were used to define full intrinsic activity for D2L receptor coupling to forskolin-stimulated cAMP synthesis and K+-stimulated release. Activities for all other agonists (10 μM) are expressed as a percentage of the effect of quinpirole. Each intrinsic activity calculation represents an average of at least three separate experiments. ∗, significantly different from same drug versus inhibition of dopamine release (P < 0.05).
Dihydrexidine and Dopamine Inhibit FSK- and VIP-Stimulated Adenylate Cyclase Activity in Lactotrophs.
Adenylate cyclase activity in isolated pituitary lactotrophs was increased markedly by the addition of 100 μM FSK or 1 μM VIP. Both DHX and DA inhibited FSK- and VIP-stimulated adenylate cyclase activity in a concentration-dependent manner (Fig. 6, A and B, respectively), although the maximal inhibition effect was different between FSK and VIP stimulation (for FSK, maximal inhibition of ca. 50%, whereas VIP was ca. 80%). The greater intrinsic activity of both DA and DHX to inhibit VIP-stimulated versus FSK-stimulated cAMP may reflect the mechanistic differences in VIP- versus FSK-mediated cyclase stimulation (a receptor-mediated stimulation of cAMP versus direct activation of the enzyme). Although DHX showed a somewhat lower potency than DA in the FSK-stimulated paradigm, no statistically significant difference was found. The D2-like selective antagonists (−)-sulpiride, (−)-eticlopride, and domperidone all blocked DA-mediated inhibition of FSK-stimulated cyclase (Fig. 6A). Likewise, the D2-like selective antagonist (−)-sulpiride significantly attenuated both the DA- and DHX-mediated inhibition of VIP-stimulated cAMP (Fig. 6B). The fact that complete blockade of the agonists effects was not observed probably reflects the known low D2 affinity of sulpiride relative to these agonists, and the very high concentrations of the agonists. In contrast, the D1-like selective antagonistR-(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine [R-(+)-SCH23390] had no effect on either DA- or DHX-mediated inhibition of adenylate cyclase, a result predicted by the fact that D1-like receptors are not coupled to inhibition of adenylate cyclase activity in these cells (Schoors et al., 1991).

Inhibition of forskolin-stimulated adenylate cyclase in primary lactotroph cultures by dopamine and DHX. Cells were treated with forskolin (A) or VIP (B) alone or in combination with DA or DHX (concentrations of 1 or 10 μM). Data are normalized to total cAMP synthesis versus cells treated only with forskolin or VIP (seeExperimental Procedures). For D2 antagonist treatment, cells were preincubated with either domperidone (30 μM; Dunnett's test, ∗, P < 0.05), (−)-eticlopride (100 μM; Student's t test, ∗∗,P < 0.001) or (−)-sulpiride (1 μM; unpaired Student's t test), before agonist treatment. The D1 antagonist SCH23390 (1 μM) produced no significant blockade of agonist-induced cyclase inhibition. Each bar represents from three to 10 independent replicates.
Dihydrexidine Has Little Intrinsic Activity at D2-Like Receptors Coupled to GIRK Channels in Lactotrophs.
DA potently activated GIRK currents as expected (Einhorn et al., 1991; Einhorn and Oxford, 1993), whereas DHX (at concentrations as high as 100 μM) had little intrinsic activity at D2-like receptors coupled to GIRK channels. Both summary and exemplary data illustrating this difference are shown in Fig. 7. Traces represent agonist activated K+-current (agonist − control records) in response to a voltage ramp over the indicated range. Dopamine (100 nM) activates a large inward current, whereas in the same cell DHX (10 μM) does not. Cumulative data for currents at −100 mV (bars) are shown for both standard whole-cell recordings (n = 12) or perforated patch recordings (n = 18) in single lactotrophs. No differences were seen in the results obtained using either recording method. Compared with a maximally effective concentration of DA in this paradigm (100 nM), DHX (pooled data from 10 and 100 μM trials) elicits potassium current responses less than one-fourth that of DA. Repetition of DA application to single cells results in identical current responses, thus the discrepancy in DHX response cannot be accounted for by desensitization of D2-like receptors coupling to GIRK channels or inactivation of GIRK channels. Drug application order was randomized revealing that the order of agonist application to a single cell did not alter the results. Finally, cells that did not respond to DA also showed no response to DHX (n = 21).

