


Abstract
We evaluated the potential of the Na+- and Cl--coupled amino acid transporter ATB0,+ as a delivery system for amino acid-based prodrugs. Immunofluorescence analysis indicated that ATB0,+ is expressed abundantly on the luminal surface of cells lining the lumen of the large intestine and the airways of the lung and in various ocular tissues, including the conjunctival epithelium, the tissues easily amenable for drug delivery. We screened a variety of β-carboxyl derivatives of aspartate and γ-carboxyl derivatives of glutamate as potential substrates for this transporter using heterologous expression systems. In mammalian cells expressing the cloned ATB0,+, several of the aspartate and glutamate derivatives inhibited glycine transport via ATB0,+. Direct evidence for ATB0,+-mediated transport of these derivatives was obtained in Xenopus laevis oocytes using electrophysiological methods. Exposure of oocytes, which express ATB0,+ heterologously, to aspartate β-benzyl ester as a model derivative induced inward currents in a Na+- and Cl--dependent manner with a Na+/Cl-/aspartate β-benzyl ester stoichiometry of 2:1:1. ATB0,+ transported not only the β-carboxyl derivatives of aspartate and the γ-carboxyl derivatives of glutamate but also valacyclovir, which is an α-carboxyl ester of acyclovir with valine. The transport of valacyclovir via ATB0,+ was demonstrable in both heterologous expression systems. This process was dependent on Na+ and Cl-. The ability of ATB0,+ to transport valacyclovir was comparable with that of the peptide transporter PEPT1. These findings suggest that ATB0,+ has significant potential as a delivery system for amino acid-based drugs and prodrugs.
ATB0,+ is a Na+- and Cl--coupled amino acid transporter that is energized by transmembrane gradients of Na+ and Cl- as well as by the membrane potential (Palacin et al., 1998; Ganapathy et al., 2001, 2003). It belongs to the neurotransmitter transporter gene family (SLC6) (Chen et al., 2003). Among the currently known mammalian amino acid transporters, ATB0,+ is the only transporter with a very broad substrate specificity that is driven by a Na+ gradient and a Cl- gradient. This transporter recognizes neutral as well as cationic amino acids as substrates. In addition to the surprisingly broad substrate specificity of ATB0,+ with regard to the naturally occurring l-amino acids, this transporter can also transport various d-amino acids (Hatanaka et al., 2002), nitric-oxide synthase inhibitors (Hatanaka et al., 2001), and carnitine and its esters (Nakanishi et al., 2001). Unlike most other amino acid transporters that exhibit a broad expression pattern, Northern blot analysis has revealed that ATB0,+ transcripts are detectable primarily in the colon, lung, and mammary gland (Sloan and Mager, 1999).
The fact that ATB0,+ recognizes neutral and cationic amino acids, but not anionic amino acids as substrates, indicates that the presence of a negative charge in the side chain of amino acids prevents their recognition by the transporter as substrates. The presence of an uncharged side chain or a positively charged side chain does not interfere with the recognition as evidenced from the transport of neutral and cationic amino acids by the transporter. This is further exemplified by the findings that although aspartate and glutamate are not substrates for ATB0,+, the amide derivatives of these anionic amino acids, namely, asparagine and glutamine, are excellent substrates. Therefore, we hypothesized that if the β-carboxyl group of aspartate and the γ-carboxyl group of glutamate are modified with substitutions such that the resulting products do not have a negatively charged side chain, such modified amino acids may be recognized by ATB0,+ as substrates. If this hypothesis is correct, it might be feasible to design prodrugs in the form of side chain derivatives of aspartate and glutamate that would be recognized as substrates by ATB0,+ to facilitate the delivery of drugs into cells. We provide evidence in this article in support of the feasibility of this approach. In addition, our results show evidence in support of even a broader applicability of this strategy than originally hypothesized. Drugs that are derivatized as the α-carboxyl esters of neutral amino acids are also recognized as substrates by ATB0,+. This broadens the spectrum of amino acid-based prodrugs that can be designed for delivery into cells via this transporter. Because the utility of any transporter as a drug delivery system is influenced by its tissue distribution, we investigated whether the distribution of ATB0,+ was suitable to provide effective drug delivery. These studies have shown that ATB0,+ is expressed abundantly on the luminal surfaces of the large intestine and bronchi and in the conjunctival epithelium. This tissue distribution pattern lends support to our hypothesis that ATB0,+ is potentially very useful for the delivery of amino acid-based prodrugs in the form of, for example, oral pills and rectal suppositories, aerosol, and eye drops.
Materials and Methods
Materials. [2-3H]Glycine (specific radioactivity, 30 Ci/mmol) and [8-3H]valacyclovir (specific radioactivity, 5 Ci/mmol) were purchased from Moravek Biochemicals (Brea, CA). Valacyclovir was from GlaxoSmithKline (Research Triangle Park, NC). Acyclovir, l-aspartate, l-asparagine, and various β-carboxyl derivatives of aspartate were purchased from Sigma-Aldrich (St. Louis, MO). Similarly, l-glutamate, l-glutamine, and various γ-carboxyl derivatives of glutamate were also purchased from Sigma-Aldrich. N-α-Benzyloxycarbonyl-l-glutamic acid α-benzyl ester (Z-Glu-OBzl) was obtained from Novabiochem (San Diego, CA). 1-Ethyl-3-(3′dimethylaminopropyl)carbodiimide, palladium 10% (w/v) on activated carbon (Pd/C), 4-dimethylaminopyridine, dimethylformamide, and CH2Cl2 were purchased from Aldrich Chemical Co. (Milwaukee, WI).
Immunofluorescent Localization of ATB0,+in Mouse Colon, Lung, and Eye. A polyclonal antibody, raised against the peptide TDHEIPTISGSTKPE corresponding to the C-terminal region of mouse ATB0,+ (amino acid position 624-637), was used for immunofluorescent detection of the protein in mouse tissues. The specificity of the antibody has been described previously (Hatanaka et al., 2002). Immunolocalization was carried out as described previously (Smith et al., 1999; Bridges et al., 2000; Ola et al., 2001). Cryosections (10 μm in thickness) of the tissues, fixed with ice-cold acetone, were blocked with 10% normal goat serum. These sections were incubated with the primary antibody for 3 h at room temperature at a dilution of 1:50 followed by an overnight incubation at 4°C. Incubation with 0.1% normal rabbit serum (preimmune) or with buffer only served as negative controls. After rinsing, all sections were incubated at 4°C with a fluorescein isothiocyanate-conjugated AffiniPure goat anti-rabbit IgG antibody at a dilution of 1:100. The sections were then optically sectioned (z series) using an MRC-600 laser scanning confocal imaging system (Bio-Rad, Hercules, CA). Images were analyzed using the COSMOS software package (Bio-Rad). Additional sections were subjected to hematoxylin-eosin staining.
