


Abstract
The present study was undertaken to investigate the effects of specific inhibitors of calmodulin-dependent protein kinase II (CamKII) on macroscopic voltage-dependent K+ current (KV) recorded from rabbit portal vein smooth muscle cells. Inhibition of L-type Ca2+ current facilitation by 1 μM KN-62, a blocker of CamKII, was first demonstrated and provided evidence for functional CamKII activity in this preparation. KN-93, another specific and more potent inhibitor of CamKII in the rat brain, suppressed KVand enhanced the rate of inactivation in a dose-dependent manner, in cells dialyzed with both low (0.1 mM) and high (10 mM) EGTA pipette solution. Prolonged dialysis with 10 μM of a synthetic peptide inhibitor of CamKII (fragment 281–301) had little effect on KV and did not prevent the inhibitory action of KN-93 on the current. The estimated IC50 for inhibiting peak and late currents during 250-ms steps to +60 mV (holding potential = −60 mV) were 2.9 and 0.27 μM, respectively. KN-93 also induced slight shifts of the steady-state activation (−7 mV) and inactivation (−6 mV) curves. KN-62, and KN-92, an inactive analog of KN-93, produced effects similar to those of KN-93. In current clamp experiments, 5 μM KN-93 depolarized the myocytes from a control resting membrane potential of −42.3 ± 2.8 mV to −28.5 ± 1.4 mV, an effect that was partially reversible after washout (−34.4 ± 1.3 mV, n = 6). In conclusion, blockers of CamKII produce nonspecific inhibitory effects on KV that warrant cautious use of these compounds in physiological experiments designed to assess the role of CamKII.
Calmodulin-dependent protein kinase II (CamKII) is a multifunctional cytosolic enzyme that plays an important role in regulating cardiac and smooth muscle Ca2+ homeostasis and contractility. This enzyme is stimulated by the binding of the Ca2+–calmodulin complex after elevation of free intracellular calcium concentration ([Ca2+]i) and by a series of autophosphorylation steps (Singer et al., 1996). CamKII is known to play a major role in the facilitation of Ca2+ current during enhanced repetitive stimulations after a period of rest in cardiac and smooth muscle cells (McCarron et al., 1992; Yuan and Bers, 1994) and to increase the activity of the cardiac and skeletal muscle sarcoplasmic reticulum Ca2+-ATPase (Hawkins et al., 1994; Li et al., 1997) and Ca2+ release channel (Takasago et al., 1991; Li et al., 1997).
Four separate classes of K+ channels have so far been identified in vascular smooth muscle cells. These include large-conductance Ca2+-dependent K+ channels (KCa), voltage-dependent or delayed rectifier K+channels (KV), ATP-sensitive K+ channels, and inwardly rectifying K+ channels (Nelson and Quayle, 1995). Among these, it is well accepted that the 4-aminopyridine (4-AP)-sensitive KV current plays a prime function in regulating resting membrane potential and vascular tone, especially at low or resting levels of [Ca2+]i(Leblanc et al., 1994; Nelson and Quayle, 1995). It has been recently reported that KV, which may be the result of several components and at least two distinct molecular entities, namely KV1.5 (Overturf et al., 1994) and KV1.2 (Hart et al., 1993), can be up- and down-regulated by phosphorylation mediated by protein kinase A (Aiello et al., 1995) and C (Clement-Chomienne et al., 1996), respectively. It is unknown at the present time whether KVchannels are modulated by phosphorylation involving CamKII.
Several novel organic compounds have recently been developed to study the biochemical and functional properties of CamKII in different biological preparations. Among these, KN-93, a methoxybenzenesulfonamide compound, has been reported to be a highly specific inhibitor of CamKII in the brain, with little or no influence on the activity of PKA, PKC, and other important protein kinases (Sumi et al., 1991). As for KN-62, another CamKII inhibitor, KN-93 was reportedly shown to bind to the Ca2+-calmodulin domain of CamKII and thus prevent the stimulation of this enzyme (Tokumitsu et al., 1990; Sumi et al., 1991). In view of the important role of KV in controlling the resting membrane potential and vascular tone in vascular myocytes (Nelson and Quayle, 1995), our main objective was to investigate the effects of KN-93, KN-92, an analog of KN-93 bearing no influence on CamKII, and those of another CamKII blocker, KN-62, on macroscopic KVcurrents recorded in freshly dissociated rabbit portal vein smooth muscle cells at 35°C. We provide evidence that the two widely used inhibitors of CamKII, KN-93 and KN-62, both inhibit whole-cell KV with a potency well within the range of their respective effects on CamKII. These inhibitory actions do not appear to be mediated by CamKII, because they could still be observed with high intracellular Ca2+ buffering or mimicked by the inactive analog of KN-93, KN-92. These results have been presented in preliminary format (Leblanc and Chartier, 1998).
Materials and Methods
Single-Cell Preparation.
Isolated vascular smooth muscle cells were enzymatically dispersed from the rabbit portal vein as previously described (Leblanc and Leung, 1995). In brief, albino rabbits of either sex weighing 2.5 to 3 kg were sacrificed by an overdose of pentobarbital sodium injected through the ear vein. After exsanguination, the portal vein was removed and immediately immersed in cold (4°C) and well oxygenated physiological salt solution (PSS) containing: 120 mM NaCl, 15 mM NaHCO3, 4.2 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 0.01 mM CaCl2, and 5.5 mM dextrose (pH = 7.4 when bubbled with 95% O2-5% CO2). The portal vein was first pinned down at the bottom of a Petri dish layered with Sylgard and containing well oxygenated PSS. Under the view of a stereomicroscope (model SMZ-1B; Nikon Corp., Tokyo, Japan), the vessel was cut open along the longitudinal axis and was freed of blood, fat, and connective tissue by using fine dissecting tools. After dissection, the vein was cut into small pieces (2 × 2 mm), which were transferred into a small beaker containing ≈10 ml of a solution similar to that described above except that it was nominally Ca2+ free (no added CaCl2), and it contained the Ca2+ chelating agent EGTA (0.1 mM). After 30 min of incubation at room temperature, the tissue pieces were transferred in the PSS solution containing the following enzymes: collagenase (0.2 mg/ml, type 1A; Sigma Chemical Co., St. Louis, MO) and protease (50 μg/ml, type XXVII; Sigma Chemical Co.). This incubation was allowed to proceed at 35°C for 20 to 25 min, based on the number of cells released during trituration of a few pieces in low Ca2+ PSS by using a Pasteur pipette. Once a large number of cells was observed under microscopic examination, all pieces were rinsed several times in enzyme-free PSS and triturated to release single spindle-shaped smooth muscle cells. The supernatant containing the single cells was collected and kept in the cold (4°C) until use. All experiments were carried out within 4 to 8 h after isolation.
