


Abstract
The adequate distribution of STI-571 (Gleevec) to the central nervous system (CNS) is critical for its effective use in CNS tumors. P-glycoprotein-mediated efflux in the blood-brain barrier may play a role in the CNS delivery of this drug. Whether STI-571 is a substrate of P-glycoprotein was determined by examining the directional flux of [14C]STI-571 in parental and MDR1-transfected Madin-Darby canine kidney (MDCK) II epithelial cell monolayers. The basolateral-to-apical flux of STI-571 was 39-fold greater than the apical-to-basolateral flux in the MDR1-transfected cells and 8-fold greater in the parental cell monolayers. This difference in directional flux was significantly reduced by a specific P-glycoprotein inhibitor (2R)-anti-5-{3-[4-(10,11-difluoromethanodibenzo-suber-5-yl)piperazin-1-yl]-2-hydroxypropoxy}quinoline trihydrochloride (LY335979). The role of P-glycoprotein in the CNS distribution of STI-571 was examined in vivo, using wild-type and mdr1a/b (−/−) knockout mice that were orally administered 25 mg/kg [14C]STI-571. In the wild-type mice, the brain-to-plasma STI-571 concentration ratio at all time points was low (1–3%); however, there was an 11-fold greater brain partitioning of STI-571 at 1 h postdose in the mdr1a/b (−/−) mice compared with the wild-type mice. When 12.5 mg/kg STI-571 was given intravenously, the brain-to-plasma ratio of STI-571 in the mdr1a/b (−/−) mice was approximately 7-fold greater than that of wild-type mice up to 120 min postdose. These data indicate that STI-571 is a substrate of P-glycoprotein, and that the inhibition of P-glycoprotein affects the transport of STI-571 across MDCKII monolayers. Moreover, P-glycoprotein plays an important role in limiting the distribution of STI-571 to the CNS.
STI-571 (Gleevec) is an inhibitor of BCR/ABL tyrosine kinase (Buchdunger et al., 1996), stem cell factor receptor (c-kit) kinase (Buchdunger et al., 2000; Heinrich et al., 2000), and platelet-derived growth factor receptor (PDGFR) kinase (Buchdunger et al., 2000). BCR/ABL tyrosine kinase is overexpressed in chronic myelogenous leukemia (CML) patients because of a chromosomal translocation (Philadelphia chromosome) and is responsible for the oncogenesis of CML (Rowley, 1973; Daley et al., 1990; Melo, 1996). STI-571 has been shown to be effective for CML patients, including those who are refractory to interferon-α treatment (Druker et al., 2001). In one study, 53 of 54 patients examined showed complete and lasting hematological remission (Druker et al., 2001). The success of this compound in the treatment of CML has led to the broader examination of its application in the treatment of other tumors, such as glioblastoma (Kilic et al., 2000; Uhrbom et al., 2000), small cell lung cancer (Krystal et al., 2000; Wang et al., 2000), and gastrointestinal stroma (Joensuu et al., 2001; Tuveson et al., 2001; van Oosterom et al., 2001), in which c-kit or PDGFR is expressed and participates in the autocrine loop (Uhrbom et al., 2000;Wang et al., 2000; Tuveson et al., 2001). Animal experiments with nude mice suggest that STI-571 may be effective against glioblastoma xenograft (Kilic et al., 2000). Currently, several clinical trials of glioblastoma patients treated with STI-571 are under way [e.g., National Cancer Institute protocol 01-C-0243, a phase I/II trial of STI571 (NSC716051) in patients with recurrent malignant gliomas]. However, the treatment of CNS tumors is often limited by low distribution of the antitumor agents into brain because of blood-brain barrier. Various efflux transporters expressed in the blood-brain barrier, including P-glycoprotein, can eliminate xenobiotics from the brain and further limit their CNS distribution (Kusuhara and Sugiyama, 2002). A limited distribution of STI-571 to the cerebrospinal fluid in humans has been reported (Leis et al., 2001; Petzer et al., 2002; Takayama et al., 2002), but factors responsible for this low distribution have not been characterized. This is an important issue not only for treatment of the primary CNS tumor such as glioblastoma, but also it may be critical for CML patients, considering that CNS relapses have been observed in CML patients even though they have showed a complete hematological response to STI-571 (Leis et al., 2001;Petzer et al., 2002; Takayama et al., 2002). The objective of this study was to determine whether STI-571 is a substrate of P-glycoprotein and to examine the role of P-glycoprotein in the distribution of STI-571 into the brain.
Materials and Methods
Chemicals.
