


Abstract
The present study examined whether zolpidem, an imidazopyridine with selectivity for benzodiazepine (BZ)/γ-aminobutyric acidAreceptors containing the α1-subunit, had discriminative stimulus effects similar to typical BZs and other sedative/hypnotic drugs in primates. Squirrel monkeys (Saimiri sciureus) were trained to discriminate zolpidem (1.0 mg/kg i.v.) from vehicle under a 10-response fixed-ratio schedule of food delivery. Under test conditions, zolpidem (0.1–3.0 mg/kg) increased responding on the drug lever to an average maximum of 90% of total responding. When pretreatment times were varied from 5 to 50 min, the discriminative stimulus effects of zolpidem were maximal at 5 min and near control levels 35 min after administration. Flumazenil antagonized both the discriminative stimulus and rate-decreasing effects of zolpidem in a dose-dependent and surmountable fashion (in vivo apparent pA2 values of 7.3 and 6.6 for the discriminative stimulus and rate-suppressing effects, respectively). The BZs triazolam, midazolam, diazepam, and N-desmethyldiazepam engendered dose-related increases in drug-lever responding that reached zolpidem-like levels (90%) in the majority of monkeys tested. In contrast, lorazepam, chlordiazepoxide, and oxazepam engendered average maximums of 70% or less and substituted fully for zolpidem in one or two monkeys only. Representative barbiturates as well as drugs that bind to non-BZ sites (muscimol, baclofen, buspirone, cyproheptadine, diphenhydramine) engendered 0 to 45% of responses on the drug lever up to doses that markedly reduced response rate. These results support the view that zolpidem's selectivity for the α1-subunit of the BZ/γ-aminobutyric acidA receptor complex confers a distinctive profile of interoceptive effects that overlaps partially with those of typical BZs but not with those of barbiturates.
Zolpidem (Ambien) is an imidazopyridine hypnotic drug that acts at the benzodiazepine (BZ) recognition site associated with the γ-aminobutyric acidA(GABAA) receptor/chloride channel complex (for review, see Rush, 1998). The receptor-binding profile of zolpidem is distinct from that of typical BZs in that it displays highest affinity at GABAA receptors expressing α1-subunits (Ki = 15–25 nM), which are thought to be associated with the BZ1 receptor subtype (Pritchett and Seeburg, 1990; Hadingham et al., 1993). Zolpidem binds with intermediate to low affinity (Kivalues ranging from 350 to > 15,000 nM) at receptors expressing α2-, α3-, and α5-subunits, which together comprise the BZ2 receptor subtype (Pritchett and Seeburg, 1990; Hadingham et al., 1993). Moreover, binding sites for zolpidem are denser in the cerebellum than in other brain regions (Dennis et al., 1988; Benavides et al., 1993), consistent with zolpidem's apparent selectivity for the BZ1 receptor subtype (for review, see Sanger et al., 1994; Lüddens et al., 1995).
The profile of behavioral effects produced by zolpidem also appears to differ from that of typical BZs. In rats trained to discriminate chlordiazepoxide from saline, for example, zolpidem engendered only partial drug-appropriate responding, whereas both chlordiazepoxide and triazolam engendered full drug-appropriate responding (Depoortere et al., 1986; Sanger et al., 1987). Zolpidem, unlike conventional BZs, also did not mimic the discriminative stimulus (DS) effects of pentobarbital in rats (Ator and Griffiths, 1989; Rowlett and Woolverton, 1997) and did not substitute for a high training dose of midazolam in rats (Sannerud and Ator, 1995b). Moreover, when zolpidem was trained as a DS in rats, BZs and pentobarbital engendered only partial drug-appropriate responding up to doses that markedly suppressed rates of responding (Sanger and Zivkovic, 1986).
In contrast to the results obtained in rodents, recent studies with monkeys and human volunteers suggest that the DS effects of zolpidem are similar to those of typical BZ receptor agonists (Griffiths et al., 1992; Rowlett and Woolverton, 1997; Rush et al., 1997). In this regard, zolpidem fully mimicked the DS effects of lorazepam in baboons (Griffiths et al., 1992). Zolpidem also engendered full drug-appropriate responding in pentobarbital-trained monkeys (Griffiths et al., 1992; Rowlett and Woolverton, 1997), as well as in human volunteers trained to discriminate pentobarbital from placebo (Rush et al., 1997). Similarly, the subject-rated effects of zolpidem were comparable to those induced by typical BZs in people (Rush and Griffiths, 1996; Rush et al., 1997). Collectively, these results raise the possibility of species differences with respect to the interoceptive effects of zolpidem. Consistent with this view, in vivo receptor-binding studies have suggested differences between rodents and primates with respect to the heterogeneity of binding sites recognized by zolpidem (Schmid et al., 1995).
Because zolpidem consistently mimics the DS effects of typical BZs and barbiturates in monkeys trained to discriminate either lorazepam or pentobarbital, it might be assumed that typical anxiolytic and hypnotic drugs would correspondingly mimic the DS effects of zolpidem in these species. To date, however, there have been no published articles on the interoceptive effects of drugs in monkeys trained to discriminate zolpidem from vehicle. The purpose of the present study, therefore, was to establish zolpidem as a DS in nonhuman primates (squirrel monkeys,Saimiri sciureus) and to evaluate the degree to which zolpidem's effects could be reproduced by typical BZ receptor agonists, barbiturates, and selected reference compounds. The procedures used in these experiments were as similar as possible to those of a previous study in which squirrel monkeys were trained to discriminate midazolam from vehicle (Spealman, 1985) to facilitate comparison of the effects of zolpidem with those of a typical BZ receptor agonist.