Functional Expression of ATB0,+in Human Retinal Pigment Epithelial (HRPE) Cells. The mouse ATB0,+ cDNA (Hatanaka et al., 2001; Nakanishi et al., 2001) was used for functional expression in HRPE cells in the analysis of its role in the transport of amino acid-based prodrugs. The vaccinia virus expression system was used for this purpose (Hatanaka et al., 2001, 2002). This procedure involves infection of the cells with a recombinant vaccinia virus carrying the gene for T7 RNA polymerase, followed by lipofectin-mediated transfection of the cells with plasmid DNA in which the cDNA insert is under the control of T7 promoter. Glycine was used as the substrate for ATB0,+ (Hatanaka et al., 2001, 2002; Nakanishi et al., 2001). Transport of 10 μM glycine (radiolabeled glycine, 0.05 μM; unlabeled glycine, 9.95 μM) in cDNA-transfected cells was measured at 37°C for 30 min. The transport was linear under these conditions. The transport buffer was 25 mM Hepes/Tris (pH 7.5) containing 140 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 0.8 mM MgSO4, and 5 mM glucose. Interaction of various amino acid-derivatives and amino acid-based prodrugs with the transporter was assessed by monitoring the ability of these compounds to inhibit ATB0,+-mediated glycine transport. Transport of 5 μM valacyclovir (radiolabeled valacyclovir, 1 μM; unlabeled valacyclovir, 4 μM) via ATB0,+ was monitored directly by measuring its uptake. Transport measurements were made in parallel in vector-transfected cells and in ATB0,+ cDNA-transfected HRPE cells to account for endogenous valacyclovir transport activity. The ATB0,+-specific transport of valacyclovir was determined by subtracting the transport values measured in vector-transfected cells from the transport values measured in cDNA-transfected cells. The dependence of ATB0,+-mediated transport of valacyclovir on Na+ was determined by comparing the transport values measured in the presence of Na+ and in the absence of Na+ (N-methyl-d-glucamine chloride replacing NaCl isoosmotically). The dependence of the transport process on Cl- was determined by comparing the transport values measured in the presence of Cl- and in the absence of Cl- (sodium gluconate replacing NaCl isoosmotically and also potassium gluconate and calcium gluconate replacing KCl and CaCl2, respectively, at equimolar concentrations).
Transport of valacyclovir via PEPT1 was measured in HRPE cells expressing the human PEPT1 cDNA (Liang et al., 1995) heterologously. Because PEPT1 is a H+-coupled transporter (Ganapathy and Leibach, 1991; Leibach and Ganapathy, 1996), the transport of 5 μM valacyclovir (radiolabeled valacyclovir, 1 μM; unlabeled valacyclovir, 4 μM) via this transporter was monitored with the transport buffer that contained 25 mM MES/Tris (pH 6), 140 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 0.8 mM MgSO4, and 5 mM glucose. Transport measurements were made at 37°C with 15-min incubation to represent initial transport rates. Transport measurements were made in parallel in vector-transfected cells and in cDNA-transfected cells to correct for endogenous transport activity in determining the PEPT1-specific valacyclovir transport.
Functional Expression of ATB0,+in Xenopus laevis Oocytes. Capped cRNA from the cloned mouse ATB0,+ cDNA was synthesized using the mMESSAGE mMACHINE kit (Ambion, Austin, TX). Mature oocytes (stage IV or V) from X. laevis were isolated by treatment with collagenase A (1.6 mg/ml), manually defolliculated, and maintained at 18°C in modified Barth's medium supplemented with 10 mg/ml gentamycin as described previously (Hatanaka et al., 2001, 2002; Nakanishi et al., 2001). On the following day, oocytes were injected with 50 ng of cRNA. Water-injected oocytes served as controls. The oocytes were used for electrophysiological studies 6 days after cRNA injection. Electrophysiological studies were performed by the two-microelectrode voltage-clamp method (Parent et al., 1992). Oocytes were perfused with a NaCl-containing buffer (100 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 3 mM Hepes, 3 mM MES, and 3 mM Tris, pH 7.5), followed by the same buffer containing different amino acid derivatives. The membrane potential was clamped at -50 mV. The differences between the steady-state currents measured in the presence and absence of substrates were considered as the substrate-induced currents. In the analysis of the saturation kinetics of substrate-induced currents, the kinetic parameter K0.5 (i.e., the substrate concentration necessary for the induction of half-maximal current) was calculated by fitting the values of the substrate-induced currents to Michaelis-Menten equation. The Na+-and Cl- activation kinetics of substrate-induced currents were analyzed by measuring the substrate-specific currents in the presence of increasing concentrations of Na+ (the concentration of Cl- kept constant at 100 mM) or in the presence of increasing concentrations of Cl- (the concentration of Na+ kept constant at 100 mM). In these experiments, the composition of the perfusion buffer was modified to contain 2 mM potassium gluconate, 1 mM MgSO4, and 1 mM calcium gluconate in place of KCl, MgCl2, and CaCl2, respectively. The data from these experiments were analyzed by the Hill equation to determine the K0.5 values for Na+ and Cl- (i.e., the concentrations of Na+ and Cl- necessary for half-maximal activation) and the Hill coefficient (nH; the number of Na+ and Cl- ions involved in the activation process). The kinetic parameters were determined using the commercially available computer program Sigma Plot, version 6.0 (SPSS Science, Inc., Chicago, IL).
Data Analysis. Experiments with HRPE cells were repeated three times with three independent transfections and transport measurements were made in duplicate in each experiment. Electrophysiological measurements of substrate-induced currents were repeated at least three times with separate oocytes. The data are presented as means ± S.E. of these replicates.