Electrophysiology.
Calcium-tolerant portal vein myocytes were either current or voltage clamped using the standard or perforated (nystatin) variant of the whole-cell patch clamp technique (Hamill et al., 1981). Large-diameter micropipettes were pulled by using a two-stage micropipette puller (model PP-83; Narishige Scientific Laboratory, Tokyo, Japan) and polished using a microforge (model FP-83; Narishige Scientific Laboratory). With tip diameters of about 1 μm, the pipette resistances were in the range of 2 to 4 MΩ when filled with the internal solutions described below. Voltage or current clamp protocols were computer driven using pClamp software (Version 5.5.1) and an Axopatch 200A integrating patch amplifier (Axon Instruments, Inc., Foster City, CA). Pipette and stray capacitances and series resistance were compensated for in all voltage clamp experiments. Membrane current was low-pass filtered at 1 or 2 kHz (four-pole bessel filter) before being acquired at a sampling rate of 2 or 5 kHz by using a PC-486 computer interfaced with a 12-bit analog-to-digital acquisition board (TL-125; Axon Instruments, Inc.). The resting membrane potential was measured in the current clamp mode by using the perforated patch clamp technique. The output voltage signal was filtered at 1 kHz and converted to digital format (sampled at 100 Hz) by using the same acquisition system and the Axotape software (Version 2.0; Axon Instruments, Inc.). Once digitized, the data were temporarily stored on the computer hard disk for later analysis (pClamp Version 6.0 or Axotape Version 2.0; Axon Instruments, Inc.) and display (Hewlett-Packard LaserJet series III).
Solutions.
With the exception of L-type Ca2+ currents that were recorded at room temperature (Fig. 1), all other experiments were carried out at 35°C. In voltage clamp experiments designed to measure K+ currents (Figs.2-8) and all current clamp protocols (Fig. 9), the solution used to superfuse the myocytes had the following composition: 130 mM NaCl, 10 mM NaHCO3, 4.2 mM KCl, 1.2 mM KH2PO4, 0.5 mM MgCl2, 1.8 mM CaCl2, 5.5 mM dextrose, and 5.0 mM HEPES-NaOH (pH 7.35). Except for the data presented in Figs. 1, 2, and 9, all other experiments were carried out in the presence of 1 μM nifedipine to inhibit L-type Ca2+ channels. To record L-type Ca2+ channels in isolation (Fig. 1), the same solution was used except that KCl and KH2PO4 were replaced by 5.4 mM tetraethylammonium chloride (TEA).

Evidence for functional CamKII activity in rabbit portal vein myocytes. A, selected L-type Ca2+ current (ICa) tracings recorded with the perforated patch technique during a train of 100-ms steps to 0 mV imposed at a rate of 0.5 Hz from HP of −70 mV, as depicted. Currents elicited during the first pulse for each condition were used to scale all currents to the same amplitude for comparison. Each train was preceded by a 2-min rest period while the myocyte was maintained at HP. For each condition, pulses 1, 4, and 30 are shown. Superimposed traces obtained in the absence (Control, leftward traces) and after 7 min of incubation with 1 μM KN-62. B, effects of KN-62 on the frequency dependence of ICa. For each condition, the magnitude of ICa currents recorded during a train of 32 pulses to 0 mV from HP = −70 mV (0.5 Hz) was normalized against that elicited during the first pulse. The graphs represent mean data of normalized currents from six experiments similar to that of A. Lines passing through the data points are least-square double-exponential fits of the following form:
 + C, where A1 andA2 are the amplitudes of the currents for the exponential components, τ1 and τ2 are the time constants for the two kinetic processes, and Cis a constant. The following parameters were derived for each set of data: Control: A1, .11; τ1, 2.29 pulses, A2, .15; τ2, 21.7 pulses, and C, .79; KN-62:A1, .38; τ1, 1.93 pulse,A2, .36; τ2, 52.2 pulses, andC, .43. All comparisons between Control and KN-62 data points revealed significant differences with at leastp < .05 (paired t test).
+ C, where A1 andA2 are the amplitudes of the currents for the exponential components, τ1 and τ2 are the time constants for the two kinetic processes, and Cis a constant. The following parameters were derived for each set of data: Control: A1, .11; τ1, 2.29 pulses, A2, .15; τ2, 21.7 pulses, and C, .79; KN-62:A1, .38; τ1, 1.93 pulse,A2, .36; τ2, 52.2 pulses, andC, .43. All comparisons between Control and KN-62 data points revealed significant differences with at leastp < .05 (paired t test).

Effects of KN-93 on the resting membrane potential recorded in current clamp mode using the perforated patch technique. A, typical experiment showing the effects of application and washout 5 μM KN-93 (indicated by the thick bar) on the resting membrane potential recorded in a whole-cell current clamped portal vein myocyte with the nystatin technique. B, bar graph displaying mean data of RMP derived from six experiments similar to that shown in A. Each data point is a mean ± S.E.M. **, significantly different from each other with p < .01; *, significantly different from each other with p < .05 (one-way ANOVA test).