STI-571 and [14C]STI-571 (both as the mesylate salt) were provided by Novartis Pharma AG (Basel, Switzerland). (2R)-Anti-5-{3-[4-(10,11-difluoromethanodibenzo-suber-5-yl)piperazin-1-yl]-2-hydroxypropoxy}quinoline trihydrochloride (LY335979) (Zosuquidar.3 HCl) was kindly provided by Eli Lilly Cancer Research Laboratory (Indianapolis, IN). Tritiated inulin was purchased from Moravek Biochemicals (Brea, CA). All other chemicals were reagent grade or better.
Cellular Flux of STI-571 across the MDR1-Transfected and Wild-Type Madin-Darby Canine Kidney (MDCK) II Monolayers.
MDCKII (wild-type and MDR1-transfected) were kindly provided by Dr. Piet Borst (Netherlands Cancer Institute, Amsterdam, The Netherlands) and maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 2.5% penicillin and streptomycin at 37°C in a humidified incubator with 5% CO2. MDCKII cells (1 × 106, wild-type and MDR1-transfected) were seeded in six-well polyester membrane inserts (Transwell, Costar Brand Tissue Culture Products; Fisher Scientific Co., Pittsburgh, PA) with a pore size of 0.4 μm and a diameter of 24.0 mm. The media were changed every 24 h, and a polarized monolayer was established in 3 days. After the monolayers were established, the directional transport of a tracer concentration (1.1 μg/ml) of [14C]STI-571 across the cellular monolayer was examined. Briefly, media were removed from the chambers and the monolayers were washed with phosphate buffer (pH 7.2). The stock solution of [14C]STI-571 was prepared in ethanol and diluted 100-fold with the assay buffer containing122 mM sodium chloride, 25 mM sodium bicarbonate, 10 mM glucose, 10 mM HEPES, 3 mM potassium chloride, 1.2 mM magnesium sulfate, 1.4 mM calcium chloride, and 0.4 mM potassium phosphate dibasic (pH 7.4). Then, 0.01 μCi of [14C]STI-571 in 2 ml of assay buffer was applied to the donor chamber (either the apical side or the basolateral side). The receiver chamber was filled with 2 ml of blank assay buffer. At 0, 15, 30, 60, 90, 120, and 180 min, 200 μl of assay buffer was sampled with replacement from the receiver chamber. The samples were mixed with 4 ml of scintillation cocktail (ScintiSafe Econo 1 cocktail; Fisher Scientific Co.) and counted for beta radioactivity using a Packard Tri-Carb 2500 liquid scintillation counter. The measured radioactivity over the initial radioactivity of the donor chamber is calculated as percentage of the radioactivity transported across the monolayer at different time points (% transported). When the apical side (upper chamber) serves as the donor chamber, the flux from apical-to-basolateral (A-to-B) side of the monolayer was measured and vice versa the basolateral-to-apical (B-to-A) flux was measured. The percentage of STI-571 transported was plotted as a function of time, the slope of which is related to the first order rate constant (K) for the steady-state flux and effective permeability (Peff) as follows: K = slope/100 andPeff = K ·V/Area, where V and Area are the volume of the donor chamber and effective cellular surface area of the insert, respectively.
When a specific inhibitor of P-glycoprotein, LY335979, was used, the MDR1-transfected MDCKII monolayer was first preincubated with 1 μM LY335979 in assay buffer for 30 min and then the A-to-B flux and the B-to-A flux were measured with 1 μM LY335979 present in both chambers.
CNS Distribution of STI-571 in mdr1a/b (−/−) Knockout Mice and Wild-Type Mice.
The mdr1a/b (−/−) knockout and wild-type mice (129/Ola × FVB) used for this study were purchased from Taconic Farms (Germantown, NY). In oral dosing experiments, the mice received 25 mg/kg [14C]STI-571 [approximately 3 μCi of tracer in a 200-μl saline solution (6.7 ml/kg)] as a single oral dose via gavage. At different times postdose (30, 60, and 120 min,n = 4 for each time point), the mice were euthanized and the plasma and total brain tissue were collected. The brain tissue was homogenized in 3 volumes of saline phosphate buffer with 5% albumin using a manual tissue homogenizer (Schinkel et al., 1996). Then 0.2 ml of plasma or brain homogenate was mixed with 4 ml of scintillation fluid and counted for radioactivity using liquid scintillation counting. In addition, the plasma and brain tissue homogenate collected at 60 min postdose were analyzed by HPLC to separate the parent drug from its metabolites. Briefly, the sample was deproteinized with 2 volumes of acetonitrile at room temperature. After centrifugation (6000g), evaporation, and reconstitution with mobile phase, 30 μl of the solution was injected onto the HPLC column (base-deactivated C18; Thermo Hypersil, Keystone Scientific Operations, Bellefonte, PA). The mobile phase was 5% acetonitrile, 4% tetrahydrofuran, and 0.5% triethylamine (w/w) in 25 mM phosphate buffer (pH 3), isocratic, at a rate of 0.25 ml/min. The column eluant was collected every 15 s for 2 min around the peak of interest (the retention time of STI-571, approximately 18 min) and every 2 min otherwise for a total of 22 min. The collected eluant was measured for radioactivity using liquid scintillation counting. The brain-to-plasma ratio is the ratio of disintegrations per minute per gram of brain tissue over disintegrations per minute per milliliter of plasma. The radiopurity of [14C]STI-571 administered was 98.3%, as stated in the certificate of analysis (Novartis Pharma AG).