Synthesis of Acyclovir Glutamic Acid γ-Ester. Acyclovir (0.888 mmol) and Z-Glu-OBzl (2.22 mmol) were dissolved in dimethylformamide (6 ml); 1-ethyl-3-(3′dimethylaminopropyl)carbodiimide (426 mmol) and 4-dimethylaminopyridine (2.22 mmol) were then added to the solution. The solution was stirred for 18 h at room temperature. Dimethylformamide was removed in vacuo, and the residue was chromatographed on NH silica gel (Chromatorex DM-1020; Fuji Silysia Chemical LTD., Japan) using 1:10 to 1:5 methanol-CH2Cl2 as the eluent to generate acyclovir Z-Glu-OBzl γ-ester. This compound was then dissolved in ethanol/methanol/CH2Cl2 [1:1:1 (v/v)], and 20 mg of Pd/C was added to the solution. The mixture was stirred under hydrogen for 3 days. The mixture was then filtered to remove the catalyst (Pd/C) followed by removal of the solvent in vacuo. The product (acyclovir Glu γ-ester) was a white amorphous solid (120 mg) with an overall yield of 38%. The compound was analyzed by 1H NMR and electrospray ionization-mass spectrometry. The purity of the final product was confirmed by reverse-phase high-performance liquid chromatography using a Dionex analytical high-performance liquid chromatography system (Dionex Corp., Sunnyvale, CA) with an analytical column (Cyclobond I 2000, 4.6 × 100 cm; Advanced Separation Technologies Inc., Whippany, NJ). The purity of the compound was 96%. The molecular weight of the compound was determined by electrospray ionization-mass spectrometry (Finnigan Mat LC-Q; Thermo Finnigan, San Jose, CA).
Results
Tissue Expression Pattern of ATB0,+ For any transporter to be useful as a drug delivery system, it should be expressed in tissues amenable for drug delivery. Northern analysis has already shown that ATB0,+ transcripts are detectable in colon and lung (Sloan and Mager, 1999), two of the tissues that are suitable for drug delivery. We have recently cloned this transporter from a mouse colon cDNA library (Hatanaka et al., 2001, 2002; Nakanishi et al., 2001). We have also shown that this transporter is expressed on the luminal membrane of the colonocytes (Hatanaka et al., 2002). We confirm this finding in the present study (Fig. 1). We have also determined the localization of ATB0,+ in the lung. The transporter is expressed most abundantly in the cells lining the bronchi, although the expression is evident in other sites in the lung as well (Fig. 1). Recent studies localizing ATB0,+ in airway epithelial cells (Sloan et al., 2003) support our findings. We also examined the expression of this transporter in ocular tissues because administration of drugs in the form of eye drops is useful for ophthalmic drug delivery. The data in Fig. 1 show that ATB0,+ is expressed abundantly in conjunctival epithelial cells, retinal ganglion cells, retinal pigment epithelium, and in the inner nuclear layer of the retina.
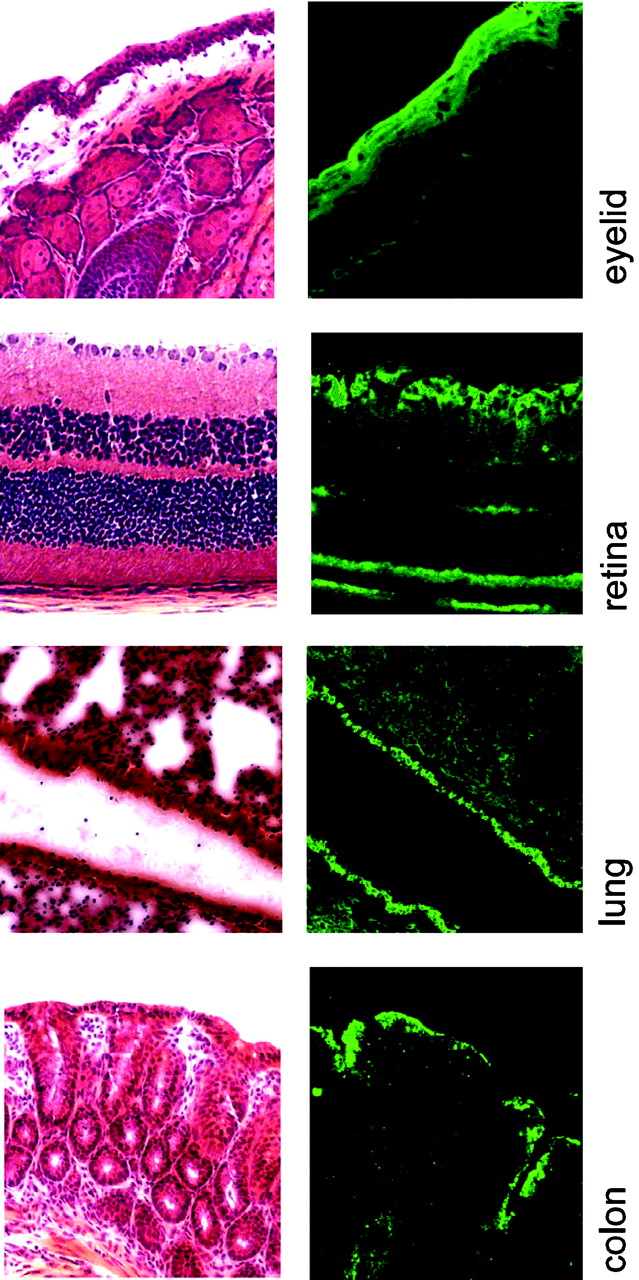
Immunolocalization of ATB0,+ in mouse colon, lung, and eye. Cryosections of colon, lung, and ocular tissues were incubated with a primary antibody (polyclonal) specific for ATB0,+ and then with a fluorescein isothiocyanate-coupled goat anti-rabbit IgG as the secondary antibody. The slides were examined under a laser scanning fluorescence confocal microscope. Additional sections from colon, lung, retina, and eye lid were stained with hematoxylin and eosin for morphological assessment. There were no detectable fluorescence signals in negative controls in which either the primary antibody was omitted or preimmune goat serum was used instead of the secondary antibody.