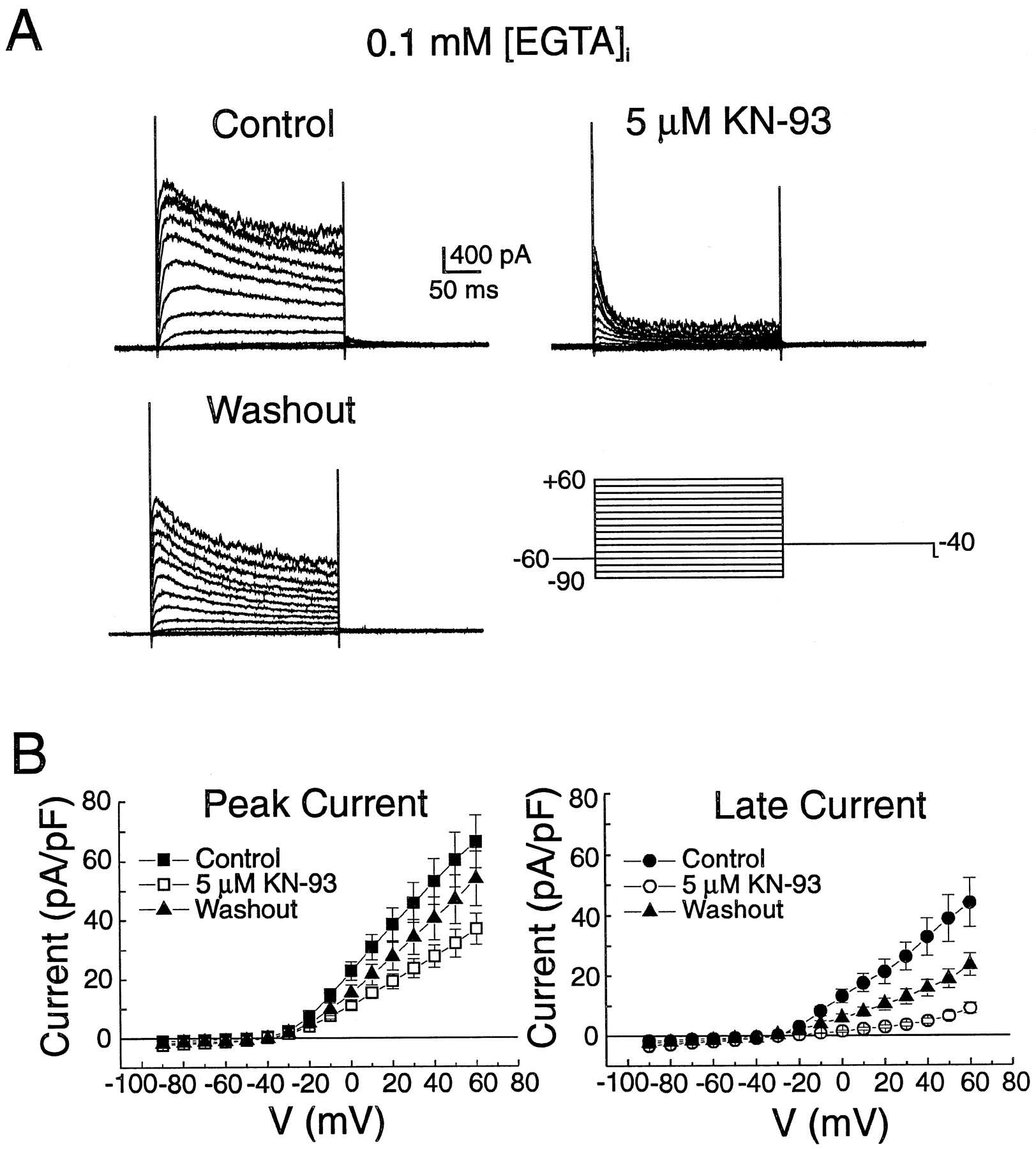
Effects of KN-93 on K+ currents recorded from portal vein smooth muscle cells dialyzed with low EGTA pipette solution. A, family of whole-cell K+currents recorded in control (Control, top left), presence of 5 μM KN-93 (top right) and after return to drug-free solution (Washout, bottom left). The currents were evoked by the protocol shown at the bottom right. B, I–V relationships for peak (left) and late (right) currents as in A in Control (▪, ●), in presence of 5 μM KN-93 (■, ○), and after Washout (▴). Each data point is a mean ± S.E.M. (n = 6). All comparisons for peak and late currents between Control and KN-93 revealed significant differences at potentials ≥ −20 mV with at least p < .05 (paired t test). Pipette solution contained 0.1 mM EGTA, as indicated.
The pipette solution used to record K+ currents (Figs. 2-8) was as follows: 110 mM K-gluconate, 30 mM KCl, 10 mM NaCl, 0.5 mM MgCl2, 5 mM ATP · Mg, 5.0 mM HEPES-KOH (pH 7.2), and 0.1 or 10 mM EGTA. In the perforated patch experiments to measure the resting membrane potential in current clamp mode (Fig. 9), EGTA and ATP · Mg were omitted from the above solution, and the pore-forming antibiotic nystatin was added [stock solution in dimethyl sulfoxide (DMSO), 60 mg/ml] to the pipette solution at a final concentration of 400 μg/ml. Whole-cell access was monitored under voltage clamp conditions by viewing the appearance of cell capacitative current transients elicited by repetitive 5 mV depolarizing steps (line frequency) from a holding potential of −60 mV. A stable access (in general series resistance <15 MΩ) was usually obtained within 5 to 10 min after formation of the gigaohm seal. The nystatin technique was also used to record L-type Ca2+ current (Fig. 1). For these experiments, the composition of the pipette solution was: 75 mM Cs2SO4, 55 mM CsCl, 5 mM MgCl2, and 10 mM HEPES-CsOH (pH 7.2).
KN-93 and KN-62 were dissolved in DMSO at a concentration of 10 mM. KN-92 was prepared as a 10-mM stock solution in distilled water. A 100-μM stock solution of the synthetic peptide inhibitor of CamKII (fragment 281–301; IC50 = 2 μM) was prepared by dissolving the powder in pipette solution; a small aliquot was subsequently diluted to reach the final concentration of 10 μM. The peptide inhibitor and all three KN compounds were purchased from Calbiochem (San Diego, CA). Nifedipine (Sigma Chemical Co.) was dissolved in DMSO at a concentration of 10 mM. The DMSO or water aliquots that were added to the PSS solution never exceeded 0.1%.
Statistical Analysis.
Except where original tracings of membrane currents or resting membrane potential are displayed, all data are reported as means ± S.E.M. Where appropriate, a paired Student’s t test was used to determine the difference between two groups. Comparisons between more than two groups were evaluated by a one-way ANOVA. Least-squares fits to mean data points by using a double-exponential formalism (Fig. 1) or the appropriate forms of the Boltzmann equation (Fig. 6) were performed by the Origin software (Version 4.1; Microcal Softwares, Inc., Northampton, MA). A probability p < .05 was accepted as the level of significance.