Four wild-type mice received 25 mg/kg LY335979 in a 200-μl saline solution (6.7 ml/kg), administered by intraperitoneal injection 30 min before the oral dosing of [14C]STI-571. The total radioactivity of STI-571 in the plasma and brain homogenates, collected at 60 min post-STI-571 dose, was measured as described above.
Determination of the Brain Vascular Space by Dual-Labeling Using [3H]Inulin and [14C]STI-571.
The brain vascular space was determined by [3H]inulin administered via tail vein together with oral dosing of [14C]STI-571. Briefly, the mice were given an oral dose of [14C]STI-571, and 10 min before sacrificing the mice, [3H]inulin (1 μCi in 0.9% saline) (molecular weight 5000–5500) was administered via the tail vein. After euthanasia by CO2 inhalation, plasma and total brain tissue homogenate were collected and counted for both 3H and 14C radioactivity using spectrum-based dual label counting.
Intravenous Administration of STI-571 to the Wild-Type and mdr1a/b (−/−) Mice.
The mice received 12.5 mg/kg STI-571 [in 100 μl of 0.9% saline (3.3 ml/kg)] via tail vein injection. The plasma and total brain tissue were collected at different times (30, 60, and 120 min postdose, n = 4 for each time point) as described above. STI-571 was measured using LC-MS as described inBakhtiar et al. (2002).
Statistical Analysis.
Groups were compared using simple one-way analysis of variance analysis with a significance level ofp < 0.05 (Microsoft Excel, 1997). To compare the brain-to-plasma ratios in the wild-type and knockout mice at 1 h postoral dose, where the variance was not equal between groups, the nonparametric alternative of a two-sample t test, the Mann-Whitney test, was used.
Results
Directional Flux across the MDCKII Monolayer.
The directional flux study measures the percentage of compound in the donor compartment transported from the A-to-B side and from the B-to-A side of MDCKII monolayers at different time points. P-glycoprotein is located in the apical side of the monolayer and it transports substrates from the basolateral to apical side, thereby decreasing the A-to-B flux and increasing the B-to-A flux of a substrate, while not affecting the flux of the nonsubstrates. As shown in Fig. 1, the B-to-A flux of STI-571 was significantly greater (p< 0.001) and the A-to-B flux was significant lower (p< 0.001) in P-glycoprotein-transfected cell monolayers compared with wild-type cell monolayers. Also, there was a significant difference between A-to-B flux and B-to-A flux in both P-glycoprotein-transfected (p < 0.001) and wild-type cell lines (p < 0.001). However, this difference is significantly greater in P-glycoprotein-transfected cell monolayer than wild-type cell monolayer (p < 0.001). The ratio of B-to-A flux over A-to-B flux is 39 in MDR1-transfected cells and about 8 in wild-type cells (Fig. 1). These data indicate that the expression of P-glycoprotein influences the transcellular transport of STI-571 across this model barrier. The difference between A-to-B and B-to-A flux in wild-type cells suggests that an endogenous active transporter that facilitates B-to-A transport, possibly P-glycoprotein, is expressed in these cells. As shown in Fig. 2, a specific inhibitor of P-glycoprotein (Starling et al., 1997; Dantzig et al., 1999), LY335979, significantly reduced the difference between A-to-B and B-to-A fluxes in the MDR1-transfected MDCKII monolayer, strongly suggesting the endogenous transporter is P-glycoprotein (Fig.2). These in vitro cellular experiments lead to the conclusion that STI-571 is a substrate of the active efflux transporter P-glycoprotein.