Intrinsic activity of dopamine agonists to activate GIRK channels in pituitary lactotrophs. Macroscopic (whole-cell) current responses to a 200-ms voltage ramp recorded during application of either DA or DHX. Data shown are subtracted current responses (drug − vehicle). The current is carried by K+ ions, as indicated by the reversal potential (current response = 0 at the calculated EK = −35 mV). The traces shown were recorded from a single lactotroph. The current amplitudes at −100 mV for drug responses in all cells were calculated and are indicated by bars on the graph (mean ± S.E.M.; n = 30; filled bar, DHX responses; striped bar, DA responses). ★★, p < 0.001 comparing current values for DA vs. DHX (paired Student'st-test).
Antagonist analysis for the partial agonist effects of DHX in this paradigm was difficult, because 65% of lactotrophs tested had an IK response to DHX ≤10 pA, a current magnitude that is difficult to distinguish unequivocally from baseline or that expected from a pure antagonist. Of cells that exhibited comparatively larger current responses to DHX, four cells tested had DA- and DHX-mediated IK reversibly antagonized by the D2-like selective antagonists (−)-sulpiride, remoxipride, and domperidone (n = 2, 1, and 1, respectively). An example of this antagonism is shown in Fig. 8A. The current traces again represent responses from a single cell challenged (in order) with DHX, DA, DHX + sulpiride, and DA + sulpiride. Clearly, D2 receptor antagonists block the action of both DA and DHX to activate GIRK currents.

Both D2 antagonist and DHX block dopamine-stimulated GIRK channel activity. A, D2-like selective dopamine receptor antagonists block both the dopamine and dihydrexidine-activated GIRK current response in pituitary lactotrophs. Current-voltage data from a single lactotroph illustrate that in the presence of (−)-sulpiride (30 μM), the GIRK responses to both DA and DHX are blocked reversibly (reversal data not shown). B, dihydrexidine competes with dopamine at D2-like receptors coupled to GIRK. Current-voltage data from a single lactotroph illustrate that in the presence of the full agonist DA DHX behaves as an antagonist, as is expected for a low intrinsic activity partial agonist interacting with the same receptor (n = 6).
DHX is known to have full intrinsic activity at D1 receptors (Brewster et al., 1990), and it is conceivable that the inability to observe significant GIRK activation reflects a functional “masking” of an effect by a compensatory action mediated by trace amounts of D1-like receptors expressed on the lactotrophs (Schoors et al., 1991). To explore this possibility, we assessed the effect of the D1-like selective antagonist SCH23390 on DHX responses. SCH23390 had no effect on either DA- or DHX-mediated GIRK currents (n = 5; data not shown), further supporting the view that DHX has little intrinsic activity at D2 receptors coupled to GIRK.
As shown in Fig. 8B, the addition of DHX (1 μM) + DA (100 nM) yielded a current response much smaller than observed for DA alone (n = 11). Again, application of a comparable concentration of DHX alone failed to elicit a significant current. In addition, when a very high concentration of DHX (100 μM) was applied before DA application alone, the DA current response was not evident for several minutes. DA recovery proceeded in a graded manner over a 6- to 10-min washout period (data not shown). This further supports a reversible, surmountable interaction between DA and DHX at the same binding site (i.e., a D2 receptor).