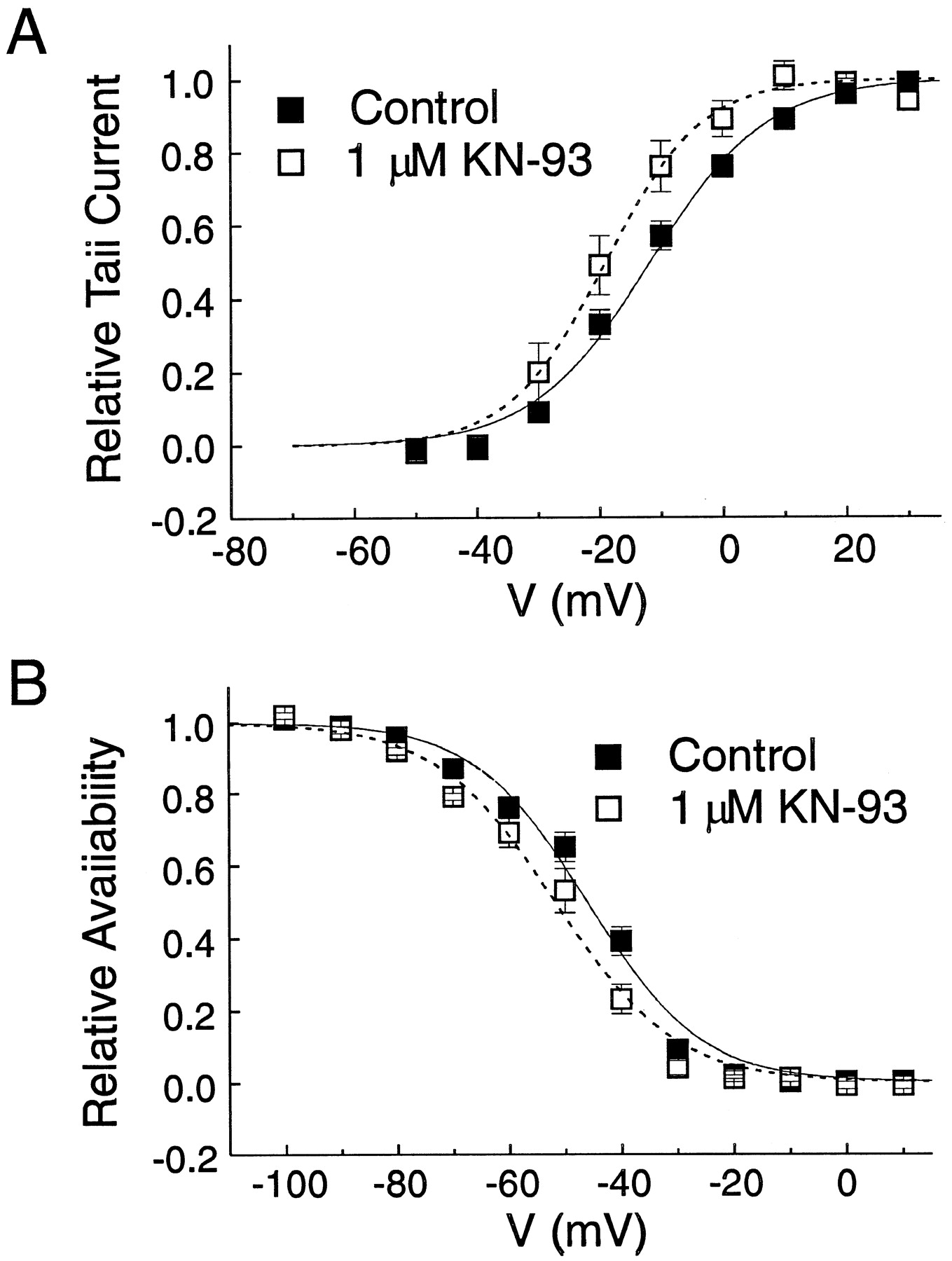
Effects of KN-93 on the steady-state activation and inactivation properties of KV. A, effects of 1 μM KN-93 on the steady-state activation curve of KV. From holding of −70 mV, 5-ms steps (P1) from −50 to +30 mV (10-mV increments) at a frequency of 0.2 Hz were used to activate KV. P1 was followed by a second constant pulse to −35 mV (P2) to record outward deactivating tail current. The amplitude of outward tail current was normalized against the maximum tail current amplitude and plotted as a function of the voltage step during P1. Each data point is a mean ± S.E.M. of normalized tail current (n = 5). The two lines are least-squares Boltzmann fits to the data using the following formulation: Y= {1 + exp[(V0.5 −V)/k]}−1, whereY is the value of the steady-state activation gating variable, V0.5 is the voltage at which KV is 50% activated, and k is the slope factor; in control: V0.5 = −11.9 andk = 9.2 mV; with KN-93:V0.5 = −19.0 mV, andk = 7.5 mV. B, effects of 1 μM KN-93 on the relative availability of KV. From holding potential of −70 mV, a 10-s variable conditioning step (P1) from −100 to −10 mV (10-mV increments) was applied at a rate of 0.1 Hz. After each of these pulses, a constant 5-ms pulse to −100 mV (P2) preceded a 45-ms step to +60 mV (P3) to record outward current. Amplitude of peak outward current recorded during P3 was normalized against the maximum outward current amplitude and plotted as function of the voltage step in P1. Each data point is a mean ± S.E.M. of normalized current (n = 5). The two lines are least-squares Boltzmann fits to the data using the following formulation: Y= {1 + exp[(V −V0.5)/k]}−1, where Y is the value of the steady-state inactivation gating variable, V0.5 is the voltage at which KV is 50% inactivated, and k is the slope factor; in control: V0.5 = −46.2 mV, and k = 9.6 mV; with KN-93:V0.5 = −51.8 mV, andk = 10.2 mV. Myocytes were dialyzed with 10 mM EGTA and exposed to 1 μM nifedipine.
Results
Activation of CamKII by elevation of [Ca2+]i has been demonstrated to up-regulate the activity of L-type Ca2+ current (ICa) in smooth (McCarron et al., 1992) and cardiac (Yuan and Bers, 1994) muscle cells. We took advantage of this relationship to provide evidence for functional CamKII activity in our preparation. Figure 1 shows the results of experiments illustrating the stimulation-dependence of ICa elicited by a train of 32 pulses to 0 mV from holding potential (HP) of −70 mV (0.5 Hz), in the absence (Control) and presence of 1 μM KN-62, a CamKII inhibitor. The more potent blocker of CamKII, KN-93, could not be used, because it inhibited ICa by more than 50%, possibly because of a direct effect on the channel. Cell exposure to 1 μM KN-62 for 5 to 7 min did not significantly influence ICa as monitored by repetitive depolarizations to 0 mV from HP = −70 mV applied every 20 to 30 s (n = 6); the amplitude of ICa in the absence and presence of KN-62 was −83 ± 28 and −76 ± 22 pA, respectively (p= .55). Each train of pulses was preceded by a 2-min rest period at the holding potential. Figure 1A shows selected ICacurrents at different times during each train. In this experiment, control ICa currents evoked by the first and fourth pulses were similar, whereas that triggered by the 30th pulse displayed a slightly reduced amplitude. In contrast, ICa declined more quickly and to a lower level in the presence of KN-62. Figure 1B shows plots of mean normalized currents for the stimulation-dependent changes of ICa in control and after incubation with 1 μM KN-62. In control condition, the decline of Ca2+current during the onset of stimulation can be explained, at least in part, by incomplete recovery from inactivation. It can be noticed that the current elicited during the second. pulse (arrow) was not significantly different from that induced during the first pulse (p = .55). The rate of decline of ICa was enhanced and the steady-state level reached at the end of the train was lower in the presence of the CamKII inhibitor. In contrast, in the presence of KN-92 (data not shown), an analog of KN-93 that does not influence CamKII, the time course of decline of ICa during a train paralleled that seen in the absence of drug (n = 4). These results are compatible with the idea that Ca2+ accumulation during repetitive opening of ICa exerts rapid positive feedback regulation of ICa likely by the involvement of a phosphorylating step mediated by CamKII, as proposed for cardiac and smooth muscle cells (McCarron et al., 1992; Yuan and Bers, 1994).