Directional flux of STI-571 across MDR1-transfected and wild-type MDCKII cell monolayers. [14C]STI-571 (0.01 μCi) in assay buffer was applied to the donor chamber. The receiver chamber was sampled with replacement at the time points as indicated and counted for radioactivity. The percentage of STI-571 transported is the measured radioactivity over the initial radioactivity of the donor chamber. The A-to-B flux is the flux from the apical-to-basolateral side of the monolayer and the B-to-A flux is from the basolateral side to the apical side. The solid lines are regression lines (A). ThePeff (see Materials and Methods) represents the overall intrinsic permeability of the cell monolayer for STI-571 (B). The effective permeability in the B-to-A direction is significantly greater than in the A-to-B direction in both wild-type and MDR1-transfected cells; ∗∗∗,p < 0.001. The values are presented as mean ± S.D.

Effect of the inhibition of P-glycoprotein on the directional flux. Control, the directional flux of STI-571 across MDR1-transfected MDCKII cell monolayer were determined as described in Fig. 1. Txt LY335979, MDCKII monolayer was preincubated with 1 μM LY335979 for 30 min and then the A-to-B and B-to-A fluxes were measured with 1 μM LY335979 present in both chambers. The solid lines are regression lines (A). The effective permeability (Peff) represents the overall intrinsic permeability of the cell monolayer for STI-571 (B). The effective permeability in the MDR1-transfected cells in the B-to-A direction is significantly greater than in the A-to-B direction in both LY335979-treated and control cells; ∗∗∗, p < 0.001. The values are presented as mean ± S.D.
Brain Penetration of STI-571 in mdr1a/b (−/−) Knockout Mice and Wild-Type Mice.
When using whole brain homogenates to determine the distribution of a drug into the brain, the drug remaining in the brain vascular space needs to be excluded from the drug in the brain tissue to accurately determine the penetration of drug across the blood-brain barrier. This is especially important when the drug of interest has a relatively low CNS distribution. To accomplish this in the current study, the volume of brain vascular space was measured using [3H]inulin, which is assumed to not penetrate the blood-brain barrier (Smith et al., 1988). Our results from total brain homogenates show that the volume of brain vascular space in mice accounts for 1.4% of the whole brain volume (Fig.3). This value is comparable with those of previous studies using inulin and in situ perfusion in the rat and mouse, where the brain distribution volume of inulin was calculated to be about 1.2% (Abbruscato et al., 1997; Murakami et al., 2000). Upon oral administration of radiolabeled STI-571, only 2.7 to 3.3% the total radioactivity is distributed into the brain, as depicted by the brain-to-plasma ratio (Fig. 3). Of this percentage of radiolabeled material, when considering the radioactivity in the brain vascular space using the inulin distribution data, it can be seen that about 40 to 50% of the total radiolabeled material in the brain is localized within the brain vascular space. Therefore, all the brain distribution values of STI-571 radioactivity reported hereafter are corrected for the brain vascular space using the value measured by [3H]inulin (1.4%).