Discussion
In the current study, we have used three independent measures of D2 receptor function: the inhibition of forskolin-stimulated cAMP accumulation, the inhibition of potassium-stimulated dopamine release, and activation of GIRK channels, to explore the pharmacological properties exhibited by DHX and its analogs at D2 receptors. The results presented herein indicate that DHX and its analogs exhibit unique D2 functional characteristics. Like dopamine and quinpirole, DHX and N-Pr-DHX inhibit forskolin-stimulated cAMP accumulation with high intrinsic activity in both MN9D-transfected cells and pituitary lactotrophs. Yet unlike quinpirole, neither DHX nor its analogs exhibited significant intrinsic activity at D2 receptors either linked to inhibition of potassium-stimulated dopamine release, or to activation of GIRK channels. Moreover, in these latter assays in both MN9D cells and lactotrophs, DHX inhibited the effects of quinpirole or dopamine in a dose-dependent manner, consistent with the action of DHX as a competitive antagonist at these functions.
These results are provocative and invite speculation about the factors that might create such a pattern of selective effector activation. Four obvious possibilities can be identified: receptor subtype heterogeneity, indirect effects or effects mediated by nondopamine receptors, sequential response amplification, and graded strength-of-agonist stimulus. Of these explanations, receptor heterogeneity can be ruled out most easily: dopamine receptors are not expressed endogenously in MN9D cells (Tang et al., 1994a), only the D2L receptors were present in these cells, and D2 antagonists had the predicted effects. Pituitary lactotrophs express only D2 dopamine receptors (Bunzow et al., 1988), but not D3 or D4 isoforms (Sokoloff et al., 1990; Landwehrmeyer et al., 1993; Levant et al., 1993). Both D2S and D2L have been shown to couple functionally to adenylate cyclase activity and to GIRK activity in transfected cell systems (Malek et al., 1993; Werner et al., 1996; Kuzhikandathil et al., 1998). Although DHX has equal affinity for D2S and D2L receptors, DHX has high intrinsic activity at D2-coupled adenylate cyclase, but antagonist activity (very low intrinsic activity) at D2-coupled GIRK channels. It is possible that native D2S and D2L receptors expressed in the lactotroph bind DHX differentially, yet if this were the case, each receptor isoform would need to couple exclusively with only one effector (cyclase versus GIRK channels) to explain the effector-selective actions of DHX. The overwhelming evidence (in transfected cells) is that both D2S and D2L receptors can alter adenylate cyclase, as well as GIRK channel activity, when activated by conventional D2 agonists. Thus, differences in affinity for D2 isoforms cannot account for the effector selectivity of DHX. The possibility of either indirect agonist effects, or actions at other receptors occurring in MN9D cells or lactotrophs, was ruled out in Mottola et al. (1992).
A third explanation that should be considered relies on the response amplification that can occur when measuring sequential responses. If two cellular responses occur at different positions in a sequential cascade then the cumulative effect of hyperbolic stimulus-response mechanisms at each step results in amplification of later responses (Kenakin, 1997). This amplification would benefit agonists of low intrinsic efficacy, with the result that they may produce a measurable effect at the second, but not the first, response element. This explanation might apply to the present data obtained with DHX if dopamine release or activation of GIRK channels initiated inhibition of adenylate cyclase. There is, however, little evidence to support a simple sequential dependence between these events (Memo et al., 1986;Starke et al., 1989; Vallar and Meldolesi, 1989; Hille, 1994; Takano et al., 1994; Zelles et al., 1995). More importantly, this explanation cannot accommodate the reversal of selectivity patterns exhibited by DHX and S-(+)-NPA in the MN9D system, or the similar potencies observed with the typical D2 receptor agonists quinpirole and R-(−)-NPA at the two effector pathways studied.