We then examined the effects on K+ currents measured in cells dialyzed with low EGTA pipette solution of a concentration of KN-93 known to produce a potent inhibition of CamKII in the brain (Ki = 370 nM; Sumi et al., 1991). Figure 2A shows typical families of membrane currents elicited by the protocol shown at the lower right, in control (top left), after incubation with 5 μM KN-93 (top right), and after washout (bottom left). In the control condition, the time-dependent outward current elicited by such a protocol is composed of two dominant K+ currents: 1) a putative voltage-dependent K+ current (KV) that displays slow inactivation at positive potentials and is sensitive to block by 4-AP (Beech and Bolton, 1989a; Hume and Leblanc, 1989; Miller et al., 1993), and a Ca2+-dependent K+ current (KCa) carried by large conductance maxi-KCa channels that are sensitive to low doses of TEA (≈1 mM), and to charybdotoxin (Beech and Bolton, 1989a) and iberiotoxin (Morales et al., 1996). As evident, KN-93 produced a potent inhibition of peak and late outward K+ currents at all potentials in the range of −20 to +60 mV. These effects were accompanied by marked increases in the rate of activation and inactivation. The KN-induced changes in outward K+ currents were partially reversible upon washout.
Figure 2B reports mean current–voltage (I–V) relationships for peak current (left graph) and current measured at the end of the 250-ms steps (right graph) in six experiments similar to that shown in Fig. 2A. KN-93 reduced peak and late currents, but the latter was more sensitive to block. For example, at +40 mV, KN-93 decreased peak (control: 53.0 ± 7.9 pA/pF; KN-93: 27.8 ± 3.83 pA/pF) and late (control: 32.8 ± 6.2 pA/pF; KN-93: 4.8 ± 0.9 pA/pF) by 48% and 85%, respectively. Consistent with the data described in Fig.2A, washout of KN-93 led to incomplete recovery of peak (51% at +40 mV) and late (39% at +40 mV) outward K+ current.
Because the above experiments did not allow us to distinguish the effects of KN-93 on KV and KCa, we carried out another series of experiments in conditions set to minimize the activity of KCaand record KV in isolation. The myocytes were dialyzed with 10 mM EGTA to buffer intracellular Ca2+ (<1 nM) and superfused with 1 μM nifedipine to block l-type Ca2+channels. Figure 3A illustrates the results of one typical experiment in which the effects of KN-93 were tested on whole-cell KV currents elicited by the voltage clamp shown at the lower right-hand side. Families of K+ currents (250 ms steps from −60 mV), which mainly consisted of transient voltage-dependent K+ currents (KV), were recorded in the absence of drug (Control), after 6 min of incubation with 5 μM KN-93, and 8 min after return to control solution (Washout). As for cells dialyzed with low EGTA, KN-93 inhibited KV at all step potentials and caused similar changes in activation and inactivation kinetics; both effects were partially reversible on drug removal.

Effects of KN-93 on the voltage-dependent K+ current. A, typical family of whole-cell voltage-dependent K+ currents recorded in absence of drug (Control), 6 min after incubation with 5 μM KN-93, and 8 min after washout. The current was elicited by the protocol shown at the lower right-hand side. B, average I–Vrelationships for peak (▪, ■) and late (●, ○) currents in absence (Control, ▪, ●) and in the presence of 5 μM KN-93 (■, ○). Each data point is a mean ± S.E.M. (n = 4). As for the data presented in Fig. 2B, all comparisons for peak and late currents between Control and KN-93 revealed significant differences at potentials ≥−20 mV with at least p< .05 (paired t test). Myocytes were dialyzed with 10 mM EGTA and incubated in the presence of 1 μM nifedipine.
The graph depicted in Fig. 3B reports the average data for theI–V relationships of peak (squares) and late (circles) KV currents measured in the absence (Control, filled symbols) and presence (empty symbols) of 5 μM KN-93. Because KCa activates in a time-dependent manner but does not inactivate, its contribution to total current should be greater at the end of a depolarizing step. Consistent with this proposal, the current at the end of steps was suppressed to a greater extent than peak current by enhanced intracellular Ca2+buffering with elevated EGTA concentration in the pipette solution (compare Figs. 2B, 3B, and 7, A and B). At +40 mV, peak current was 53.0 ± 7.9 and 34.2 ± 5.2 pA/pF with low- and high-EGTA pipette solutions, respectively, a difference near the limit of significance (p = .123); late current was 32.8 ± 6.2 and 15.8 ± 3.2 pA/pF (p = .011) with low- and high-EGTA pipette solutions, respectively. KN-93 blocked peak and late KV current but with more prominent effects on the latter. The magnitude of block of the peak and late KV current components by KN-93 displayed little voltage dependence for step potentials ranging from −20 to +60 mV (for peak and late current components, percentage block ranged from 50 to 54% and from 87 to 93%, respectively). The percentage of block produced by KN-93 in the presence of nifedipine and 10 mM EGTA was similar to that measured in control conditions (Fig. 2) for peak (+40 mV: 48 versus 54% for low and high EGTA, respectively) and late (+40 mV: 85 versus 92% for low and high EGTA, respectively).

The putative CamKII inhibitor KN-62 and KN-92, a structural analog of KN-93, also inhibit KV. (A, left) Sample traces of KV currents evoked by 250-ms steps to +60 mV in absence (Control, ▪) and in presence of 5 μM KN-62 (○). Right, mean I–V relationships (n = 4) for peak (▪, ■) and late (●, ○) outward currents elicited by protocol as in Figs. 2 and 3, in Control (▪, ●) and in the presence of 5 μM KN-62 (■, ○). All comparisons for late currents between Control and KN-62 revealed significant differences at potentials ≥0 mV with at leastp < .05 (paired t test). Comparisons for peak current indicated no significant difference at all potentials examined. (B, left) Sample traces of KV currents evoked by 250-ms steps to +60 mV in absence (Control, •) and in presence of 5 μM KN-92 (○). Right, meanI–V relationships (n= 9) for peak (▪, ■) and late (●, ○) currents elicited by protocol as in A, in absence (Control, ▪, ●) and presence of 5 μM KN-92 (■, ○). All comparisons for peak and late currents between Control and KN-92 revealed significant differences at potentials ≥0 mV with at least p < .05 (paired ttest). C, bar graph reporting the relative potency of block by KN-93 (expressed as percentage of block) of peak and late KVmeasured at +60 mV. Myocytes were dialyzed with 10 mM EGTA and exposed to 1 μM nifedipine.