Interaction of l-Aspartate and l-Glutamate Derivatives with ATB0,+in Mammalian Cell Expression System. Although aspartate is not a substrate for ATB0,+, asparagine in which the β-carboxyl group of aspartate is amidated is recognized by the transporter as a substrate. This is evident from the ability of asparagine, but not aspartate, to inhibit glycine transport via ATB0,+ (Table 1). Based on this, we screened several additional β-carboxyl derivatives of aspartate for their ability to interact with ATB0,+ to determine whether the transporter tolerates substitutions other than the amide group on the β-carboxyl group. This screening showed that aspartate β-methyl ester and aspartate β-benzyl ester are able to interact with ATB0,+ as assessed by their ability to inhibit glycine transport via the transporter. At 1 mM, these two esters caused >90% inhibition of ATB0,+-mediated glycine transport (Table 1). Two other derivatives, aspartate β-hydroxamate and N-β-aspartyl-phenylalanine methyl ester, were also able to inhibit glycine transport via ATB0,+, although to a lesser extent. These two derivatives, at 1 mM, caused 40 to 60% inhibition of glycine transport. Aspartate β-(7-amido-4-methylcoumarin) did not inhibit glycine transport at a concentration of 0.25 mM. This derivative could not be used at concentrations higher than 0.25 mM due to limited solubility; therefore, it is not known at present whether this compound can inhibit ATB0,+-mediated glycine transport at higher concentrations. We also screened several γ-carboxyl derivatives of glutamate for their interaction with ATB0,+ as assessed by their ability to inhibit glycine transport via the transporter (Table 1). The results were essentially similar to those obtained with the β-carboxyl derivatives of aspartate. We did not determine the stability of these compounds under the experimental conditions. Because the incubation time used in these studies was 30 min, questions may arise as to whether these compounds stayed intact in the incubation medium under these conditions. Therefore, we determined the inhibitory potencies of these compounds using a much shorter incubation period (2 min). The results of these studies were essentially the same as those in Table 1 (data not shown).
Inhibition of ATB0,+ -mediated glycine uptake in HRPE cells by the derivatives of aspartate and glutamate
Mouse ATB0,+ cDNA was expressed functionally in HRPE cells by the vaccinia virus expression technique. The transport activity of ATB0,+ was monitored by the uptake of 10 μM glycine in the presence of NaCl. Uptake was measured in the absence (control, 100%) and presence of 1 mM aspartate, glutamate, or their derivatives.
Because the benzyl esters of aspartate and glutamate showed excellent ability to inhibit glycine transport via ATB0,+, we used these esters for further analysis of their interaction with the transporter. Figure 2 describes the dose-response relationship for the inhibition of ATB0,+-mediated glycine transport by l-aspartate β-benzyl ester [Asp(OBzl)] in comparison with asparagine and aspartate and by l-glutamate γ-benzyl ester [Glu(OBzl)] in comparison with glutamine and glutamate. As expected, aspartate and glutamate did not inhibit glycine transport. In contrast, asparagine and glutamine were able to inhibit glycine transport with IC50 values (i.e., the concentration of the inhibitor necessary to cause 50% inhibition) of 0.34 ± 0.06 and 0.31 ± 0.03 mM, respectively. Asp(OBzl) and Glu(OBzl) were even more potent than asparagine and glutamine in inhibiting glycine transport. The IC50 values for Asp(OBzl) and Glu(OBzl) were 0.07 ± 0.01 and 0.15 ± 0.01 mM, respectively.

Dose-response relationship for the inhibition of ATB0,+-mediated glycine transport by l-aspartate and l-glutamate and their side-chain derivatives. Mouse ATB0,+ cDNA was expressed heterologously in HRPE cells, and the transport function of ATB0,+ was measured by the uptake of 10 μM glycine. Glycine uptake in control cells transfected with vector alone was less than 2% of uptake measured in cDNA-transfected cells. ATB0,+-specific uptake of glycine, measured in the absence of inhibitors, was taken as the control (100%).
Direct Evidence for the Transport of Asp(OBzl) via ATB0,+in X. laevis Oocyte Expression System. The results obtained with the mammalian cell expression system show that the β-carboxyl derivatives of l-aspartate and the γ-carboxyl derivatives of l-glutamate interact with ATB0,+ as assessed by their ability to inhibit glycine transport via the transporter. However, these data suggest, but do not prove, that these derivatives are actually transported by ATB0,+. It is possible that these compounds may block the transport by competing with glycine for binding to the substrate-binding site of the transporter without themselves being transported across the membrane. To determine whether these compounds are transportable substrates for ATB0,+, direct measurements of their transport must be monitored rather than merely demonstrating their ability to compete with glycine. This would require use of these compounds in radiolabeled form so that their transport can be measured directly in mammalian cells. Because these compounds are not readily available in radiolabeled form from commercial sources, we used an alternative method for direct measurement of their transport via ATB0,+. We used the X. laevis oocyte expression system for this purpose. The cloned mouse ATB0,+ was functionally expressed in oocytes by injection of cRNA, and the transport of Asp(OBzl) via the transporter was monitored by inward currents induced by this compound using the two-microelectrode voltage-clamp technique. This approach is feasible because ATB0,+ is an electrogenic transporter (Sloan and Mager, 1999; Hatanaka et al., 2001, 2002; Nakanishi et al., 2001). Induction of inward currents in ATB0,+-expressing oocytes upon exposure to a test compound in the presence of NaCl under voltage-clamped conditions (holding potential, -50 mV) indicates depolarization of the oocyte as a result of transport of the compound into the oocyte. Water-injected oocytes served as negative controls in these experiments. The results of these experiments show that exposure of ATB0,+-expressing oocytes to aspartate induces very little inward currents (Fig. 3). In contrast, exposure of the oocytes to asparagine or Asp(OBzl) induces marked inward currents [245 ± 20 nA for 1 mM asparagine and 93 ± 12 nA for 1 mM Asp(OBzl) in four different oocytes], providing evidence for the transport of asparagine and Asp(OBzl) in these oocytes. Water-injected oocytes do not show any detectable inward currents upon exposure to asparagine or Asp(OBzl) (data not shown).