A final explanation that must be considered more carefully involves possible inherent differences in the competency of D2L receptor coupling to multiple pathways in MN9D cells. In the present case, if D2L receptors coupled more efficiently to adenylate cyclase than to dopamine release, agonists with low intrinsic efficacy would exhibit an apparent functional selectivity (i.e., they would induce a signal sufficient only for activating efficiently coupled pathways). Two aspects of our data (vide infra) again argue against this hypothesis. First, similar potencies were measured for the D2 agonists quinpirole and R-(−)-NPA for inhibition of dopamine release and adenylate cyclase. This finding would be unexpected if large inherent differences in stimulus-response coupling existed (Kenakin, 1995a). Second, S-(+)-NPA more effectively inhibited dopamine release than adenylate cyclase, the opposite pattern from that of DHX and N-Pr-DHX. Such a reversal of effector selectivity implies that the functional selectivity we observed derives from drug- not cell-specific parameters.
The failure of the aforementioned hypotheses to provide a satisfactory explanation of the present findings prompts consideration of alternative schemes. Our interpretation of these data derives from what we have termed the functional selectivity hypothesis outlined previously (Mailman et al., 1998; Lawler et al., 1999). This hypothesis is derived in part from the promiscuity of receptor-G protein effector relationships. The most critical assumption of our model, however, is that there are differences among ligands in the conformational effects induced after these ligands bind to the receptor, and that these receptor conformations can differ qualitatively in their ability to serve as a signal for activating specific G proteins. In the present case, the endogenous neurotransmitter dopamine and other typical D2 receptor agonists such as quinpirole are viewed as inducing one or more “versatile” conformations that are sufficient to activate multiple G proteins. In contrast, certain atypical ligands [e.g., DHX and S-(+)-NPA] induce unique conformations that are favorable for activating only a subset of available G proteins. From this perspective, the functional effects of agonist-receptor interaction are determined not only by the second messenger system/G protein complement with which the receptor is associated but also by the agonist-specific three-dimensional changes within the receptor that occur upon binding of the ligand. By these criteria, functional selectivity conforms to recent G protein-coupled receptor models that seek to reconcile selective agonist activity (Kenakin, 1995b; Leff et al., 1997). There are, however, key features of the functional selectivity hypothesis that differ from these models (vide infra).
Nearly all of the effector pathways linked to D2receptor signal transduction have been associated with the Gαi family of G proteins, which includes Gαi1–3, GαoA-D, and Gαz. Within this sphere, however, the cellular constitution in which the receptor is expressed exerts some coupling variability (Huff, 1997). In many cell types and tissues, there is significant evidence that adenylate cyclase coupling to the D2L receptor commonly segregates with Gαi2 (Albert et al., 1990; Montmayeur et al., 1993; Liu et al., 1994; Guiramand et al., 1995). Furthermore, O'Hara et al. (1996a) have provided direct evidence for a Gαi2 role in D2Lreceptor-mediated inhibition of adenylate cyclase in MN9D cells. Using pertussis toxin-insensitive mutants, they demonstrated rescue of the D2L receptor inhibition of adenylate cyclase by pertussis toxin-insensitive Gαi2 but not Gαo. The identity of the G protein pathways mediating inhibition of DA release by D2receptors in MN9D cells is less clear. Multiple pathways have been identified for the modulation of neurotransmitter release, including inhibition of Ca2+ channels, activation of K+ channels, and regulation of vesicle release complexes (Miller, 1998). In a variety of cell types possessing D2 receptors, membrane-delimited calcium channel inhibition has been found to be the result of the activation of the G protein Gαo (Baertschi et al., 1992; Wolfe and Morris, 1999; Wolfe et al., 1999). The βγ subunits of these G proteins play a significant role in Ca2+ channel modulation (Garcia et al., 1998; Meza and Adams, 1998). In addition, recent studies have demonstrated a direct role for Gαo in inhibiting the ATP-dependent priming reaction of exocytosis, thus interfering with catecholamine secretion (Gasman et al., 1997).