Separate experiments were performed to quantify the potency of KN-induced block of KV. Figure4A shows superimposed tracings of KV currents elicited by the voltage clamp protocol shown at the bottom. The currents were recorded in the same cell in the absence (Control) and presence of 0.1, 0.5, 1, and 10 μM KN-93, as depicted. As shown above, KN-93 inhibited both peak and late currents, but it was more potent on late current. It is also evident that KN-93 produced an apparent increase in the rate of development of inactivation, an effect that was also dose dependent.

Concentration-dependent inhibition of KVby KN-93. A, whole-cell KV currents elicited by the protocol shown at the bottom were all recorded in the same cell in absence (Control, ●) and presence of 0.1, 0.5, 1, and 10 μM KN-93, as depicted by the various open symbols. B, concentration–response relationships expressed in percentage of block of KV for peak (▪) and late (○) outward currents recorded at +60 mV. Each data point is a mean ± S.E.M. (n = 6). The two smooth lines for the block of peak and late KV currents by KN-93 were obtained by nonlinear least-squares fitting of the mean data points to a Logistic function of the following form:Y = 100/{1 + (IC50/[KN-93])p}, whereY represents percentage block, [KN-93] is the concentration of KN-93 in μM, IC50 is the concentration (in micromolar concentrations) of KN-93 producing 50% inhibition of the current, and p is the power factor. For peak current (▪), the IC50 (as labeled) and p values are 2.9 μM and .64, respectively, and for late current (○), the IC50 (as labeled) and p parameters are .27 μM and 1.301, respectively. Myocytes were dialyzed with 10 mM EGTA and exposed to 1 μM nifedipine.
Figure 4B shows the concentration–response relationships for KN-93-induced block of KV for peak (filled squares) and late (empty circles) currents recorded at +60 mV. The two smooth lines are nonlinear least-squares fits to a Logistic function. As evident from the IC50 values indicated, KN-93 was about 10 times more potent at inhibiting late versus peak currents. These results show that KN-93 inhibits KV in a dose-dependent manner.
Figure 5 displays graphs illustrating the voltage dependence of the fast (τfast, A) and slow (τslow, B) time constants of inactivation in the absence (Control, filled squares) and presence (empty circles) of 5 μM KN-93. In the absence of drug, both τfast and τslowdecreased from −10 to +10 mV but displayed little, if any, voltage dependence at potentials ≥+20 mV. KN-93 produced an ≈2- to 3-fold reduction of τfast and τslow.

Effects of KN-93 on inactivation kinetics of KV (A and B). Voltage dependence of the fast (τfast, A) and slow (τslow, B) time constants of inactivation in control (▪) and in presence of 5 μM KN-93 (○). For A and B, all comparisons revealed significant differences between time constants measured in Control and KN-93 at all potentials with p < .05 (paired ttest). Myocytes were dialyzed with 10 mM EGTA and exposed to 1 μM nifedipine.
To obtain information on the possible mechanism of block of KV by KN-93, we also examined its effects on the steady-state activation and inactivation properties of KV. For these experiments, a lower concentration of KN-93 (1 instead of 5 μM) was tested to record larger KV currents to facilitate the analysis. A double-pulse protocol was used to construct the steady-state activation curves shown in Fig. 6A. From holding potential of −70 mV, 5-ms steps (P1) from −50 to +30 mV were applied in 10-mV increments at a frequency of 0.2 Hz to activate KV; each of these steps was followed by a constant pulse (P2) to −35 mV to record outward tail current. The amplitude of each tail current recorded during P2 was normalized against the maximum tail-current amplitude and plotted as a function of the voltage step during P1. KN-93 shifted the activation curve by −7 mV (V0.5: from −12 ± 2 to −19 ± 3 mV, p < .05) and enhanced the steepness of the relationship (slope factor k: from 8.8 ± 0.4 to 6.3 ± 0.5 mV, P< .01).
A three-step protocol was used to construct the steady-state inactivation curve of KV in control and presence of 1 μM KN-93. From HP = −70 mV, the membrane was first conditioned for 10 s to potentials varying from −100 to +10 mV (P1) applied in 10-mV increments at a rate of 0.1 Hz; this was followed by a constant 5-ms step to −100 mV (P2), itself followed by a 45-ms step to +60 mV (P3) to record KV. The amplitude of KV during P3 for each conditioning voltage step was normalized against the maximum amplitude of KV and plotted against P1 voltage. KN-93 shifted the steady-state inactivation curve by −6 mV (V0.5: from −45 ± 2 mV to −51 ± 3,p < .05) with little effect on the slope of the relationship (slope factor k: from 9.3 ± 1.1 to 9.4 ± 1.3 mV, p = .86).
We also explored whether the effects on KVreported above are unique to KN-93. Fig.7 reports the effects of another blocker of CamKII, KN-62, and of KN-92, the structural analog of KN-93. Figure7A shows sample recordings of whole-cell currents elicited by the protocol shown at the bottom and obtained in control condition and after exposure to 5 μM KN-62. Although less potent than KN-93, KN-62 also inhibits KV in a manner similar to that produced by KN-93: 1) the current at the end of the step was more sensitive to block by KN-62, and 2) the kinetics of inactivation were accelerated in the presence of the drug. To the right of Fig. 7A are depicted the mean I–V relationships for peak (squares) and late (circles) currents recorded in response to a protocol identical with those described in Figs. 2 and 3, in the absence (Control) and presence of 5 μM KN-62. On average, KN-62 did not significantly reduce peak current, although a small tendency was noticeable (≈10% at +60 mV). However, the CamKII inhibitor produced 40% inhibition of late current at +60 mV.
Figure 7B shows that 5 μM KN-92 produced a more potent inhibition of KV than KN-62. The effects of KN-92 on inactivation kinetics were also more prominent than those caused by KN-62. Figure 7C shows a bar graph comparing the potency of inhibition of KV at +60 mV by KN-93, KN-62, and KN-92 at a concentration of 5 μM. These results indicate a similar profile of inhibition of peak and late currents with the following order of potency: KN-93 > KN-92 > KN-62. In summary, KN-62 inhibits and affects the kinetics of KV in a manner similar to KN-93, although less potent than the latter at equivalent concentrations.