Materials and Methods
Subjects and Apparatus.
Four adult male squirrel monkeys, weighing between 720 and 900 g at the beginning of the study, were used as subjects. All monkeys were experimentally naive at the beginning of the study. Between experimental sessions, the monkeys lived in individual cages with water available continuously. Each animal was fed a nutritionally balanced diet (Teklad monkey diet; Teklad, Inc., Monmouth, IL) supplemented with fresh fruit in amounts sufficient to maintain them at ∼85 to 90% of their free-feeding body weights. Animals in this study were maintained in accordance with the guidelines of the Committee on Animals of the Harvard Medical School and the “Guide for Care and Use of Laboratory Animals” of the Institute of Laboratory Animal Resources. Research protocols were approved by the Harvard Medical School Institutional Animal Care and Use Committee.
Monkeys were surgically prepared with chronic venous catheters following the general procedure described by Carey and Spealman (1999). Under isoflurane anesthesia and in aseptic conditions, one end of a polyvinyl catheter (0.38-mm i.d.; 0.76-mm o.d.) was passed by way of a femoral vein to the level of the right atrium. The distal end of the catheter was passed s.c. and exited through the skin in the midscapular region. Catheters were flushed daily with 0.9% saline solution containing heparin (150 U/ml) and were sealed with stainless steel obturators when not in use. Monkeys wore nylon-mesh jackets (Lomir Biomedical, Toronto, Canada) at all times to protect the catheter.
During experimental sessions, monkeys were seated in a Plexiglas chair identical to the chairs used previously to study the discriminative stimulus effects of midazolam (Spealman, 1985). Two response levers (BRS/LVE; model 121-05) were mounted 15 cm apart on the wall of the chair in front of the monkey. A press of either lever produced an audible click and was recorded as a response. Food pellets (Formula F sucrose pellets, 190 mg; P. J. Noyes Co. Inc., Lancaster, NH) could be delivered to a tray located between the levers. Lights, mounted at eye level above the levers, were illuminated during the session except during timeout periods (see below). The chair was enclosed in a ventilated, sound-attenuating chamber with white noise to mask extraneous sounds.
Drug Discrimination Procedure.
Procedures used to establish the DS effects of zolpidem were similar to those described previously for midazolam (Spealman, 1985). Before surgery, each monkey was trained to respond on both levers under a 10-response fixed-ratio (FR 10) schedule of food reinforcement. Once stable response rates were obtained (no increasing or decreasing trends for at least three sessions on either lever), the monkeys were prepared with catheters and drug discrimination training started 2 to 4 days later. After i.v. injections of zolpidem (1.0 mg/kg), 10 consecutive responses on one lever produced a food pellet, whereas after i.v. injections of saline 10 consecutive responses on the other lever produced a pellet. For half of the monkeys, responding on the right lever following an injection of zolpidem resulted in pellet delivery, whereas responding on the left lever following injection of zolpidem was reinforced for the other monkeys. Delivery of each pellet was followed by a 10-s timeout period. Responses on the incorrect lever (e.g., the saline-appropriate lever when zolpidem was injected) reset the FR requirement. Training sessions consisted of a variable number of components (n = 1–4) of the FR schedule. Each component ended after the completion of the 10th FR 10 or after 5 min had elapsed, whichever occurred first. A 10-min timeout period, during which the lights were off and responses had no programmed consequences, preceded each component. During most training sessions, saline was injected during timeout periods preceding the first n − 1 components, and zolpidem was injected before the last component of the session. Periodically, saline was injected before all components of a training session to prevent an invariant association between the last component and zolpidem. Injections of zolpidem or saline were administered outside the chamber via a catheter extension and were given during the fifth min of the 10-min timeout periods. Each injection was followed by a 1-ml injection of saline to clear the catheter of any residual drug solution.
Drug-Testing Procedure.
Drug test sessions were conducted once or twice per week with training sessions scheduled on intervening days. Test sessions were conducted only if ≥80% of responses were made on the injection-appropriate lever during at least four of the preceding five training sessions. Test sessions consisted of four FR components, each preceded by a 10-min timeout period. In each component, completion of 10 consecutive responses on either lever produced food. In most experiments, dose-response functions were determined for test drugs with the cumulative dosing procedure described by Spealman (1985). Under this procedure, incremental doses were injected i.v. during the fifth min of the 10-min timeout periods that preceded sequential FR components, permitting a four-point cumulative dose-response function to be determined in a single session. Each dose-response function was determined at least twice in each subject: the first determination consisted of four cumulative doses, and the second determination consisted of the same doses except that drug vehicle was given first instead of the lowest dose of drug. A third determination, consisting of higher doses of the test drug, was conducted if <90% drug-lever responding was observed or if rate of responding was not reduced to or below 10% of saline values. The drugs studied with the cumulative-dosing procedure were: zolpidem (0.1–3.0 mg/kg); the conventional BZ agonists triazolam (0.001–0.1 mg/kg), midazolam (0.1–3.0 mg/kg), lorazepam (0.03–10 mg/kg), diazepam (0.1–3.0 mg/kg), chlordiazepoxide (1.0–30 mg/kg),N-desmethyldiazepam (0.1–3.0 mg/kg), and oxazepam (1.0–30 mg/kg); the BZ antagonist flumazenil (0.01–1.0 mg/kg); the barbiturates pentobarbital (0.3–18 mg/kg), barbital (3.0–56 mg/kg), and amobarbital (1.0–18 mg/kg); the 5-hydroxytryptamine (5-HT) antagonist cyproheptadine (0.3–5.6 mg/kg); the 5-HT1aagonist buspirone (0.03–0.56 mg/kg); the histamine H1antagonist diphenhydramine (0.3–5.6 mg/kg); the GABAAagonist muscimol (0.03–0.56 mg/kg); and the GABAB agonist baclofen (0.3–10 mg/kg).