Transport of l-aspartate and its side-chain derivatives via ATB0,+ in X. laevis oocytes. Oocytes were injected with mouse ATB0,+ cRNA, and 6 days after injection, they were used in electrophysiological studies. The oocytes were perfused with 1 mM l-aspartate, l-asparagine, or Asp(OBzl), and the substrate-induced inward currents were monitored at -50 mV under voltage-clamp conditions. Water-injected oocytes were used as control and perfusion of these oocytes with l-aspartate, l-asparagine, and Asp(OBzl) did not show any detectable inward currents under identical conditions (data not shown).
ATB0,+ is a Na+- and Cl--coupled transporter and accordingly there were no detectable inward currents in ATB0,+-expressing oocytes when these oocytes were perfused with asparagine or Asp(OBzl) in the absence of either Na+ or Cl- in two different oocytes (data not shown). This shows that the transport of Asp(OBzl) via ATB0,+ is a Na+- and Cl--dependent process. We analyzed the Na+- and Cl--activation kinetics for the transport of Asp(OBzl) via ATB0,+. The activation of Asp(OBzl)-induced inward currents by Na+ followed sigmoidal kinetics (Fig. 4A), indicating the involvement of more than one Na+ ion in the activation process. The nH for the process was 2.1 ± 0.5. In contrast, the activation of Asp(OBzl)-induced inward currents by Cl- was hyperbolic, indicating involvement of one Cl- in the activation process (Fig. 4B). The Hill coefficient, calculated from the Cl--activation kinetics, was 0.9 ± 0.3. This shows that the Na+/Cl-/Asp(OBzl) stoichiometry for the transport process is 2:1:1. This stoichiometry is the same as was previously calculated for other amino acid substrates (Sloan and Mager, 1999). Because Asp(OBzl) does not carry a net charge, this stoichiometry provides the basis for the electrogenic nature of the transport process.

Na+-activation kinetics and Cl--activation kinetics of Asp(OBzl)-induced inward currents in oocytes expressing mouse ATB0,+. A, oocytes expressing mouse ATB0,+ were perfused with 1 mM Asp(OBzl) in the presence of increasing concentrations of Na+, and the substrate-induced inward currents were monitored at -50 mV under voltage-clamp conditions. The osmolality of the perfusion buffer was maintained by isoosmotically substituting NaCl with N-methyl-d-glucamine chloride. Inset, Hill plot. B, oocytes expressing mouse ATB0,+ were perfused with 1 mM Asp(OBzl) in the presence of increasing concentrations of Cl- and the substrate-induced inward currents were monitored at -50 mV under voltage-clamp conditions. The osmolality of the perifusion buffer was maintained by isoosmotically substituting NaCl with sodium gluconate. Inset, Hill plot.
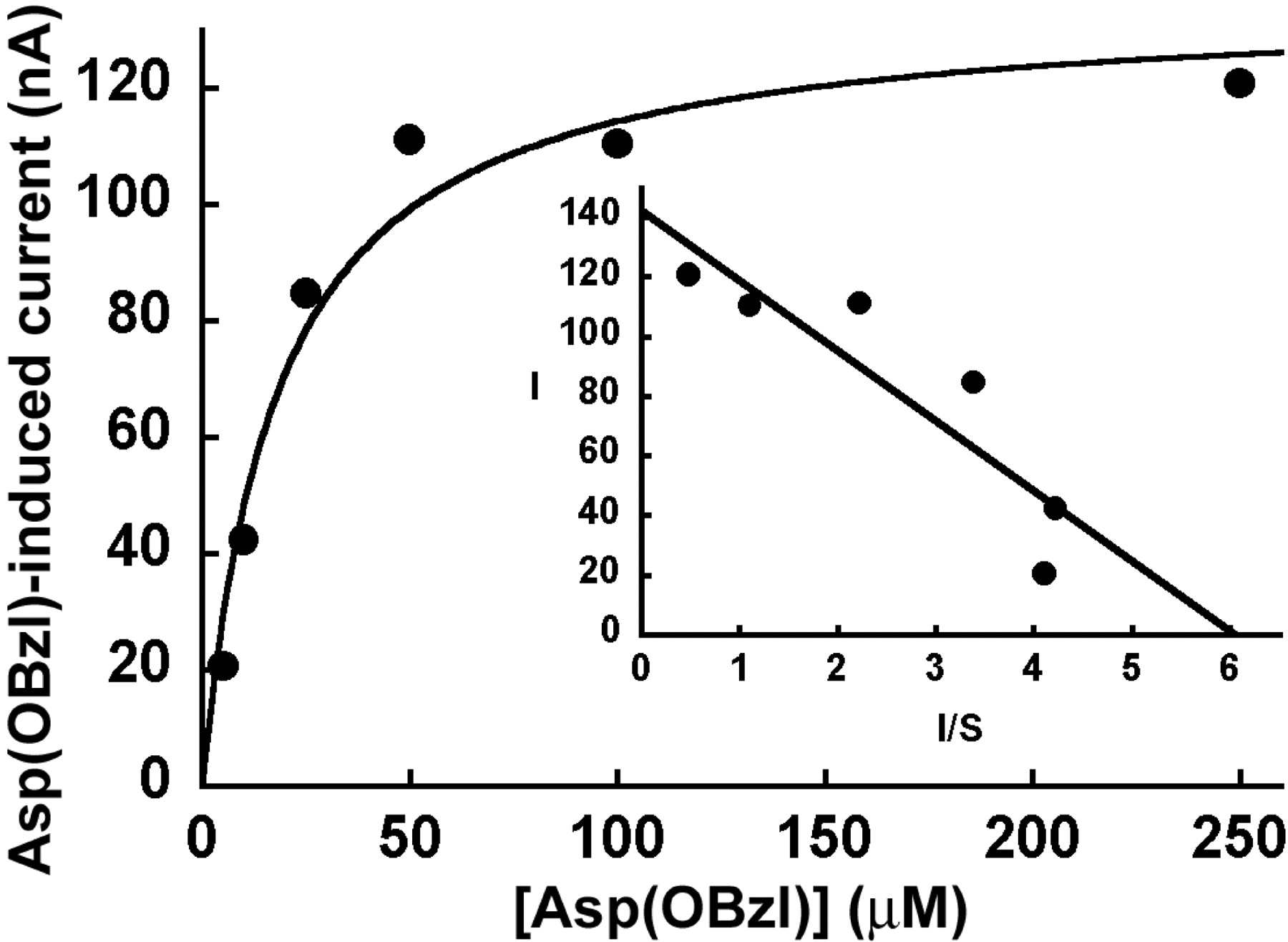
The inward currents induced by Asp(OBzl) were saturable (Fig. 5). The K0.5 value [i.e., the concentration of Asp(OBzl) necessary for the induction of half-maximal current] was 18 ± 4 μM. This value is significantly different from the IC50 value obtained in HRPE cells for the inhibition of ATB0,+-mediated glycine transport by Asp(OBzl). The most likely reason for this difference is that whereas the inward currents in oocytes were measured under voltage-clamp conditions, uptake measurements in HRPE cells were not made under similar conditions. Membrane potential may have significant influence on the affinity of the transporter for its substrates.