Brain-to-plasma ratio of [14C]STI-571 and [3H]inulin. Wild-type mice were given an oral dose of [14C]STI-571 and an intravenous dose of [3H]inulin (administered 10 min before sacrifice, for simultaneous determination of cerebral intravascular space). At 90 and 120 min after STI-571 oral administration, the plasma and total brain tissue homogenate were collected and counted for both 3H and 14C radioactivity using spectrum-based dual label counting. The results are expressed as mean ± S.D.
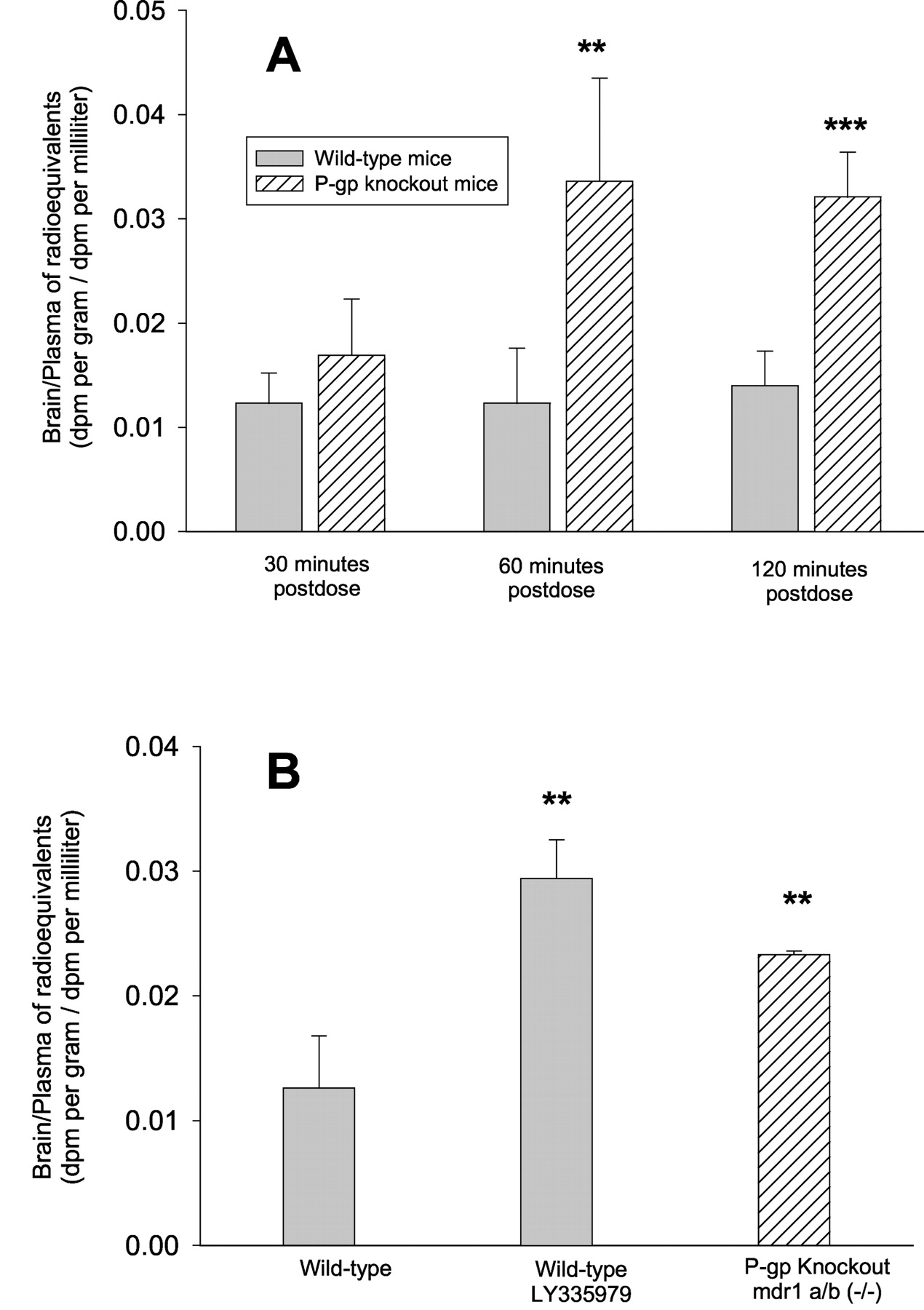
It is well known that P-glycoprotein is expressed in blood-brain barrier and can limit the distribution of its substrates into brain (Kusuhara and Sugiyama, 2002). It has been shown in our in vitro directional flux studies that STI-571 is a substrate of P-glycoprotein and it can therefore be hypothesized that the CNS distribution of STI-571 will be greater in the mdr1a/b (−/−) knockout mice. When measuring the total radioactivity in brain homogenates and plasma after oral dosing, the penetration of STI-571 radioequivalents into brain tissue (brain-to-plasma ratio) in mdr1a/b (−/−) knockout mice was 1.37-fold greater at 30 min, which is not statistically significant (p > 0.05), and significantly greater at 60 min (2.73-fold, p < 0.01) and 120 min (2.3-fold,p < 0.001), compared with that of wild-type mice (Fig.4). Treatment of the wild-type mice with the P-glycoprotein-specific inhibitor LY335979 increased the brain penetration of [14C]STI-571 total radioactivity (p < 0.01) to a similar extent as seen in mdr1a/b (−/−) knockout mice (Fig. 4).

Effect of P-glycoprotein on the brain penetration of [14C]STI-571 radioequivalents in mice. A, mdr1a/b (−/−) knockout and wild-type mice received an oral dose of 25 mg/kg [14C]STI-571. At indicated times postdose (n = 4), the plasma and total brain tissue homogenate were collected and counted for radioactivity. Results are expressed as mean ± S.D. The brain/plasma ratio was significantly greater in the knockout mice at 60 (∗∗, p < 0.01) and 120 min postdose (∗∗∗, p < 0.001). B, mdr1a/b (−/−) knockout and wild-type mice received an oral dose of 25 mg/kg [14C]STI-571. Of eight wild-type mice, four received 25 mg/kg LY335979 via intraperitoneal injection 30 min before the oral dosing. The radioactivity of [14C]STI-571 in the plasma and brain homogenates was measured at 60 min post-STI-571. The brain-to-plasma concentration ratio has been corrected for the brain vascular space and is expressed as mean ± S.D. The brain/plasma ratio in both the LY335979-treated wild-type and the untreated knockout mice was significantly greater than the control wild-type mice; ∗∗,p < 0.01. The values are presented as mean ± S.D.