Additional studies with zolpidem were conducted with a conventional single-dose testing procedure to determine the comparability of dose-response functions obtained by the two methods and by varying pretreatment times (5–50 min) to determine the time course of zolpidem's effects. The ultrashort-acting barbiturate methohexital (0.3–3.0 mg/kg) also was evaluated with a conventional single-dose procedure, in which the drug was administered immediately before the test sessions and the length of the session was reduced by decreasing the number of FRs per component from 10 to 5. For all drug substitution experiments, the order of drug testing was different for each monkey, and all drugs were tested in at least three monkeys.
Antagonism studies were conducted by administering flumazenil (0.03–1.0 mg/kg i.v.) immediately before the session, followed by cumulative doses of zolpidem as described above. Rightward shifts in the zolpidem dose-response function were evaluated by testing higher doses of zolpidem in combination with the higher doses of flumazenil. The dose-response functions in the antagonism studies were determined once in three monkeys. At the end of the study, the cumulative dose-response function for zolpidem was redetermined in all monkeys to identify any changes in sensitivity to the training drug that may have developed over the course of the experiments.
Analysis of Drug Effects.
Percentage of zolpidem-lever responding was computed for individual subjects in each component of a test session by dividing the number of responses on that lever by the total number of responses on both levers and multiplying by 100. Percentage of zolpidem-lever responding was calculated for an individual monkey only if the response rate was >0.1 responses/s during the component. Mean percentage of zolpidem-lever responding and S.E.M. were then calculated for the group of monkeys at each dose. An additional analysis of percentage drug-lever responding was conducted to evaluate individual differences in the DS effects of the test compounds. For individual animals, the ability of a drug to substitute fully for zolpidem was analyzed based on the average maximum for percentage of drug-lever responding for zolpidem at the 1.0 mg/kg training dose. The first and second determinations of this dose of zolpidem, obtained from cumulative dose-response functions, were averaged for each monkey and a group mean with 95% confidence interval (CI) was computed. For each animal, the highest percentage of zolpidem-lever responding for a test compound, irrespective of dose, was compared with the lower limit of the CI. A drug was considered to substitute fully for zolpidem in an individual monkey if the maximum percentage of drug-lever responding fell within the lower limit of the CI value for 1.0 mg/kg zolpidem.
The overall rate of responding in each component was computed by dividing the total number of responses in a component (regardless of lever) by the total component duration. Rate of responding data were converted to percentage of control by dividing an individual animal's response rate after drug or vehicle by that animal's average response rate during saline training sessions (average of 2 saline sessions immediately before the test session), and multiplying by 100. Mean response rates (% control ± S.E.M.) were then calculated for the group at each dose.
The doses of drug needed to engender 50% zolpidem-appropriate responding or suppression of response rate (ED50) were calculated with nonlinear regression analysis. The nonlinear regression analysis used was an iterative curve-fitting technique for sigmoidal dose-response functions with variable slopes. The equation used for the analyses was the four-parameter logistic equation: y = min + max(max/[1 + eslope(dose − ED50)]), where min equals the lowest value for percentage of drug-lever responding or reduction of response rate, and max equals the highest values obtained for these measures. All parameters in the nonlinear regression analysis were free to vary. For all curves, standard errors of the coefficient (SEC) were obtained for each estimate as a measure of variability analogous to S.E.M. In addition, apparent pA2 analysis was conducted according the method of Arunlakshana and Schild (1959), with drug dose substituted for drug concentration (Takemori, 1974). A Schild plot was constructed by calculating the relationship between dose of flumazenil (expressed as −log[mol/kg]) and the logarithm of the dose ratio-1 (ED50 for zolpidem plus flumazenil/ED50 for zolpidem alone). Linear regression analysis was conducted to obtain the slope with 95% CIs to test whether the slope differed reliably from −1.0, which could indicate a violation of the assumption of unity (Tallarida et al., 1979). The apparent pA2 value was obtained as thex-intercept computed via linear regression analysis. The regression estimate of slope was assessed for deviation from zero by the t ratio of the slope coefficient to the SEC. For comparisons with previous antagonism results with midazolam-trained squirrel monkeys (Spealman, 1985), in vivo apparent pKB values were obtained for 0.1 and 1.0 mg/kg flumazenil according to the calculation described by Rowlett and Woolverton (1996). In vivo pKB analysis is based on the same theoretical framework as apparent pA2 analysis, the primary difference being that the former value is obtained with a single antagonist concentration (Negus et al., 1993).
Drugs.
The base forms of zolpidem, triazolam, diazepam, lorazepam, N-desmethyldiazepam, oxazepam, muscimol, and baclofen as well as chlordiazepoxide HCl, midazolam maleate, diphenhydramine HCl, sodium pentobarbital, and sodium amobarbital were purchased from commercial sources (Research Biochemicals Inc., Natick, MA; Sigma Chemical Co., St. Louis, MO). Other drugs were gifts from the manufacturers, including buspirone HCl (Mead Johnson Laboratories, Evansville, IL), cyproheptadine HCl and sodium barbital (Merck Sharp & Dohme, West Point, PA), sodium methohexital (Eli Lilly and Co., Indianapolis, IN), and flumazenil (Hoffman-La Roche Inc., Nutley, NJ). Zolpidem was mixed in a 45% (w/v) solution of hydroxypropyl-β-cyclodextrin (Research Biochemicals Inc.). Triazolam, diazepam, lorazepam, N-desmethyldiazepam, oxazepam, flumazenil, pentobarbital, and barbital were dissolved in propylene glycol and then diluted to a 50% propylene glycol/50% saline solution. Chlordiazepoxide, amobarbital, cyproheptadine, buspirone, muscimol, and baclofen were dissolved in 0.9% saline solution. All drugs were injected i.v. in volumes of 0.5 to 1.0 ml/kg.