Saturation kinetics of Asp(OBzl)-induced inward currents. Oocytes expressing mouse ATB0,+ were perfused with increasing concentrations of Asp(OBzl) in the presence of NaCl and the substrate-induced inward currents were measured at -50 mV under voltage-clamp conditions. Inset, Eadie-Hofstee plot. I, inward current in nanoamperes; S, concentration of Asp(OBzl) in micromolar.
Transport of Acyclovir Glutamic Acid γ-Ester by ATB0,+ The ultimate goal of this investigation is to determine whether ATB0,+ holds potential as a drug delivery system for amino acid-based prodrugs. Our studies with β-carboxyl derivatives of aspartate and γ-carboxyl derivatives of glutamate strongly suggest that pharmacologically active drugs may be coupled to the side chain of aspartate or glutamate in the form of prodrugs, consequently making these prodrugs substrates for ATB0,+ and facilitating their delivery into cells. To provide direct evidence in support of this approach, we chose the antiviral agent acyclovir as a model drug. To our knowledge, this drug is not recognized as a substrate by any of the amino acid transport systems present in the plasma membrane of mammalian cells. Therefore, we wished to test whether this drug, if coupled to the side chain of glutamate in the form of an ester, can be transported by ATB0,+. For this purpose, we synthesized acyclovir glutamic acid γ-ester (Acv-Glu) and compared the interaction of acyclovir and Acv-Glu with ATB0,+ in the mammalian cell expression system by assessing the ability of these two compounds to inhibit glycine transport via the transporter (Fig. 6). As expected, acyclovir failed to inhibit ATB0,+-mediated glycine transport, showing that this drug is not recognized by the transporter. In contrast, Acv-Glu was able to inhibit glycine transport via ATB0,+ to a significant extent. The IC50 value for this inhibition was 3.3 ± 0.7 mM. These data show that although acyclovir is not a substrate for ATB0,+, Acv-Glu is recognized by the transporter with significant affinity. To provide direct evidence for the transport of Acv-Glu via ATB0,+, we used the X. laevis oocyte expression system and assessed the transport of this derivative by the electrophysiological approach. As shown in Fig. 7, exposure of ATB0,+-expressing oocytes to acyclovir did not induce detectable inward currents. In contrast, exposure of ATB0,+-expressing oocytes to Acv-Glu induced marked inward currents, providing evidence for the transport of this derivative via ATB0,+. The inward currents induced by this derivative were dependent on the presence of Na+ and Cl- (Fig. 7), further confirming its transport via ATB0,+. We examined the saturation kinetics of the inward currents induced by Acv-Glu by measuring the magnitude of the induced currents with varying concentrations of the derivative (data not shown). The K0.5 value (i.e., concentration of the compound necessary for the induction of half-maximal current), calculated by fitting the data to Michaelis-Menten equation, was 2.2 ± 0.7 mM.

Dose-response relationship for the inhibition of ATB0,+-mediated glycine uptake by acyclovir and Acv-Glu. Mouse ATB0,+ was expressed in HRPE cells heterologously, and the transport function of ATB0,+ was monitored by measuring the uptake of 10 μM glycine. The uptake of glycine, measured in the absence of inhibitors, was taken as the control (100%).

Transport of acyclovir and Acv-Glu via ATB0,+ in X. laevis oocytes. Oocytes were injected with mouse ATB0,+ cRNA, and 6 days after injection, they were used in electrophysiological studies. The oocytes were perfused with either 1 mM acyclovir or 1 mM Acv-Glu, and the substrate-induced inward currents were monitored at -50 mV under voltage-clamp conditions. In the case of Acv-Glu, inward currents were monitored in the presence of NaCl, in the absence of Na+ but in the presence of Cl- (-Na+), and in the absence of Cl- but in the presence of Na+ (-Cl-). A representative tracing of the inward currents from a single oocyte is shown. Similar results were obtained with three other oocytes from different batches. Water-injected oocytes were used as control and perfusion of these oocytes with acyclovir or Acv-Glu did not induce any detectable inward currents under identical conditions (data not shown).
Transport of Valacyclovir by ATB0,+ Acyclovir has poor oral bioavailability due to lack of transport systems in the intestinal tract that can recognize this drug as a substrate. This prompted the design of prodrugs in which acyclovir was modified as α-carboxyl esters of amino acids and one of these amino acid-based prodrugs, valacyclovir (l-valyl ester of acyclovir), was found to have 3- to 5-fold greater oral bioavailability (Beutner et al., 1995; Soul-Lawton et al., 1995; Wang et al., 1996). The comparatively more efficient oral absorption of valacyclovir than acyclovir is believed to be due to the recognition of the prodrug, but not the parent drug, by intestinal transport systems as a substrate. The peptide transporter PEPT1, which is expressed abundantly in the intestine, has been shown to transport valacyclovir efficiently (de Vrueh et al., 1998; Han et al., 1998; Ganapathy et al., 1998; Anand et al., 2003). In contrast, acyclovir is not recognized by PEPT1. Similar results have been obtained with valganciclovir which is the l-valyl ester of ganciclovir, another effective antiviral agent (Sugawara et al., 2000). Conversion of ganciclovir as an α-carboxyl ester of l-valine makes it a substrate for PEPT1 and consequently enhances its oral bioavailability. Based on our findings that Acv-Glu is recognized as a substrate by ATB0,+, we asked whether the valine α-ester of acyclovir can be transported by this amino acid transporter. Acv-Glu contains unmodified α-amino and α-carboxyl groups on the glutamate backbone with acyclovir attached to the γ-carboxyl group as an ester. In contrast, valacyclovir is an ester of acyclovir at the α-carboxyl group of valine and consequently valacyclovir possesses a free α-amino group but not a free α-carboxyl group. Therefore, initially we thought that valacyclovir would not be recognized as a substrate by ATB0,+, assuming that an amino acid transporter would require the presence of a free α-amino group and a free α-carboxyl group on its substrate for optimal recognition. When we examined the interaction of acyclovir and valacyclovir with ATB0,+ using the mammalian cell expression system, the results were unexpected (Fig. 8). As seen before, acyclovir failed to inhibit glycine transport via ATB0,+. But, valacyclovir was able to inhibit ATB0,+-mediated glycine uptake very effectively, with an IC50 value of 0.7 ± 0.1 mM. This was a surprising finding not only because valacyclovir is able to interact with ATB0,+ but also because the interaction seems to be of significantly higher affinity than that seen with Acv-Glu. The potency of valacyclovir to inhibit ATB0,+-mediated glycine transport is at least 3-fold higher than that seen with Acv-Glu.