Of the K+ currents that are modulated by D2 receptors, the inwardly rectifying channels GIRK2 and GIRK3 are of particular interest. These channels predominate in rat midbrain, and couple to D2 receptors in this tissue and in many clonal cell lines (Kim et al., 1995; Dascal, 1997). The activation of GIRK currents by G proteins is membrane-delimited, and is mediated by the βγ subunits of the Gαi2 and Gαi3 subtypes (Dascal, 1997). All D2-like receptors (D2, D3, and D4 receptors) couple to GIRK activity presumably through Gβγ (Clapham and Neer, 1993; Werner et al., 1996). The pituitary lactotroph expresses Gαs, Gαi1, Gαi2, Gαi3, Gαo, as well as two distinct Gβ subunits (Cussac et al., 1993). Thus, the G protein heterogeneity necessary to support a mechanism for differential agonist activity by receptor-G protein precoupling certainly is present in the lactotroph. This explanation, although an attractive hypothesis, may be oversimplified. For example, not only is receptor-mediated GIRK activation believed to occur preferentially through Gβγ dimers but also GIRK channels may be largely indiscriminate with respect to Gβγisoforms (Wickman et al., 1994). Thus, one might expect that any agonist that triggers dissociation of inhibitory heterotrimeric G proteins (including DHX-triggered inhibition of adenylate cyclase) would yield a pool of Gβγ dimers available to activate GIRK. In like manner, if D2 agonists interact with Gαi2 (or any other G protein) to inhibit adenylate cyclase via α subunit actions in the MN9D cells then one might expect that the βγ units released would lead necessarily to either activation of K+, or inhibition of Ca2+ channels, and a corresponding inhibition of dopamine release. The results obtained with DHX indicate, however, that these events are not inexorably linked in either lactotroph or MN9D cells. This lack of linkage may reflect selectivity of release-associated channels for particular βγ subunits and/or compartmentalization of receptors and their effectors, such that βγ subunits released from the adenylate cyclase effector pathway are not in proximity to the relevant proteins controlling dopamine release or GIRK channel activation.
Any or all of the mechanisms mentioned above may be used by D2 receptors to modulate dopamine release in MN9D cells. It should be noted that the D2-mediated inhibition of K+-stimulated release in these cells is sensitive to the application of nonselective K+ channel inhibitors (Tang et al., 1994b), suggesting that K+ currents figure prominently in this cascade. Gαi2, GαoA, GαoB, and Gαz have been identified in MN9D cells (Tang et al., 1994a; O'Hara et al., 1996a), and there is a reasonable basis for the hypothesis that distinct G proteins or G protein subunits mediate D2L receptor effects on dopamine release and adenylate cyclase in MN9D cells. Gαi2 is known to mediate adenylate cyclase inhibition, whereas Gαo or Gαi is capable of mediating D2 receptor effects on dopamine release. Development of tentative schema for G protein-effector segregation requires qualitative and quantitative assessment of the ability of various agonists to activate specific G proteins.
Our findings provide the first clear demonstration of functional selectivity at dopamine receptors. There is ample evidence to indicate that a similar phenomenon occurs for a variety of G protein-coupled receptors (Spengler et al., 1993; Chabre et al., 1994; Gettys et al., 1994; Gurwitz et al., 1994; Journot et al., 1994, 1995; Robb et al., 1994; Berg et al., 1998; Brink et al., 2000). These previous findings can be accommodated easily by the functional selectivity hypothesis. Two previous models that have been proposed and can also reconcile such phenomena are that of “agonist trafficking” (Kenakin, 1995b) and the “three-state model” (Leff et al., 1997); these differ primarily in the number of required receptor conformational states for activation of effector pathways (Berg et al., 1998). Neither the present data nor any extant data provide a means for favoring one of these three models, because all describe ligand-specific effector signaling through a single receptor. The functional selectivity mechanism that we maintain, however, bears a significant distinction with respect to the origin of selective activation. Kenakin (1995b) and Leff et al. (1997) have proposed models that seem to link selective activation to the conformations of the receptor presented to the ligand. In these systems, the active states of the receptor exist before interaction with the ligand, and are merely stabilized by the binding of the ligand. This “conformational selection” theory proposes that apparent agonist-induced states are very rare, but natural unliganded receptor conformations are not, although it was acknowledged that the data could not definitively rule out conformational induction (Kenakin, 1995a). In our view, however, functional selectivity posits that control of selective activation lies with the structure of the ligand. Upon binding to the receptor/G protein complex, functionally selective drugs actuate a conformational change in the receptor that may or may not result in G protein activation (“conformational induction”). It is not required that the receptor exist naturally in several active states, although the presence of active states resulting from energy landscapes (Kenakin, 1995b, 1997) is not excluded. Rather, it is assumed only that the receptor can adopt different conformations based on the structure of the ligand in the binding pocket (Fig.9, see schematic). Unfortunately, evaluating the merits of these ideas must await the development of methodologies that discriminate between ligand initiation and maintenance of receptor conformations.