Results
Zolpidem Discrimination.
Monkeys acquired the zolpidem discrimination after 42 to 118 sessions (median sessions-to-criteria, 70.5). During training sessions conducted over the course of the study, individual monkeys made between 80 and 100% of responses (mean ± S.E. = 89 ± 0.7) on the zolpidem-associated lever after injections of zolpidem and 0 to 15% (mean ± S.E. = 1.0 ± 0.03) of responses on the zolpidem-associated lever after injections of saline. Rates of responding during training sessions over the course of the study were typically lower after injections of zolpidem (mean responses/s = 1.42 ± 0.018) than after injections of saline (mean responses/s = 2.31 ± 0.019).
During initial determination of the dose-response function for zolpidem, increasing cumulative doses engendered corresponding increases in drug-lever responding (Fig.1, top, filled circles), reaching an average maximum of ≥90% at doses of 1.0 mg/kg or higher. Nearly identical dose-response functions were obtained with a conventional single-dose testing procedure (Fig. 1, top, triangles) and again when cumulative doses of zolpidem were tested at the end of the study, ∼1 year later (open circles). ED50 values determined for the three zolpidem dose-response functions were 0.23, 0.25, and 0.30 mg/kg, respectively. Zolpidem suppressed the average rate of responding in a dose-related fashion at doses >0.10 mg/kg (Fig. 1, bottom). Again, no marked differences were apparent in the rate-decreasing effects of zolpidem in the initial cumulative dose-response determination, single dose tests, or the final cumulative dose-response determination (ED50 value = 0.87, 0.88, and 1.05 mg/kg, respectively).
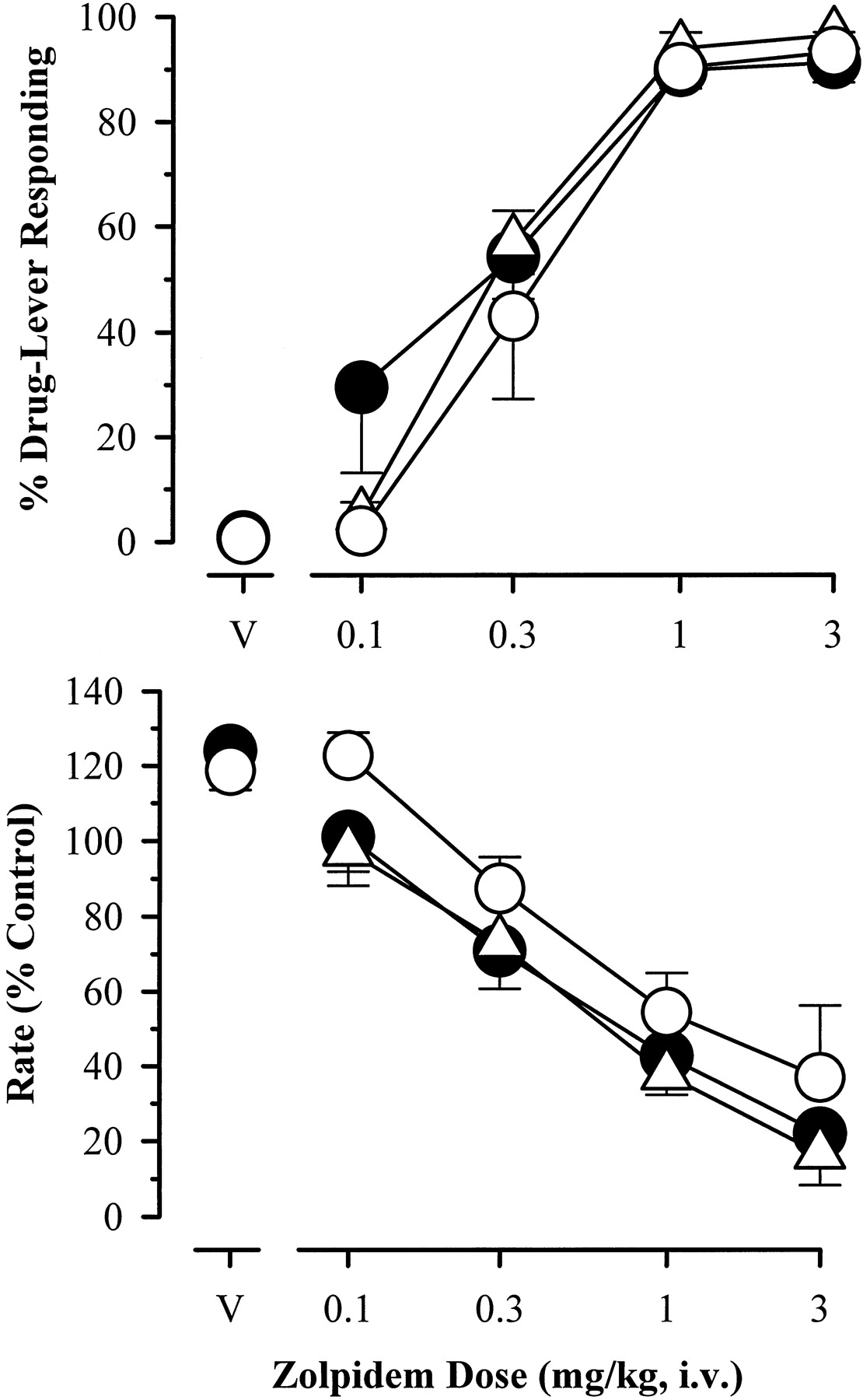
Percentage of drug-lever responding (means ± S.E.) and response rate (mean percentage of saline control ± S.E.) for a range of doses of zolpidem in squirrel monkeys (n = 4) trained to discriminate zolpidem (1.0 mg/kg) from saline. Points above “V” represent data from test sessions with the zolpidem vehicle (45% hydroxypropyl-β-cyclodextrin). The second determination of the cumulative dose-response function was obtained ∼1 year after the initial determination. ●, cumulative dosing; ▵, single dosing; ○, second determination (cumulative).