Although it appears unlikely that KN-93 and KN-62 influenced KV through an inhibitory effect of CamKII in myocytes dialyzed with high EGTA, we nevertheless further tested this hypothesis by examining the effects of cell dialysis with a synthetic peptide fragment (281–301) that inhibits CamKII by interacting with the calmodulin-binding domain of CamKII (IC50 = 2 μM). Over a 15-min period, cell dialysis with a pipette solution containing 10 μM of the CamKII peptide inhibitor produced little, if any, effect on the amplitude and kinetics of KV(Fig. 8A, upper and lower left families of currents). Under these conditions, cell exposure to 5 μM KN-93 for 5 min decreased KV in a manner consistent with that described previously in Figs. 2, 3, and 7 (Fig. 8A, bottom right traces). Figure 8B reports mean I–V relationships for peak (left graph) and late (right graph) KV currents recorded after 3 and 15 min of cell dialysis and after a subsequent exposure and washout of KN-93. The KN-93-induced block of KV does not appear to be influenced by the presence of the inhibitor.

Effects of KN-93 on KV in the presence of a peptide inhibitor of CamKII. A, families of whole-cell KVcurrents recorded after 3 min (upper left), 10 min (upper right), and 15 min after membrane rupture. The cell was dialyzed with 10 mM EGTA and 10 μM of the synthetic peptide inhibitor of CamKII (fragment 281–301) and incubated with 1 μM nifedipine. The voltage clamp protocol is displayed at the lower left-hand side. The currents at the bottom right-hand side were obtained after 5-min exposure to 5 μM KN-93 or 20 min of cell dialysis. B, mean I–Vrelationships for peak (left graph) and late (right graph) KV currents derived from four experiments similar to that described in A. Pooled data for currents measured after 3 min (■) and 15 min (▪) of cell dialysis and after exposure to 5 μM KN-93 (●, ≈20 min of cell dialysis) and washout of the drug (▴, ≈25 min of cell dialysis). The data at 10 min of cell dialysis are not shown for the sake of clarity. All comparisons between 3 and 15 min revealed no statistically significant difference. All other comparisons showed significant differences for currents elicited by steps ≥−10 mV.
Because of the known important function of KV in regulating resting membrane potential (RMP) in vascular smooth muscle cells, our data would predict that KN-93 may induce membrane depolarization. We directly tested this hypothesis by examining the effects of KN-93 on RMP of portal vein myocytes measured in current clamp mode with the perforated patch clamp technique. Figure9A shows a sample trace of a typical membrane potential recording before, during, and after exposing the myocyte to 5 μM KN-93. In the absence of the compound, a stable RMP of −44 mV was recorded. Application of KN-93 induced a 13-mV depolarization to −31 mV. As for the effects of KN-93 on KV, RMP only partially recovered after washout of the drug (−39 mV). Similar effects were consistently observed in a total of six experiments, and mean data are shown in Fig. 9B.
Discussion
Our study reports for the first time that KN-93, a specific inhibitor of CamKII, is a potent blocker of voltage-dependent K+ current in vascular smooth muscle cells. The effects of KN-93, as those of the other CamKII inhibitor KN-62, appeared to be independent of changes in [Ca2+]i and CamKII activity. These compounds were more potent at inhibiting late versus peak current during step depolarizations and they all enhanced the rate of inactivation of KV. Finally, current clamp experiments revealed that 5 μM KN-93 reversibly depolarized portal vein smooth muscle cells by ≈15 mV, a result consistent with a KN-93-induced block of KV.
KN-93 Primarily Inhibits Voltage-Dependent K+Channels.
Four distinct K+ currents have been described in freshly isolated rabbit portal vein smooth muscle cells: 1) an iberiotoxin- or TEA-sensitive Ca2+-dependent K+ current (KCa) that is carried by large conductance K+ channels (≈200 pS in symmetrical K+ gradient; Beech and Bolton, 1989a; Hume and Leblanc, 1989; Miller et al., 1993; Morales et al., 1996); 2) a fast transient voltage-dependent outward K+ current (Ito) (Beech and Bolton, 1989b); 3) a 4-AP-sensitive voltage-activated delayed rectifier K+ current (KV); and 4) a glibenclamide-sensitive voltage-independent ATP- and GDP-sensitive K+ current (KATP) (Kajioka et al., 1991).
Because Ito represents a very small fraction of total outward peak current and will be completely inactivated at the end of 250-ms test pulses and near RMP in current clamp (Beech and Bolton, 1989b), it appears unlikely that the major effects of either KN compounds on outward K+ current during step or ramp protocols or membrane potential in current clamp are caused by block of this current. Because the three compounds all inhibited a voltage- and time-dependent outward current, a potential block of KATP can be ruled out on the basis of the following arguments: 1) KATP channels are voltage- and time-independent channels (Kajioka et al., 1991); and 2) cells in our experiments were dialyzed with 5 mM ATP, which would lead to a strong inhibition of KATP (Kajioka et al., 1991).
Although we cannot rule out the possibility that KN-93 may inhibit KCa channels, several lines of evidence suggest that the major target of KN-93, KN-62, and KN-92, is the voltage-dependent delayed rectifier K+ current. Inhibition of Ca2+ entry through ICa and intracellular buffering of Ca2+ by EGTA significantly reduced outward current, a result consistent with an effective inhibition of Ca2+-dependent outward current that is mainly composed of KCa; for example, outward current measured at the end of the step was reduced 76% at 0 mV and 81% at +60 mV when the EGTA concentration was increased from 0.1 to 10 mM. If KCa was the current predominantly affected by KN-93, EGTA-induced buffering of intracellular Ca2+ would tend to abolish or reduce its inhibitory action on total membrane current. However, opposite to this proposal, the inhibition of outward K+ current by KN-93, if anything, tended to be more potent in cells dialyzed with 10 versus 0.1 mM EGTA.
Furthermore, a significant block of outward current was apparent at voltages as negative as −20 mV, a voltage at which the contribution of KCa current would be very small in cells buffered with high EGTA (Beech and Bolton, 1989a; Miller et al., 1993), because lowering [Ca2+]i induces a rightward shift of the steady-sate activation curve of KCa (Carl and Sanders, 1989). A second argument in favor of a block of KV is the fact that KN-93 influenced a K+ current exhibiting slow voltage-dependent inactivation, a property that is characteristic of KV (Beech and Bolton, 1989a; Remillard and Leblanc, 1996) but not of KCa (Beech and Bolton, 1989a; Carl and Sanders, 1989; Leblanc et al., 1994; Morales et al., 1996). We therefore conclude that the major target of all three KN compounds tested is the voltage-dependent delayed rectifier K+ current.