Model illustrating a possible mechanism to explain the functional selectivity hypothesis. For parsimony, it is assumed that the “functional complex” is made only of the D2receptor (D2R) and various G proteins, independent of effector system (although the same model works equally well if the functional complex includes the effector). As shown in A and C, we hypothesize that dopamine (and other “typical” agonists) will cause activation of all functional complexes for the D2L isoform. This is illustrated by the binding of dopamine (dark rectangle) to the D2 receptor, followed by dissociation of the G protein heterotrimers. The result is inhibition of both adenylyl cyclase (A) and activation of GIRK (C) due to G protein activation. Conversely, when DHX (stippled trapezoid) binds to the D2 receptor, it causes a different conformational change in the receptor depending upon the functional complex of which the receptor is a component. The consequence of this is that activation of the adenylyl cyclase-linked G protein (B), but not the release-regulatory or GIRK-related G protein (D), occurs. This model also predicts that DHX would still occupy the receptor linked to the release-regulatory or GIRK-coupled receptor, and thus would cause competitive antagonism.
In summary, the present data provide an unambiguous demonstration of the ability of DHX and other hexahydrobenzo[a]phenanthridines to activate selectively specific effectors linked to a single receptor subtype. These data provide a basis for interpreting the unusual functional effects of these ligands observed in brain (Mottola et al., 2002), and may also explain the rather different behavioral profile of this class of drugs at D2-like receptors (Darney et al., 1991; Smith et al., 1997). The existence of ligands that are functionally selective within a specific receptor subtype, based upon the receptor's environment and cellular location, could be of great pharmacological utility in the treatment of disease states. The ability to activate selectively one signaling pathway or effector system over another would greatly reduce the complexities inherent in drug treatment, and potentially alleviate unwanted side effects (Lawler et al., 1999).
Footnotes
-
This work was supported by National Institutes of Health Grants MH-53356, MH-40537, MH-42705, and NS-18788, by Training Grants DA-07244, ES-07126, and MH-14277, and by Center Grants HD-01130 and MH-03327. Portions of this work have been presented in abstract form during the Annual Meeting of the Society of Neuroscience(1994–1996).
-
J.D.K. and H.S.C. contributed equally to this work.
- Abbreviations:
- DHX
- dihydrexidine, [(±)-trans-10,11-dihydroxy-5,6,6a,7,8,12b-hexahydrobenzo[a]phenanthridine]
- GIRK
- G protein-coupled inwardly rectifying potassium channel
- 3-PPP
- 3-(3-hydroxyphenyl)-N-n-propylpiperidine
- IBMX
- isobutylmethylxanthine
- NPA
- N-propylnorapomorphine
- HCMF
- Hanks' calcium- and magnesium-free balanced salt solution
- FSK
- forskolin
- VIP
- vasoactive intestinal peptide
- KRS
- Krebs-Ringer solution
- DA
- dopamine
- N-Pr-DHX
- N-n-propyldihydrexidine (trans-10,11-dihydroxy-6-n-propyl-5,6,6a,7,8,12b-hexahydrobenzo[a] phenanthridine)
- LY171555
- quinpirole
- Received August 21, 2001.
- Accepted March 5, 2002.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}