Dose-response relationship for the inhibition of ATB0,+-mediated glycine uptake by acyclovir and valacyclovir. Mouse ATB0,+ was expressed in HRPE cells heterologously, and the transport function of ATB0,+ was monitored by measuring the uptake of 10 μM glycine in the presence of NaCl. The uptake of glycine, measured in the absence of inhibitors, was taken as the control (100%).
To determine whether valacyclovir is a transportable substrate for ATB0,+, we used two different approaches. First, we used radiolabeled valacyclovir to examine directly its transport via ATB0,+ using the mammalian cell expression system. For this purpose, we compared the transport of valacyclovir in vector-transfected HRPE cells and in HRPE cells expressing the cloned mouse ATB0,+ heterologously. Transport measurements were made in the presence of NaCl. The data given in Fig. 9A show that the transport of valacyclovir is significantly higher (2.5-fold) in ATB0,+-expressing cells than in control cells. This demonstrates that valacyclovir is a transportable substrate for ATB0,+. For comparison, we examined the transport of valacyclovir via human PEPT1 using the same expression system. Because PEPT1 is an H+-coupled transporter, we measured transport in the presence of an inwardly directed H+ gradient (extracellular pH, 6). Under these conditions, the transport of valacyclovir was significantly higher in PEPT1-expressing cells than in control cells (2.7-fold) (Fig. 9B). Thus, the abilities of ATB0,+ and PEPT1 to transport valacyclovir are comparable. The transport of valacyclovir via ATB0,+ was dependent on the presence of Na+ and Cl- as expected from the Na+- and Cl- dependence of ATB0,+ (Fig. 9A). The transport of valacyclovir via ATB0,+ was further confirmed by the electrophysiological approach using the X. laevis oocyte expression system (Fig. 10). Exposure of ATB0,+-expressing oocytes to valacyclovir induced marked inward currents when examined in the presence of NaCl in the perfusion medium. These currents were, however, not detectable if the perfusion medium lacked either Na+ (N-methyl-d-glucamine chloride replacing NaCl) or Cl- (sodium gluconate replacing NaCl) (data not shown). Interestingly, the magnitude of inward currents induced by 1 mM valacyclovir was 3- to 4-fold greater than that induced by 1 mM Acv-Glu. This is most likely related to the comparatively higher affinity of valacyclovir for ATB0,+ than that of Acv-Glu as evident from the mammalian cell expression system (IC50 values for the inhibition of glycine transport via ATB0,+: valacyclovir, 0.7 ± 0.1 mM; Acv-Glu, 3.3 ± 0.7 mM). Because the transport process is not saturated with either of these substrates at 1 mM, the amount of the substrate transported per unit time is expected to be much greater for the high-affinity substrate than for the low-affinity substrate. These results show that ATB0,+ is able to transport not only the β-carboxyl derivatives of aspartate and the γ-carboxyl derivatives of glutamate but also the α-carboxyl esters of l-valine.

Comparison of valacyclovir uptake via mouse ATB0,+ (A) and human PEPT1 (B). HRPE cells were transfected with either vector alone, mouse ATB0,+ cDNA, or human PEPT1 cDNA. Uptake of [3H]valacyclovir (5 μM) was measured for 15 min. In the case of ATB0,+, the uptake buffer (pH 7.5) contained either 140 mM NaCl, 140 mM N-methyl-d-glucamine chloride (-Na+), or 140 mM sodium gluconate (-Cl-). In the case of PEPT1, the uptake buffer contained NaCl at pH 6.

Comparison of inward currents induced by Acv-Glu and valacyclovir in oocytes expressing mouse ATB0,+. Oocytes were injected with mouse ATB0,+ cRNA, and 6 days after injection, they were used in electrophysiological studies. The oocytes were perfused with either 1 mM Acv-Glu or 1 mM valacyclovir, and the substrate-induced inward currents were monitored at -50 mV under voltage-clamp conditions. For comparison of the transport of these two acyclovir derivatives via ATB0,+ under identical conditions, the tracings of the inward currents from a single oocyte are shown. Similar results were obtained with another oocyte from a different batch. Water-injected oocytes were used as control, and perfusion of these oocytes with acyclovir or Acv-Glu did not induce any detectable inward currents under identical conditions (data not shown).