Because it is likely that STI-571 is metabolized in the mouse, which would confound the interpretation of total radioactivity levels, deproteinized plasma and brain homogenate samples from the 60-min postdose time point were injected onto the HPLC column and eluants were collected for radioactivity measurement. It was found that a significant amount of radioactivity (about 60–65%) was not associated with the parental STI-571 peak, indicating STI-571 is extensively metabolized in mice when given orally. When considering the radioactivity associated with parent STI-571, the brain penetration of STI-571 in mdr1a/b (−/−) knockout mice is 11.2-fold higher than that of wild-type mice 1 h after oral dosing (p < 0.005) (Fig. 5).

Brain distribution of the radioactivity associated with parent STI-571 in mdr1a/b (−/−) knockout and wild-type mice at 60 min postoral dose. The mdr1a/b (−/−) knockout and wild-type mice received 25 mg/kg [14C]STI-571 orally. At 60 min postdose (n = 4), the plasma and brain homogenate were collected, deproteinized, evaporated, and reconstituted with mobile phase. The samples were analyzed by HPLC to determine the radioactivity that was associated with the retention time of the parent drug. The results are expressed as mean ± S.D, and the brain/plasma ratio in the knockout mice is significantly greater than in the wild-type mice; ∗, p < 0.05. The values are presented as mean ± S.D.
To further examine the CNS distribution of STI-571 in wild-type and P-glycoprotein knockout mice, nonradiolabeled STI-571 (12.5 mg/kg) was administered via tail vein injection and concentrations of parent STI-571 in the plasma and brain at 30, 60, and 120 min postadministration were measured by LC-MS (Bakhtiar et al., 2002). The results indicate that the STI-571 level in the brain is significantly greater in the knockout mice compared with the wild-type mice, even though the plasma levels of STI-571 were similar in wild-type and the knockout mice (Fig. 6). The brain-to-plasma ratio of STI-571 in mdr1a/b (−/−) knockout mice is 6- to7-fold greater than that of wild-type mice (p < 0.001). These data clearly indicate that P-glycoprotein is an important factor in limiting the distribution of STI-571 into the CNS.

Brain distribution of STI-571 in the mdr1a/b (−/−) knockout and wild-type mice after intravenous dosing. The mdr1a/b (−/−) knockout and wild-type mice received 12.5 mg/kg STI-571 via tail vein injection. The plasma and total brain tissue were collected at different times (30, 60, and 120 min postdose, n= 4 each) and analyzed for STI-571 using LC-MS. A, plasma concentration of STI-571 versus time in mdr1a/b (−/−) knockout and wild-type mice. B, brain concentration of STI-571 versus time in mdr1a/b (−/−) knockout and wild-type mice. C, ratio of brain penetration of STI-571 in mdr1a/b (−/−) knockout mice versus wild-type mice at different time points. The values are presented as mean ± S.D.
Discussion
STI-571 is the first molecularly targeted antitumor agent, exerting its antitumor effect by inhibiting signal transduction pathways (Buchdunger et al., 1996; Druker et al., 1996; Deininger et al., 1997; Heinrich et al., 2000). Approximately 95% of patients with CML and some patients with acute lymphoid leukemia or acute myeloid leukemia, exhibit the Philadelphia chromosome, a translocation between chromosome 9 and 22. This results in the fusion gene BCR/ABL that encodes a 210-kDa protein that has deregulated tyrosine kinase activity (Rowley, 1973; Daley et al., 1990; Melo, 1996). This mutant tyrosine kinase activates downstream signal transduction pathways that lead to CML (Konopka et al., 1984; Kelliher et al., 1990). STI-571 can inhibit the BCR/ABL-encoded tyrosine kinase and subsequently the downstream transduction pathway (Buchdunger et al., 1996; Druker et al., 1996). As a result, it inhibits the proliferation of the tumor cells (Druker et al., 1996; Deininger et al., 1997) and induces apoptosis (Gambacorti-Passerini et al., 1997). Because it attacks a specific target, STI-571 has so far been shown to have mild side effects in contrast with conventional nontargeted cytotoxic agents (Druker et al., 2001). In addition to BCR/ABL, STI-571 has inhibitory effects on c-kit and PDGFR kinase (Buchdunger et al., 2000; Heinrich et al., 2000). These two enzymes are involved in the development of glioblastoma, gastrointestinal stromal tumor, and small cell lung carcinoma (Uhrbom et al., 2000; Wang et al., 2000; Tuveson et al., 2001). Thus, it is possible that STI-571 may also have therapeutic effects on these tumors. However, the therapeutic effect of a drug will depend largely on the targeted bioavailability of the drug at the site of action.