Experiments in which the zolpidem pretreatment time was varied showed that the percentage of drug-lever responding after 0.30 and 1.0 mg/kg zolpidem was maximal when administered 5 min before the session and decreased with increasing pretreatment intervals (Fig.2, top). For both doses, drug-lever responding was at or below 20% drug-lever responding by 35 min. Similarly, the rate-decreasing effects of zolpidem declined as a function of pretreatment time and were no longer apparent 20 (0.3 mg/kg) or 35 min (1.0 mg/kg) after administration (Fig. 2, bottom).

Percentage of drug-lever responding (means ± S.E.) and response rate (mean percentage of saline control ± S.E.) for zolpidem after a range of presession times in squirrel monkeys (n = 3) trained to discriminate zolpidem (1.0 mg/kg) from saline. ○, 0.30 mg/kg zolpidem; ●, 1.0 mg/kg zolpidem.
Flumazenil Antagonism.
Pretreatments with flumazenil resulted in dose-dependent shifts to the right in the dose-response function for both the DS and rate-decreasing effects of zolpidem (Fig.3; dose-response function for zolpidem alone was the average of the initial and second determinations with cumulative-dosing procedures). In general, increasing the dose of zolpidem could surmount the antagonism by flumazenil, although full substitution was not always observed after 0.1 or 0.3 mg/kg flumazenil. In vivo apparent pA2 analyses conducted on these data revealed pA2 values of 7.3 (± .47 SEC) for percentage of drug-lever responding and 6.6 (± 1.3) for response rate (Fig.4). The slope of the Schild function for percentage of drug-appropriate responding (−0.89) was reliably different from zero [t(1) = −11.9;p < .01], indicating a statistically reliable relationship between log(DR-1) and the dose of flumazenil. This slope also was not reliably different from −1.0, indicating that the assumption of unity was met and that the antagonism reflected a single receptor population. In contrast to the slope obtained for drug-appropriate responding, the slope of the Schild function for response rate (−0.69) was reliably different from −1.0. Moreover, this slope was not reliably different from zero [t(1) = −3.6; p = 0.068], indicating a relatively high level of variability for this estimate.

Percentage of drug-lever responding (means ± S.E.) and response rate (mean percentage of saline control ± S.E.) for zolpidem in the presence and absence of the benzodiazepine receptor antagonist flumazenil in squirrel monkeys (n = 3) trained to discriminate zolpidem (1.0 mg/kg) from saline. Each dose of flumazenil was administered i.v. immediately before sessions in which cumulative doses of zolpidem were tested. ●, zolpidem alone; ▿, +0.03 flumazenil; ⋄, +0.1 flumazenil; ■, +0.3 flumazenil; ▵, +1.0 flumazenil.

Schild plot for flumazenil antagonism of the discriminative stimulus and rate-altering effects of zolpidem in squirrel monkeys trained to discriminate zolpidem (1.0 mg/kg) from saline. DR, dose-ratio, computed by dividing the ED50 of zolpidem plus flumazenil by the ED50 for zolpidem alone. ●, % drug-lever responding; ○, response rate.
In vivo apparent pKB values for flumazenil antagonism of the DS and rate-altering effects of zolpidem were computed based on two doses of flumazenil used by Spealman (1985) and are shown in Table 1. For DS effects, the CI associated with apparent pKB values for zolpidem overlapped with pKB values obtained after reanalysis of flumazenil antagonism of the DS effects of midazolam. Similarly, for response rate suppression the CI associated with apparent pKB values for zolpidem overlapped with the pKB values for flumazenil antagonism of midazolam's rate-suppressing effects.
In vivo apparent pKB analysis of antagonism of the discriminative stimulus and rate-altering effects of zolpidem and midazolam in squirrel monkeys
Effects of Benzodiazepines.
Dose-related increases in drug-lever responding and decreases in response rate were observed after cumulative doses of triazolam, midazolam, diazepam, andN-desmethyldiazepam (Fig.5) as well as lorazepam, chlordiazepoxide, and oxazepam (Fig. 6). Based on the ED50 values (Table2) for all BZs (except oxazepam, which did not engender >50% drug-lever responding), the rank order of potency for percentage of drug-lever responding was triazolam > lorazepam ≅ zolpidem > midazolam >N-desmethlydiazepam ≅ diazepam > chlordiazepoxide. The rank order for response rate suppression differed from that of percentage of drug-lever responding: triazolam > zolpidem > diazepam ≅ lorazepam = midazolam ≅N-desmethyldiazepam > chlordiazepoxide (Table 2). The most notable difference in the rank order of potencies was for lorazepam, which was 15-fold more potent in engendering zolpidem-lever responding than suppressing rate of responding, compared with the 1.4- to 4.0-fold difference observed with the other compounds.

Percentage drug-lever responding (means ± S.E.) and response rate (mean percentage of saline control ± S.E.) for a range of doses of typical benzodiazepine receptor agonists in squirrel monkeys (n = 3–4) trained to discriminate zolpidem (1.0 mg/kg) from saline. Dose-response functions were determined via cumulative-dosing procedures. Dotted lines represent the group mean (n = 4) for zolpidem-lever responding after test sessions with 1.0 mg/kg of zolpidem (training dose).