Discussion
The significance of the present studies is 2-fold. First, these studies provide evidence for the first time in support of the therapeutic potential of ATB0,+ as a delivery system for amino acid-based prodrugs. Our previous studies have shown that ATB0,+ can transport a variety of nitric-oxide synthase inhibitors (Hatanaka et al., 2001). These inhibitors are structurally related to arginine, lysine, citrulline, or ornithine, all of which are transportable substrates for ATB0,+. The data presented in this article show that the therapeutic potential of ATB0,+ as a drug delivery system may be much greater than originally envisaged. The present findings suggest that it is possible to chemically modify drugs by coupling them to the side chain of aspartate or glutamate so that these modified drugs become substrates for ATB0,+, thus enhancing their delivery into cells where the transporter is expressed. Evidence is provided in the present study in support of this possibility with a potential prodrug Acv-Glu. The data showing that valacyclovir is also a substrate for ATB0,+ broadens the scope of this transporter for drug delivery even further because these findings suggest that when drugs are coupled to any of the neutral or cationic amino acids in an ester linkage to their α-carboxyl groups, these modified drugs may become substrates for ATB0,+. Because ATB0,+ recognizes almost all of the naturally occurring neutral and cationic amino acids as substrates, there is a wide range of choices for the chemical modification of drugs to ensure optimal drug delivery. It has to be pointed out, however, that we analyzed the interaction of some of the compounds with ATB0,+ only by electrophysiological methods. Even though substrate-induced currents represent a fairly reliable indication for transport, this may not constitute the absolute proof. Furthermore, the present studies only address the issue of entry of prodrugs into cells via ATB0,+. What happens to these prodrugs after they enter the cells has not been determined in the present study. Successful delivery of therapeutic drugs in the form of prodrugs would require information on how and where these prodrugs might be converted into therapeutically active drugs and on the transport processes that might mediate the exit of these drugs after their entry via ATB0,+.
The second important finding with potential clinical and therapeutic significance is the expression of ATB0,+ in tissues that are ideal for drug delivery. The expression of the transporter in the colon, lung, and conjunctiva makes this transporter suitable for use as a drug delivery system. The large intestine is amenable for drug delivery in the form of oral pills or rectal suppositories. Similarly, drugs and prodrugs can be aerosolized for delivery via the lung. We initially examined the expression of ATB0,+ in the lung and colon because of the evidence in the literature for the presence of ATB0,+ transcripts in these tissues. Subsequently, we wanted to examine the expression of this transporter in ocular tissues. This was prompted by three reasons. First, ophthalmic drug delivery is challenging because of the blood-retinal barrier (Duvvuri et al., 2003); administration of drugs in the form of eye drops requires knowledge of the transport mechanisms available in ocular tissues for delivery of drugs. Second, there is functional evidence in the literature for the expression of ATB0,+ in some of the ocular tissues such as the conjunctiva (Hosoya et al., 1997), and therefore it would be desirable to confirm these observations by immunolocalization of the ATB0,+ protein. Third, cytomegalovirus infection, which occurs commonly in the retina in AIDS patients, is treated with ganciclovir, a drug structurally related to acyclovir, and our findings that ATB0,+ can transport the prodrug valacyclovir is clinically relevant to this disease. It is therefore important to know the expression pattern of this transporter in the ocular tissues to determine whether it has potential for delivery of antiviral drugs to the retina and other areas within the eye by routes other than oral or intravenous administration. The data in Fig. 1 show that ATB0,+ is expressed abundantly in the conjunctival epithelial cells, confirming the functional studies by Hosoya et al. (1997). In addition, the transporter is expressed in the retinal ganglion cells, retinal pigment epithelium, and the inner nuclear layer. These findings suggest that it may be possible to administer antiviral agents such as valganciclovir and valacyclovir in the form of eye drops for delivery into ocular tissues for the treatment of viral infection.
PEPT1 has received considerable attention for its potential as a drug delivery system because of its broad substrate specificity and abundant expression in the intestinal tract (Tsuji and Tamai, 1996; Inui and Terada, 1999; Rubio-Aliaga and Daniel, 2002; Ganapathy and Miyauchi, 2003). We believe that the potential of ATB0,+ as a drug delivery system is at least comparable, if not greater, than that of PEPT1. The substrate specificity of ATB0,+ is also very broad. It transports all of the neutral and cationic amino acids in their l-isomeric form. It also accepts many of these amino acids in their d-isomeric form. In addition, the transport system does not seem to have very stringent structural requirements with respect to the side chain of these amino acids as long as the side chain is not negatively charged. Significantly, these two transporters differ in their energetics. PEPT1 is an H+-coupled transporter that is driven by the microclimate acid pH that naturally exists on the luminal surface of the mucosal cells in the intestinal tract (Ganapathy and Leibach, 1985, 1991; Leibach and Ganapathy, 1996). In contrast, ATB0,+ is energized by transmembrane gradients for Na+ and Cl-. This makes ATB0,+ more capable of concentrative transport than PEPT1. The differences in the localization of these two transporters in the mammalian intestinal tract are also of relevance to their role in drug delivery. PEPT1 is expressed primarily in the small intestine. Because the diet-derived peptides, which are natural substrates for PEPT1, are abundant in the small intestine, absorption of drugs via PEPT1 has to occur in competition with these naturally occurring peptides. This might compromise the efficiency of drug absorption. In contrast, ATB0,+ is expressed most predominantly in the large intestine where the entry of diet-derived amino acids is minimal. Thus, competition with the natural substrates would likely be minimal, hence enhancing the efficiency of drug absorption via this transporter. When the other potential sites of drug delivery such as the lung and conjunctiva are considered, ATB0,+ has clear advantages over PEPT1. Peptide transporters, both PEPT1 and PEPT2, are expressed in these sites but at much lower levels (Basu et al., 1998; Groneberg et al., 2001; Anand and Mitra, 2002; Sloan and Mager, 2003). In contrast, the expression of ATB0,+ at these sites is abundant. Furthermore, there is no evidence for the presence of microclimate acid pH at these sites to provide the necessary driving force for the energization of PEPT1. The transmembrane gradients for Na+ and Cl- exist at these sites due to the secretion of NaCl into the airway lumen of the lung and tear fluid in the eye, and thus ATB0,+ is expected to function optimally under these conditions. Thus, our data suggest that it would be advantageous to target ATB0,+ as a drug delivery system in the colon, lung, and conjunctiva.
Footnotes
-
This work was supported by the National Institutes of Health Grant GM65344.
-
DOI: 10.1124/jpet.103.057109.
-
ABBREVIATIONS: ATB0,+, amino acid transporter B0,+; Z-Glu-OBzl, N-α-benzyloxycarbonyl-l-glutamic acid α-benzyl ester; Pd/C, palladium 10% (w/v) on activated carbon; HRPE, human retinal pigment epithelial; PEPT1, peptide transporter 1; MES, 4-morpholineethanesulfonic acid; Asp-OBzl, l-aspartate β-benzyl ester; Glu-OBzl, l-glutamate γ-benzyl ester; Acv-Glu, acyclovir l-glutamate γ-ester.
- Received July 16, 2003.
- Accepted November 5, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}