A major challenge in the treatment of brain tumors, including secondary brain tumors, is the effective delivery of antitumor compounds across blood-brain barrier into the brain (Lesniak et al., 2001). It is possible that the blood-brain barrier is disrupted by the disease process in brain tumor (Davies, 2002), although the role of the blood-brain barrier in drug delivery to brain tumors has been controversial (Groothuis, 2000). In some cases, it may be that a drug such as STI-571 can enter the brain parencyhma through a “leaky” blood-brain barrier in some areas of the tumor. Nevertheless, it is important to recognize that the barrier of interest may be the blood-brain barrier in the brain around tumor, or the growing edge of a brain tumor, where the blood-brain barrier may have a full complement of anatomical and physiological features to limit the transport of drugs (Levin et al., 1975). It has been shown that various efflux transporters are expressed in blood-brain barrier and blood-cerebrospinal fluid barrier that eliminate compounds from the brain or cerebrospinal fluid to the blood (Kusuhara and Sugiyama, 2002). P-glycoprotein is expressed on the apical side of the brain capillary endothelium (Cordon-Cardo et al., 1989; Thiebaut et al., 1989; Beaulieu et al., 1997) and can transport substrates from the basolateral side (brain) to the apical side (blood) of the blood-brain barrier and thus limit the brain distribution and decrease the specific brain tissue bioavailability of many therapeutic agents (Kusuhara and Sugiyama, 2002), including those used for brain tumors (Regina et al., 2001). Therefore, for rational use of STI-571 in brain tumor, it is important to know whether STI-571 is a substrate of P-glycoprotein and whether this active efflux transporter is an effective limiting determinant of STI-571 distribution to the brain in vivo. Such information would provide valuable insight for the various ongoing clinical trials of STI-571 in the treatment of brain tumor.
The cellular accumulation and directional flux of a compound across a polarized cell monolayer, particularly those transfected with a specific transporter, are useful in vitro methods to determine the substrate status of a drug. Compared with cellular accumulation studies, one of the advantages of the directional flux method is that it gives information about whether the compound is transported by a particular active transporter if one knows the orientation of that transporter. The directional flux results shows that the B-to-A flux of STI-571 is significantly greater than the A-to-B flux in the MDCKII monolayer, indicating STI-571 is transported from the basolateral-to-apical side by an active transporter, which is consistent with the localization and orientation of P-glycoprotein. This is more evident in the MDR1-transfected MDCKII cellular monolayer than the wild-type monolayer. Moreover, when a specific inhibitor of P-glycoprotein is used (LY335979), the difference between the B-to-A flux and A-to-B flux in the mdr1-transfected cells is significantly reduced. These data clearly demonstrate that STI-571 is a substrate of P-glycoprotein.
The CNS distribution of antitumor agents can be limited by many factors, including 1) the physicochemical properties of the compound (hydrophilicity or size), 2) plasma protein binding, and 3) efflux transport by CNS efflux transporters. A compound with limited brain distribution may be a substrate of competing active transport systems, which would make it difficult to sort out the significance of various contributing factors that may limit its CNS distribution. The use of knockout mice, however, will give us an important tool to achieve this goal. Because the mdr1a/b genes are absent in mdr1a/b (−/−) knockout mice, the contribution of P-glycoprotein in limiting the brain distribution of STI-571 can be clearly demonstrated. In this study, by using the mdr1a/b (−/−) knockout mice, it is shown that lacking P-glycoprotein leads to a severalfold increase in the CNS penetration of STI-571, indicating P-glycoprotein plays a significant role in the distribution of STI-571 into the brain. The results in this study are the first direct evidence to show that P-glycoprotein transports STI-571 and limits its distribution into the brain.
When [14C]STI-571 was given orally, it is found that only 35 to 40% of the total radioactivity is associated with the STI-571 parent compound in the plasma at 60 min postdose, as indicated by HPLC. Under such a situation, the brain-to-plasma ratio of total radioactivity of [14C]STI-571 will not reflect the true brain-to-plasma ratio of parental STI-571. The increase in the brain-to-plasma ratio of total radioactivity of [14C]STI-571 in the knockout versus the wild-type mice is only about 2.7 at 1 h postdose and it is about 11-fold after correcting for the radioactivity associated with the STI-571 peak. However, when the drug is given intravenously, the brain-to-plasma ratio of STI-571 in knockout mice is about 7-fold greater than that of wild-type. It is possible that this difference may be due to different plasma and brain concentration-time profiles of STI-571 after different routes of administration. However, with both dosing modalities, STI-571 has a greater CNS penetration in the mice deficient in P-glycoprotein than the wild-type mice, indicating the importance of P-glycoprotein in the CNS distribution of STI-571.