Percentage of drug-lever responding and response for a range of doses of typical benzodiazepine receptor agonists in squirrel monkeys (n = 3–4) trained to discriminate zolpidem (1.0 mg/kg) from saline. Other details as in Fig. 5.
Potencies (mean ED50) and number of monkeys reaching 90% drug-appropriate responding for zolpidem and typical BZ receptor agonists in zolpidem-trained squirrel monkeys
As can be seen in Figs. 5 (top) and 6 (top), none of the BZs reached an average maximum for percentage of drug-lever responding equal to the level engendered by zolpidem (90%, represented by the dotted lines in Figs. 5 and 6). This observation is most notable for the BZs shown in Fig. 6: lorazepam, chlordiazepoxide, and oxazepam engendered average maximums of 70, 61, and 37% drug-lever responding, respectively. All BZs suppressed responding at the highest doses to <30% of control. At the dose of 30 mg/kg oxazepam, responding was eliminated completely, and hematuria was noted in one monkey that persisted for ∼2 weeks after the injection.
Analysis of the data from individual monkeys suggested that the average maximums for percentage of drug-lever responding were the result of appreciable levels of zolpidem-appropriate responding for some monkeys, but not for others. To assess these individual differences quantitatively, the number of monkeys for which percentage of drug-lever responding fell within the lower CI for responding engendered by 1.0 mg/kg zolpidem was determined. Triazolam, midazolam, diazepam, and N-desmethyldiazepam engendered full substitution in three of four monkeys, one monkey in each case responded on the zolpidem lever below 50% (Table 2). For lorazepam, chlordiazepoxide, and oxazepam, only one or two monkeys showed full substitution, consistent with the finding that these drugs engendered lower average maximums for percentage of drug-lever responding than the other BZs. It is noteworthy that these results generally reflected different animals responding <90% after different drugs.
Effects of Barbiturates and Other Drugs.
Of the four barbiturates tested, none engendered zolpidem-lever responding up to doses that markedly suppressed responding (Fig.7). The average maximums for percentage of drug-lever responding engendered by pentobarbital, amobarbital, barbital, and methohexital were 1.0, 3.0, 36, and 18%, respectively. Cyproheptadine, baclofen, muscimol, buspirone, diphenhydramine, and flumazenil similarly did not engender a majority of responses on the zolpidem lever at any dose tested (Table3). All of these latter compounds suppressed response rate to less than or equal to 25% of control, except for muscimol, which suppressed the rate to 56% of control at the highest dose tested. Higher doses of muscimol were not tested due to previous reports of seizure-like activity in squirrel monkeys given this compound via the i.v. route (Spealman, 1985).
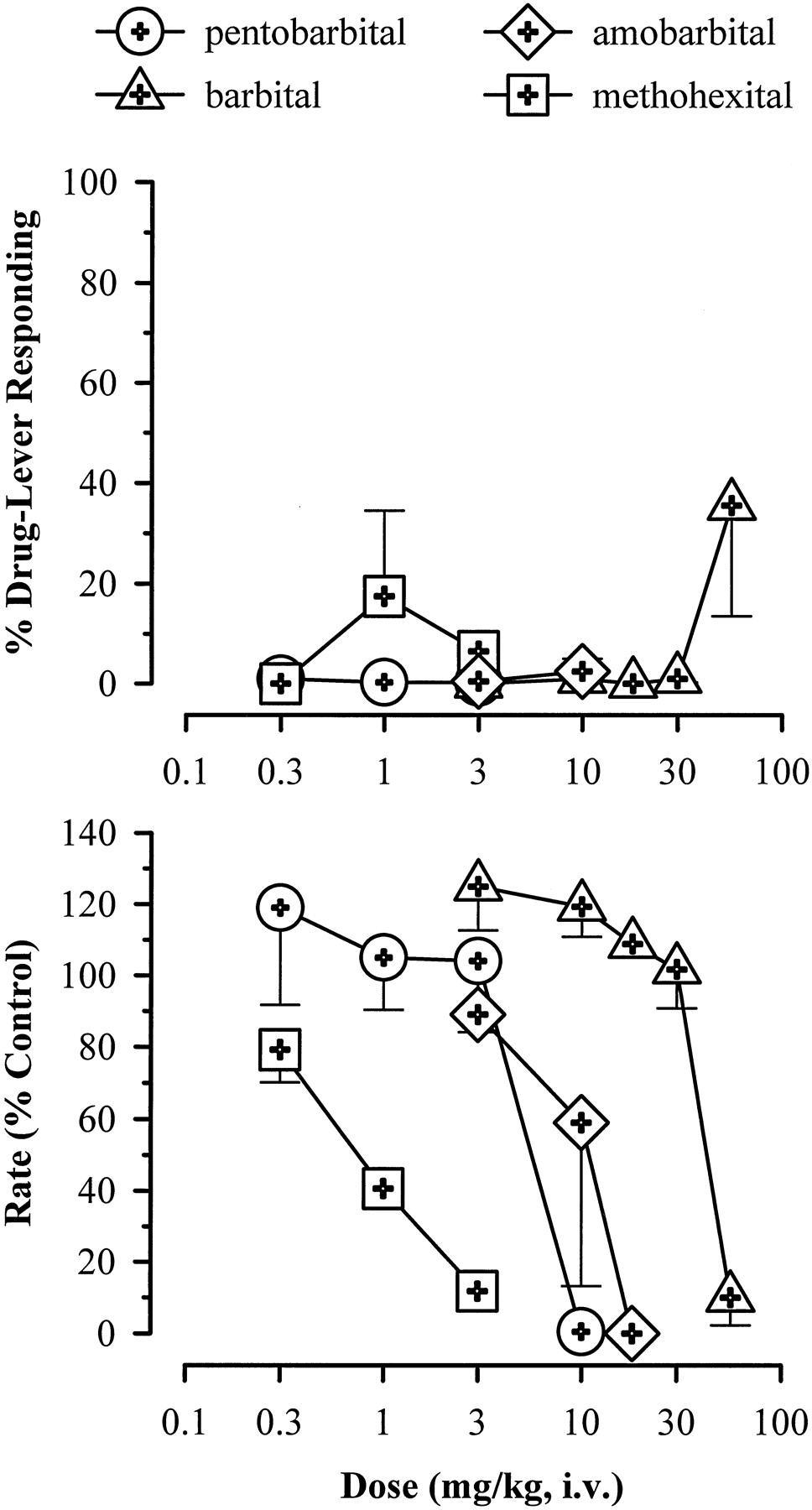
Percentage of drug-lever responding (means ± S.E.) and response rate (mean percentage of saline control ± S.E.) for a range of doses of barbiturates in squirrel monkeys (n = 3–4) trained to discriminate zolpidem (1.0 mg/kg) from saline. Dose-response functions were determined via cumulative-dosing procedures, except for methohexital, which was determined via conventional single-dosing procedures.