Finally, application of 1 mM TEA, a potent blocker of KCa in smooth muscle cells (Nelson and Quayle, 1995), had little effect on the resting membrane potential of rabbit portal vein smooth muscle cells dialyzed with 0.1 mM EGTA (Miller et al., 1993). In contrast, 2 mM 4-AP, which inhibits KV, induces a depolarization (≈10 mV; ref.Miller et al., 1993) that is equivalent to that elicited by 5 μM KN-93 in the present study (13 mV).
Potential Mechanisms of Block of KV by KN-93.
Ca2+-calmodulin and CamKII have been reported to regulate ion channel activity in several tissues. Activation of CamKII during repetitive stimulations that elevate [Ca2+]i is responsible for the positive feedback control of Ca2+-induced enhancement of Ca2+ channels in rabbit cardiac (Anderson et al., 1994), toad stomach (McCarron et al., 1992), and rabbit portal vein (this study) smooth muscle cells and inactivation of Ca2+-dependent Cl−channels in equine tracheal smooth muscle cells (Wang and Kotlikoff, 1997). Certain types of Ca2+-dependent K+ channels have been reported to be activated by Ca2+-calmodulin in renal medulla (Klaerke et al., 1987) and neurones (Onozuka et al., 1987). In contrast, other studies indicated that the inhibition of K+ channels mediated by calmodulin antagonists (McCann and Welsh, 1987; Kihira et al., 1990) may be caused by a direct interaction with the channels, thus ruling out a role for Ca2+-calmodulin in the gating process. In our study, KN-93 dose-dependently blocked KV current with a potency (IC50 = 270 nM for inhibition of late current) that was similar to its inhibitory action on rat brain CamKII (Ki = 370 nM; Sumi et al., 1991), and this effect was shown to be independent of [Ca2+]i because cell dialysis with 10 mM EGTA could not prevent the inhibition. Although we cannot rule out the possibility that EGTA may have been unable to effectively lower subsarcolemmal Ca2+ levels or that a Ca2+-independent form of regulation of KV by CamKII might be involved (Sumi et al., 1991; Singer et al., 1996), a role for CamKII seems unlikely because KN-92, an inactive substitute of KN-93 often used as a negative control, induced similar effects on KV. Moreover, and in contrast to a preliminary study in murine colonic smooth muscle cells (Koh et al., 1999), prolonged cell dialysis with a specific peptide inhibitor of CamKII did not influence the magnitude and kinetics of KV and failed to prevent the KN-93-induced inhibitory action on this current.
Analysis of the amount of block produced by KN-93 revealed little voltage dependence. These results suggest that binding of KN-93 is not influenced by the transmembrane electric field and that the block may involve a state-dependent mechanism. The more potent reduction of late versus peak currents, combined with the apparent acceleration of inactivation, is consistent with an open-state mechanism of block whereby binding of the drug to the channel is enhanced during transition from closed to open states. Similar effects were reported for the inhibition of the cloned K+ channels KV1.5 and KV1.2 by clofilium (Malayev et al., 1995) and 4-AP (Russell et al., 1994b;Yamane et al., 1995), respectively. A similar leftward shift of the inactivation curve to that produced by KN-93 was also reported for the block of KV1.5 by 4-AP (Yamane et al., 1995). Opposite to our study, a closed-state interaction of 4-AP shifted the steady-state activation and inactivation curves to more positive potentials and slowed activation kinetics of delayed rectifier K+ current recorded in rabbit coronary myocytes without influence on inactivation and recovery kinetics (Remillard and Leblanc, 1996).
A limitation of our study is that our experimental approach does not allow us to identify the K+ channel subunit(s) that might be influenced by KN-93 and related compounds. It is becoming increasingly evident that macroscopic voltage-dependent K+ currents of smooth muscle cells can be separated into multiple components (Carl, 1995) whose biophysical and pharmacological profiles may depend on the functional expression of several distinct K+ channel subunits that may coassemble into homotetramers or heterotetramers of various composition (Hart et al., 1993; Overturf et al., 1994; Yuan et al., 1998) and proportions (Russell et al., 1994a). The rabbit portal vein expresses at least two members of the Shaker or Kv1 subfamily of K+ channel genes, predominantly KV1.5 (Overturf et al., 1994) and smaller levels of KV1.2 (Hart et al., 1993) and possibly other subfamily members. Future experiments should be designed to test the effects of KN-93 on the various K+ channel clones that are expressed in native smooth muscle cells to delineate the target and specific mechanisms of interaction.
In conclusion, the CamKII inhibitors KN-93 and KN-62 and the inactive form of KN-93, KN-92, all inhibit voltage-dependent K+ current of vascular smooth muscle cells with the following order of potency: KN-93 > KN-92 > KN-62. Our study urges caution when using these compounds to assess the role of CamKII in physiological experiments, because they have the ability to depolarize the myocytes by a direct CamKII-independent inhibition of voltage-dependent K+ channels in the voltage range favoring enhanced Ca2+ entry throughl-type Ca2+ channel activity and contraction. However, our study may pave the way for developing new therapeutic agents designed to modulate the function of KV and its contribution to the resting membrane potential and tone. Indeed, it should be emphasized that KN-93 is more potent than 4-AP at blocking sustained KV by at least two orders of magnitude.
Acknowledgments
We thank Marie-Andrée Lupien for her technical assistance in isolating vascular smooth muscle cells and preparing solutions and Dr. Marc Courtemanche for fruitful discussions related to the possible mechanisms of block of KV by KN-93.
Footnotes
-
Send reprint requests to: Normand Leblanc, Ph.D., Research Centre, Montreal Heart Institute, 5000 East Bélanger St., Montréal, Québec, Canada H1T 1C8. E-mail:leblancn{at}alize.ere.umontreal.ca.
-
↵1 This work was supported by grants awarded to N.L. from the Heart and Stroke Foundation of Québec, the Medical Research Council of Canada, and funds from the Fonds pour la Formation de Chercheurs et l’Aide à la Recherche (FCAR) and Montreal Heart Institute. N.L. is a Fonds de la Recherche en Santé du Québec Senior Scholar.
- Abbreviations:
- CamKII
- calmodulin-dependent protein kinase II
- HP
- holding potential
- 4-AP
- 4-aminopyridine
- [Ca2+]i
- free intracellular calcium concentration
- DMSO
- dimethyl sulfoxide
- ICa
- L-type calcium current
- Ito
- transient outward K+current
- KATP
- ATP-dependent K+ channels
- KCa
- Ca2+-dependent K+ channels
- KV
- voltage-dependent K+ channels
- PSS
- physiological salt solution
- RMP
- resting membrane potential
- TEA
- tetraethylammonium chloride
- Received November 4, 1998.
- Accepted May 19, 1999.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}