It is interesting to note that the concentration-time profile of STI-571 in the brain parallels that of the drug in plasma as seen in Fig. 6. One question that arises is how this rapid equilibrium is established in the face of a relatively low permeability. We have considered this in regard to the present sparse data, and we feel that one explanation may be that for a drug with a small volume of distribution in the CNS, and a rapid efflux, the concentration-time profile in the brain will quickly reflect changes in the concentration-time profile in the plasma because the small volume will quickly respond to a change in driving force as long as the drug can exit easily. Further studies need to be performed to fully describe the distributional kinetics of STI-571 in the brain.
Also of interest, in case of intravenous administration, the brain-to-plasma ratio is higher than that of oral dosing (compare Figs.4 and 6). There could be several reasons for this difference, and we do not want to speculate too much at this stage of the work. However, one possibility is that the free concentration of STI-571 after i.v. bolus dosing is higher than after oral dosing. Bearing in mind that STI-571 is a highly plasma protein-bound drug, it is possible that a higher concentration of STI-571 could cause saturation of protein binding and lead to a greater free fraction, which would result in a greater driving force for drug in the plasma to enter the brain. Our future experimental studies will be to quantitatively describe the kinetics of unbound STI-571 in both plasma and brain using microdialysis. Also, at this stage we cannot rule out other possibilities such as different concentration-time profiles due to different routes of administration of drug, or saturation of other efflux transporters by the high plasma concentration in case of i.v. administration. No matter what the reason may be, however, the conclusion regarding the P-glycoprotein substrate status of STI-571 is not affected.
It is expected that a limited distribution of STI-571 into the CNS would decrease the efficacy of this peripherally effective compound in the treatment of CNS tumor, either primary glioblastoma or secondary CML in the CNS. Indeed, it is reported that isolated CML relapse has occurred in the brain even after the peripheral leukemia has been successfully treated (Leis et al., 2001; Petzer et al., 2002; Takayama et al., 2002). It has also been established that resistance to STI-571 has occurred in some patients due to a point mutation in the ATP-binding site of the enzyme or multiplication of the BCR/ABL gene (Gorre et al., 2001), and it has been shown that long-term exposure to a suboptimal concentration of STI-571 could lead to the above-mentioned mutation or gene multiplication (Mahon et al., 2000). Thus, it is likely that low concentrations of STI-571 in CNS may also lead to a mutation or overexpression of BCR/ABL in the CNS tumor cells, which in turn would lead to resistance to this drug. Therefore, elevating the STI-571 concentration in the brain by inhibiting P-glycoprotein will not only increase the efficacy in primary and secondary brain tumors but also may possibly reduce the development of resistance. It is worth noting that P-glycoprotein may also be expressed in the cytoplasmic membrane of the tumor cells, including glioblastoma (Tews et al., 2000;Demeule et al., 2001) and gastrointestinal stroma (Plaat et al., 2000), and as such may limit the penetration of the drug into the cell. Inhibition of P-glycoprotein would further increase the targeted bioavailability of STI-571 into the tumor cells.
In summary, the results reported here conclusively show that STI-571 is a substrate of P-glycoprotein and that this efflux transporter is an important determinant of distribution of STI-571 to the central nervous system. The functional evidence, both in vitro and in vivo, indicates that inhibition of P-glycoprotein may enhance the CNS delivery of STI-571. Additional studies are needed to quantitatively characterize the role of drug efflux in limiting the targeted bioavailability of this important drug to the brain.
Acknowledgments
We thank Eli Lilly Cancer Research Laboratory and Dr. Piet Borst (Netherlands Cancer Institute) for generously providing the Pgp inhibitor LY335979 and the MDCKII cell lines, respectively.
Footnotes
-
This project was partially supported by National Institutes of Health Grant CA75466, a grant from Novartis Pharma, and by a fellowship (to H.D.) from the graduate school of University of Nebraska Medical Center.
-
DOI: 10.1124/jpet.102.045260
- Abbreviations:
- PDGFR
- platelet-derived growth factor receptor
- CML
- chronic myelogenous leukemia
- CNS
- central nervous system
- MDR1
- multidrug resistance-1 gene
- MDCK
- Madin-Darby canine kidney
- A-to-B
- apical-to-basal
- B-to-A
- basal-to-apical
- HPLC
- high-performance liquid chromatography
- LC-MS
- liquid chromatography-mass spectrometry
- Received October 7, 2002.
- Accepted November 25, 2002.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}