Maximum percentage of drug-lever responding and maximum suppression of response rate by drugs in zolpidem-trained squirrel monkeys
Discussion
The imidazopyridine hypnotic zolpidem was successfully established as a DS in nonhuman primates. The onset of the DS effects of zolpidem was rapid, and the effects dissipated quickly, consistent with zolpidem's reportedly rapid onset and short duration of action (Sanger and Zivkovic, 1986; Trenque et al., 1994; Rowlett and Woolverton, 1997). Direct comparison of the DS effects of zolpidem and the typical BZ midazolam (Spealman, 1985), with similar procedures and the same species, revealed similarities based on both antagonism and drug substitution profiles. With regard to antagonism, the DS effects of both zolpidem (present study) and midazolam (Spealman, 1985) were antagonized by flumazenil in a surmountable fashion. Schild analysis of the flumazenil data was consistent with competitive antagonism at a single receptor population (Tallarida et al., 1979), and both the in vivo apparent pA2 and pKB values for zolpidem were similar to pKB values for flumazenil antagonism of the DS effects of midazolam. These findings suggest that the same population of receptors played a significant role in mediating the DS effects of both drugs.
With the exception of oxazepam, all BZs evaluated in the present study engendered a dose-related increase in responding on the zolpidem lever, with full substitution observed in at least half the subjects studied. Similarly, effective doses of midazolam, diazepam,N-desmethyldiazepam, and chlordiazepoxide engendered nearly exclusive drug-lever responding in midazolam-trained squirrel monkeys (Spealman, 1985). Comparison of the four BZs evaluated in both studies revealed a similar rank order of potency for zolpidem and midazolam, further supporting the view that the DS effects of the two drugs were mediated similarly.
Comparisons also can be made between potencies to engender zolpidem-lever responding and binding affinities at certain subtypes of BZ receptors. The rank order of potency for zolpidem compared with triazolam, lorazepam, and diazepam in zolpidem-trained monkeys was more similar to the order for displacing in vivo [3H]flumazenil binding in the cerebellum (predominantly BZ1 sites) than in the spinal cord (predominantly BZ2 sites) of rats (Sanger and Benavides, 1993). Moreover, comparison of affinities of zolpidem and triazolam for displacing [3H]flumazenil from cloned GABAA receptors reveals that zolpidem is ∼50-fold less potent than triazolam at α1-containing receptors (Hadingham et al., 1993), similar to the ∼30-fold difference observed in the present study. By contrast, zolpidem was 600- to 22,000-fold less potent than triazolam at subunit combinations associated with the BZ2 site (Hadingham et al., 1993). Collectively, these results support the view that zolpidem's effects were mediated predominantly at the BZ1 receptor subtype. Furthermore, because of the similar rank order of potency of BZs in both zolpidem- and midazolam-trained monkeys, the DS effects of midazolam also may be mediated predominantly by BZ1 receptor stimulation.
Although there were similarities in the results from the zolpidem and midazolam training conditions, notable differences also were evident. One difference was the lack of full substitution by chlordiazepoxide, resulting from substantial intersubject variability in the maximum level of drug-lever responding. Recent evidence suggests that the DS effects of chlordiazepoxide may differ from other typical BZ receptor agonists. For example, chlordiazepoxide did not substitute fully in either rats or baboons trained to discriminate lorazepam (Ator and Griffiths, 1989, 1997). Moreover, Sanger and Benavides (1993) found a relationship between chlordiazepoxide-like DS effects and receptor binding affinities consistent with BZ2 rather than BZ1 activation. These findings raise the possibility that the DS effects of zolpidem and chlordiazepoxide differ with respect to their underlying transduction mechanisms, perhaps due to differential effects at BZ receptor subtypes (Depoortere et al., 1986; Sanger and Zivkovic, 1986, 1987; Sanger, 1987).
The finding that oxazepam and lorazepam failed to engender full substitution for zolpidem is more difficult to interpret. Previous drug discrimination studies have revealed no obvious differences between oxazepam and other typical BZ receptor agonists (Hendry et al., 1983;De la Garza et al., 1987), and lorazepam was found to share DS effects with zolpidem in both lorazepam-trained baboons (Griffiths et al., 1992) and zolpidem-trained rats (Sanger and Zivkovic, 1986). Available evidence also suggests that the binding profiles of oxazepam and lorazepam are similar to those of other typical BZ agonists, although lorazepam may have moderate selectivity for BZ1 versus BZ2 receptors (Sieghart and Schuster, 1984; Maksay et al., 1991; Sanger and Benavides, 1993). This latter finding, however, is difficult to reconcile with the failure of lorazepam to substitute fully for zolpidem in the present study. It is possible, therefore, that training zolpidem explicitly as a DS in monkeys reveals effects of oxazepam and lorazepam related to a unique interaction of these compounds with BZ receptor subtypes.
Based on substitution results with several reference GABA agonists and modulators, it seems unlikely that non-BZ sites associated with the GABA receptor system mediated the DS effects of zolpidem. This conclusion is based on findings that the barbiturates pentobarbital, barbital, amobarbital, and methohexital, as well as the direct GABAA agonist muscimol and the GABAB agonist baclofen, did not engender appreciable zolpidem-lever responding at any dose tested. Pentobarbital and barbital also did not substitute for midazolam in squirrel monkeys (Spealman, 1985; but see Lelas et al., 1999). Ator and colleagues have demonstrated that pentobarbital does not substitute for lorazepam in rats or baboons (Ator and Griffiths, 1989, 1997) or for a high dose of midazolam in rats (Sannerud and Ator, 1995a). Nonetheless, for most BZ training conditions, barbiturates and typical BZs share DS effects (for review, see Ator and Griffiths, 1989). Moreover, zolpidem engendered full substitution for pentobarbital in monkeys and human volunteers (Griffiths et al., 1992; Rowlett and Woolverton, 1997; Rush et al., 1997). Although the conditions under which barbiturates either do or do not mimic the DS effects of zolpidem and other BZ receptor agonists have not been characterized completely, the present results suggest that the shared DS effects of barbiturates and BZs, when observed, may reflect a prominent BZ2 component of action.
The DS effects of zolpidem were not mimicked by diphenhydramine, a histamine H1 antagonist often used as a sedative-hypnotic, or buspirone, a 5-HT agonist commonly prescribed as an anxiolytic. These findings suggest that there was little contribution of histamine H1 or 5-HT receptor mechanisms in zolpidem's DS effects. Interestingly, the 5-HT antagonist cyproheptadine engendered primarily saline-lever responding in the present study, but engendered up to 90% midazolam-lever responding in a study by Spealman (1985). Although the role of 5-HT mechanisms in the effects of midazolam is not well understood, it is possible that some of the differences in the DS effects of zolpidem and midazolam reflect either direct or indirect differences in the effects of the two drugs on 5-HT processes.
Previously, Sanger and Zivkovic (1986) noted that the DS effects of zolpidem in rats characteristically emerged only at doses that produced decreases in response rate, implying that the two effects may be mediated by similar mechanisms. As in rats, the training dose of zolpidem used in the present study reliably decreased response rate under both training and testing conditions. There is, however, evidence from our study that the DS and rate-decreasing effects of zolpidem involved different mechanisms. For example, the rank order of potency for BZ receptor agonists for engendering zolpidem-like DS effects differed from the rank order of potency of the same drugs for decreasing response rate. Moreover, Schild analysis revealed a lower pA2 value for antagonism of zolpidem's rate-decreasing effects than for antagonism of zolpidem's DS effects, as well as a slope estimate for the former that was different from −1.0. Slopes in Schild analyses may deviate from −1.0 for several reasons, including involvement of multiple receptor populations, lack of steady-state conditions, and interfering effects of the antagonist (for review, see Kenakin, 1997). Given that the slope did not differ from unity for the DS effects of zolpidem, and that flumazenil had no effects on response rate when tested alone, the latter two possibilities seem unlikely. Although necessarily speculative, the findings therefore suggest that the rate-decreasing effects, in contrast to the DS effects, of zolpidem may involve multiple receptor populations.
Previous studies with both monkeys and human volunteers suggest that the behavioral effects of zolpidem are in some respects similar to those of other BZ receptor agonists (Griffiths et al., 1992; Rush and Griffiths, 1996; Rowlett and Woolverton, 1997; Rush et al., 1997). Based on the present study, however, it is clear that zolpidem has a profile of DS effects in squirrel monkeys that differs from the profiles observed with other BZ receptor agonists, such as midazolam (Spealman, 1985; Lelas et al., 1999), a finding perhaps revealed only when zolpidem is trained explicitly as a DS. Along with emerging evidence of certain qualitative differences between zolpidem and typical BZs in subject-rated and DS effects in people (Evans et al., 1990; Mintzer et al., 1998), the present results suggest that the interoceptive effects of zolpidem are not identical with those of typical BZ receptor agonists. These differences could reflect the drug's greater selectivity at α1-subunit-containing GABAA receptors compared with most conventional BZ agonists.
Acknowledgments
We thank Dr. D. Platt for comments on an earlier version of this manuscript and E. Lipman for technical assistance.
Footnotes
-
Send reprint requests to: James K. Rowlett, Ph.D., Harvard Medical School, New England Regional Primate Research Center, One Pine Hill Dr., Box 9102, Southborough, MA 01772-9102.
-
↵1 This research was supported by U.S. Public Health Service Grants DA11792 and RR00168.
- Abbreviations:
- BZ
- benzodiazepine
- GABA
- γ-aminobutyric acid
- DS
- discriminative stimulus
- FR
- fixed ratio
- 5-HT
- 5-hydroxytryptamine
- CI
- confidence interval
- SEC
- standard error of the coefficient
- Received June 29, 1999.
- Accepted September 7, 1999.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}